
Hallmarks for Thrombotic and Hemorrhagic Risks in Chronic Kidney Disease Patients


Abstract
:1. Introduction
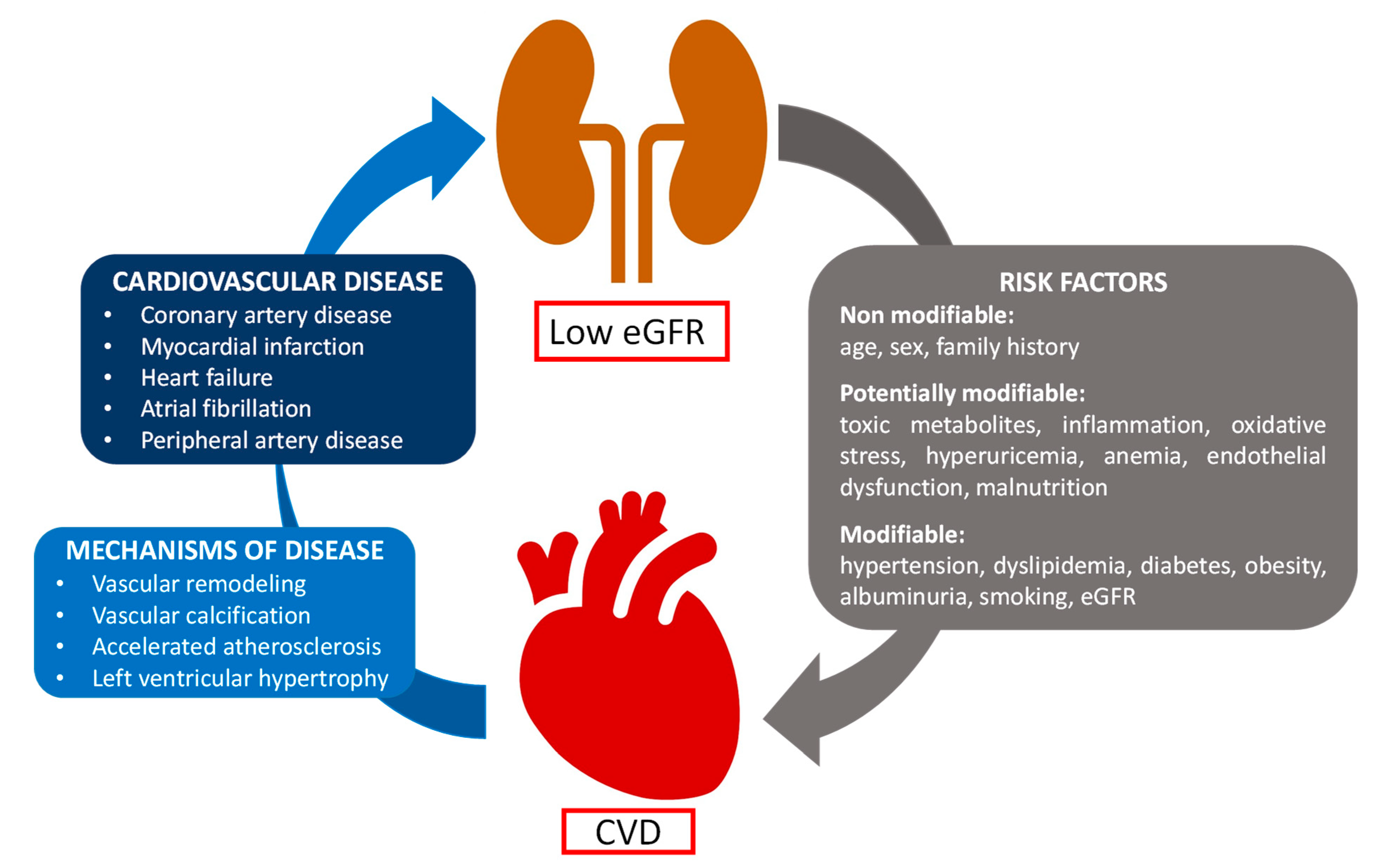
2. Cardiovascular Diseases in CKD
3. Mechanisms of Hemostatic Dysfunction in CKD
3.1. Endothelial Dysfunction
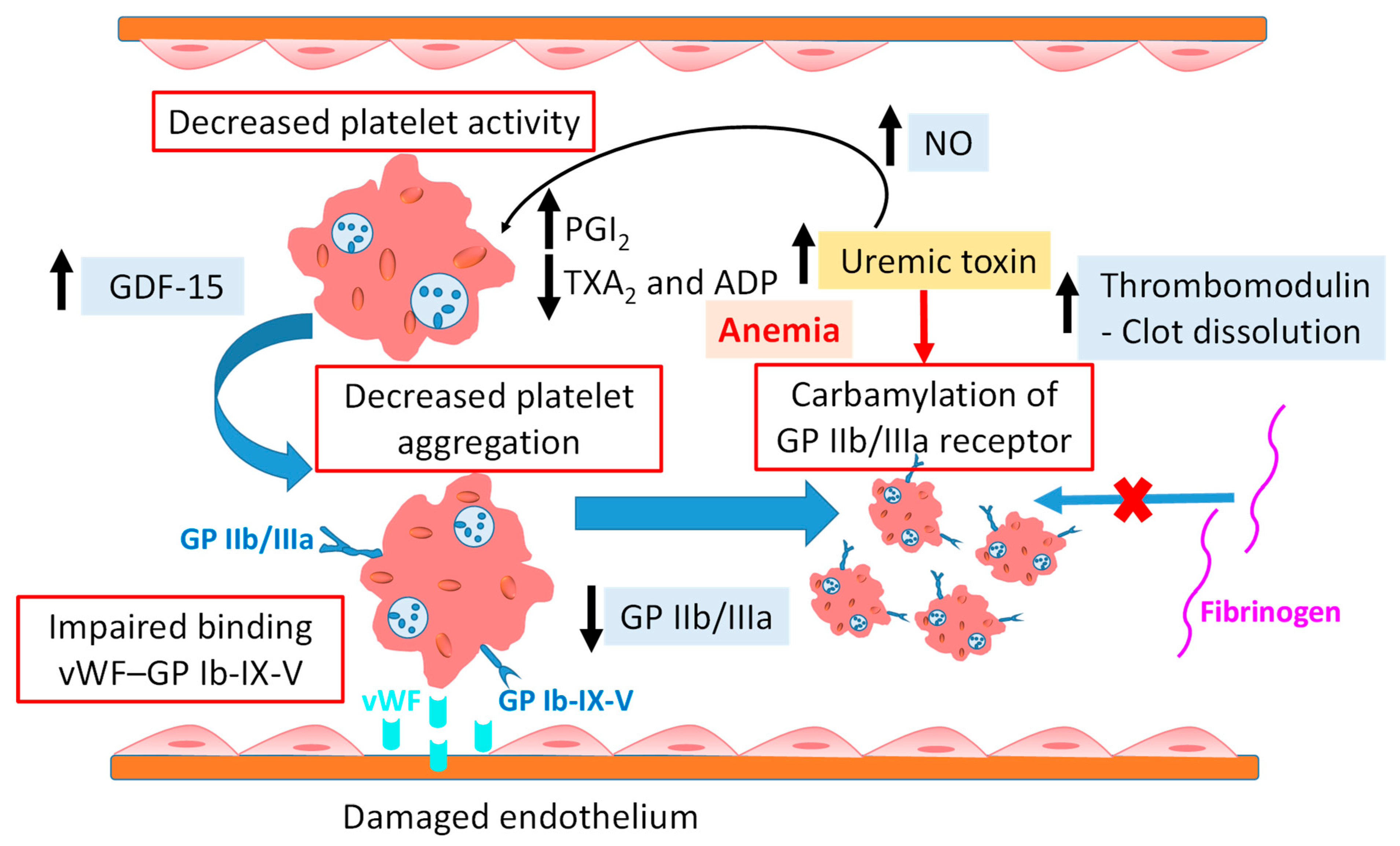
3.2. Platelet Dysfunction
3.3. Microparticles
3.4. Coagulation Abnormalities
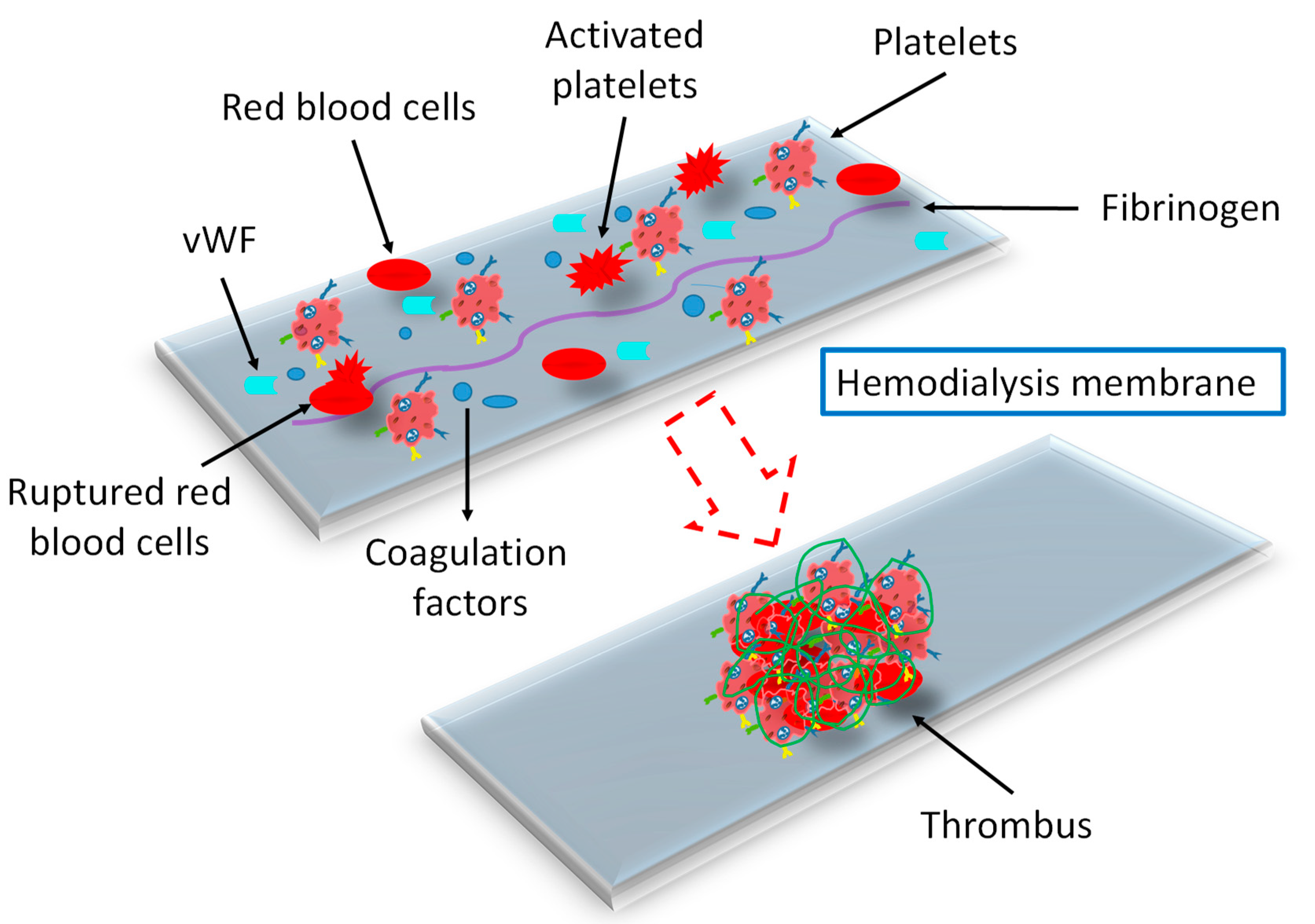
3.5. Effect of Dialysis Membranes and Modalities
3.6. Comorbidities
4. Strategies to Mitigate Thrombosis in CKD
4.1. Antiplatelet Therapy
4.2. Anticoagulant Therapy
4.3. Biocompatible Hemodialyzer Membrane
5. Bioavailability of Drugs
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, T.K.; Knicely, D.H.; Grams, M.E. Chronic Kidney Disease Diagnosis and Management: A Review. JAMA 2019, 322, 1294–1304. [Google Scholar] [CrossRef]
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; Francisco, A.L.M.D.; Jong, P.E.D.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Lamb, E.J.; et al. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar] [CrossRef]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Int. 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide Access to Treatment for End-Stage Kidney Disease: A Systematic Review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Yu, A.S.; Pak, K.J.; Zhou, H.; Shaw, S.F.; Shi, J.; Broder, B.I.; Sim, J.J. All-Cause and Cardiovascular-Related Mortality in CKD Patients with and without Heart Failure: A Population-Based Cohort Study in Kaiser Permanente Southern California. Kidney Med. 2023, 5, 100624. [Google Scholar] [CrossRef] [PubMed]
- Gäckler, A.; Rohn, H.; Lisman, T.; Benkö, T.; Witzke, O.; Kribben, A.; Saner, F.H. Evaluation of Hemostasis in Patients with End-Stage Renal Disease. PLoS ONE 2019, 14, e0212237. [Google Scholar] [CrossRef] [PubMed]
- Mitic, B.P.; Dimitrijevic, Z.M.; Hosokawa, K.; Cvetkovic, T.P.; Lazarevic, M.V.; Tasic, D.D.; Jovanovic, A.; Jancic, N.; Vrecic, T.; Ågren, A.; et al. Platelet Thrombus Formation in Patients with End-Stage Renal Disease before and after Hemodialysis as Measured by the Total Thrombus-Formation Analysis System. Int. Urol. Nephrol. 2022, 54, 2695–2702. [Google Scholar] [CrossRef]
- Gomchok, D.; Ge, R.L.; Wuren, T. Platelets in Renal Disease. Int. J. Mol. Sci. 2023, 24, 14724. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Xie, R.; Zhang, Y.; Liang, H.; Hou, L.; Yu, C.; Zhang, J.; Dong, Z.; Tian, Y.; Bi, Y.; et al. Phosphatidylserine on Microparticles and Associated Cells Contributes to the Hypercoagulable State in Diabetic Kidney Disease. Nephrol. Dial. Transplant. 2018, 33, 2215–2227. [Google Scholar] [CrossRef]
- Kaw, D.; Malhotra, D. HEMATOLOGY: ISSUES IN THE DIALYSIS PATIENT: Platelet Dysfunction and End-Stage Renal Disease. Semin. Dial. 2006, 19, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Almajdi, A.; Almutairi, S.; Alharbi, M. Safety and Efficacy of Apixaban versus Low-Molecular Weight Heparin or Vitamin-K Antagonists for Venous Thromboembolism Treatment in Patients with Severe Renal Failure: A Systematic Review and Meta-Analysis. Thromb. Res. 2023, 229, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Natale, P.; Palmer, S.C.; Saglimbene, V.M.; Ruospo, M.; Razavian, M.; Craig, J.C.; Jardine, M.J.; Webster, A.C.; Strippoli, G.F.M. Antiplatelet Agents for Chronic Kidney Disease. Cochrane Database Syst. Rev. 2022, 2022, CD008834. [Google Scholar] [CrossRef]
- Huang, C.H.; Chao, J.Y.; Ling, T.C.; Wu, J.L.; Sung, J.M.; Sun, C.Y.; Cheng, Y.Y.; Chang, Y.T. Effect of Dialysis Modalities on Risk of Hospitalization for Gastrointestinal Bleeding. Sci. Rep. 2023, 13, 52. [Google Scholar] [CrossRef]
- Chunduri, S.; Folstad, J.E.; Vachharajani, T.J. Antithrombotic Therapy in End-Stage Renal Disease. Hemodial. Int. 2017, 21, 453–471. [Google Scholar] [CrossRef]
- Yang, I.H.; Szabó, L.; Sasaki, M.; Uto, K.; Henzie, J.; Lin, F.H.; Samitsu, S.; Ebara, M. Biobased Chitosan-Derived Self-Nitrogen-Doped Porous Carbon Nanofibers Containing Nitrogen-Doped Zeolites for Efficient Removal of Uremic Toxins during Hemodialysis. Int. J. Biol. Macromol. 2023, 253, 126880. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, G.; Han, Q.; Lin, H.; Deng, G.; Li, Q.; Liu, F. Designing Adsorptive Membranes for Removing Protein-Bound Uremic Toxins via π-π and Cation-π Interaction. J. Membr. Sci. 2023, 676, 121584. [Google Scholar] [CrossRef]
- Azhar, O.; Jahan, Z.; Sher, F.; Niazi, M.B.K.; Kakar, S.J.; Shahid, M. Cellulose Acetate-Polyvinyl Alcohol Blend Hemodialysis Membranes Integrated with Dialysis Performance and High Biocompatibility. Mater. Sci. Eng. C 2021, 126, 112127. [Google Scholar] [CrossRef]
- Burton, J.O.; Hamali, H.A.; Singh, R.; Abbasian, N.; Parsons, R.; Patel, A.K.; Goodall, A.H.; Brunskill, N.J. Elevated Levels of Procoagulant Plasma Microvesicles in Dialysis Patients. PLoS ONE 2013, 8, e0072663. [Google Scholar] [CrossRef]
- Santos-Gallego, C.G.; Bayón, J.; Badimón, J.J. Thrombi of Different Pathologies: Implications for Diagnosis and Treatment. Curr. Treat. Options Cardiovasc. Med. 2010, 12, 274–291. [Google Scholar] [CrossRef]
- Matsushita, K.; Coresh, J.; Sang, Y.; Chalmers, J.; Fox, C.; Guallar, E.; Jafar, T.; Jassal, S.K.; Landman, G.W.D.; Muntner, P.; et al. Estimated Glomerular Filtration Rate and Albuminuria for Prediction of Cardiovascular Outcomes: A Collaborative Meta-Analysis of Individual Participant Data. Lancet Diabetes Endocrinol. 2015, 3, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Eikelboom, J.; Floege, J.; Thadhani, R.; Weitz, J.I.; Winkelmayer, W.C. Anticoagulation in Patients with Kidney Failure on Dialysis: Factor XI as a Therapeutic Target. Kidney Int. 2021, 100, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Ballew, S.H.; Wang, A.Y.-M.; Kalyesubula, R.; Schaeffner, E.; Agarwal, R. Epidemiology and Risk of Cardiovascular Disease in Populations with Chronic Kidney Disease. Nat. Rev. Nephrol. 2022, 18, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Mukku, V.; Ahmad, M. Coronary Artery Disease in Patients with Chronic Kidney Disease: A Clinical Update. Curr. Cardiol. Rev. 2013, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Cobo Marcos, M.; de la Espriella, R.; Gayán Ordás, J.; Llàcer, P.; Pomares, A.; Fort, A.; Ponz de Antonio, I.; Méndez, A.; Blázquez-Bermejo, Z.; Caravaca Pérez, P.; et al. Prevalence and Clinical Profile of Kidney Disease in Patients with Chronic Heart Failure. Insights from the Spanish Cardiorenal Registry. Rev. Esp. Cardiol. Engl. Ed. 2024, 77, 50–59. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the Diagnosis and Management of Atrial Fibrillation Developed in Collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Di Lullo, L.; Mariani, M.V.; Ronco, C.; Bellasi, A.; Lavalle, C.; Chimenti, C.; Paoletti, E.; Ravera, M.; Zanella, M. Atrial Fibrillation and Anticoagulant Treatment in End-Stage Renal Disease Patients: Where Do We Stand? CardioRenal Med. 2022, 12, 131–140. [Google Scholar] [CrossRef]
- Goel, N.; Jain, D.; Haddad, D.B.; Shanbhogue, D. Anticoagulation in Patients with End-Stage Renal Disease and Atrial Fibrillation: Confusion, Concerns and Consequences. J. Stroke 2020, 22, 306–316. [Google Scholar] [CrossRef]
- Ocak, G.; Khairoun, M.; Khairoun, O.; Bos, W.J.W.; Fu, E.L.; Cramer, M.J.; Westerink, J.; Verhaar, M.C.; Visseren, F.L.; UCC-SMART Study Group. Chronic Kidney Disease and Atrial Fibrillation: A Dangerous Combination. PLoS ONE 2022, 17, e0266046. [Google Scholar] [CrossRef]
- Song, W.; Wu, L.; Sun, C.; Kong, X.; Wang, H. New-Onset Atrial Fibrillation Following Arteriovenous Fistula Increases Adverse Clinical Events in Dialysis Patients with End-Stage Renal Disease. Front. Cardiovasc. Med. 2024, 11, 1386304. [Google Scholar] [CrossRef]
- Cofer, L.B.; Soomro, Q.H.; Xia, Y.; Luttrell-Williams, E.; Myndzar, K.; Charytan, D.M.; Berger, J.S. Platelet Activity and Cardiovascular Risk in CKD and Peripheral Artery Disease. Kidney Int. Rep. 2022, 7, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Bourrier, M.; Ferguson, T.W.; Embil, J.M.; Rigatto, C.; Komenda, P.; Tangri, N. Peripheral Artery Disease: Its Adverse Consequences with and without CKD. Am. J. Kidney Dis. 2020, 75, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Rojas, Á.G.; Martínez, A.V.; Benítez, P.R.; Estébanez, S.A.; Moreno, E.V.; Barrios, A.A.; de Pablo, J.C.L.; de Morales, A.M.; Antonova, A.M.; Colombina, A.B.; et al. Peripheral Arterial Disease in Hemodialysis Patients 10 Years Later. Nefrol. Engl. Ed. 2023, 43, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Hopley, C.W.; Kavanagh, S.; Patel, M.R.; Ostrom, C.; Baumgartner, I.; Berger, J.S.; Blomster, J.I.; Fowkes, F.G.R.; Jones, W.S.; Katona, B.G.; et al. Chronic Kidney Disease and Risk for Cardiovascular and Limb Outcomes in Patients with Symptomatic Peripheral Artery Disease: The EUCLID Trial. Vasc. Med. 2019, 24, 422–430. [Google Scholar] [CrossRef]
- Lim, H.Y.; Lui, B.; Tacey, M.; Barit, D.; Patel, S.K.; Donnan, G.; Nandurkar, H.; Burrell, L.M.; Ho, P. Global Coagulation Assays in Patients with Chronic Kidney Disease and Their Role in Predicting Thrombotic Risk. Thromb. Res. 2023, 226, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Chelluboina, B.; Vemuganti, R. Chronic Kidney Disease in the Pathogenesis of Acute Ischemic Stroke. J. Cereb. Blood Flow Metab. 2019, 39, 1893–1905. [Google Scholar] [CrossRef]
- Tsuyuki, Y.; Yamashita, Y.; Morimoto, T.; Amano, H.; Takase, T.; Hiramori, S.; Kim, K.; Oi, M.; Kobayashi, Y.; Ono, K.; et al. Renal Dysfunction and Long-Term Clinical Outcomes in Patients with Venous Thromboembolism: From the COMMAND VTE Registry. Thromb. Res. 2020, 187, 39–47. [Google Scholar] [CrossRef]
- Ponchia, P.I.; Ahmed, R.; Farag, M.; Alkhalil, M. Antiplatelet Therapy in End-Stage Renal Disease Patients on Maintenance Dialysis: A State-of-the-Art Review. Cardiovasc. Drugs Ther. 2023, 37, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Ocak, G.; Rookmaaker, M.B.; Algra, A.; de Borst, G.J.; Doevendans, P.A.; Kappelle, L.J.; Verhaar, M.C.; Visseren, F.L.; van der Graaf, Y.; Grobbee, D.E.; et al. Chronic Kidney Disease and Bleeding Risk in Patients at High Cardiovascular Risk: A Cohort Study. J. Thromb. Haemost. 2018, 16, 65–73. [Google Scholar] [CrossRef]
- Cheung, C.Y.S.; Parikh, J.; Farrell, A.; Lefebvre, M.; Summa-Sorgini, C.; Battistella, M. Direct Oral Anticoagulant Use in Chronic Kidney Disease and Dialysis Patients with Venous Thromboembolism: A Systematic Review of Thrombosis and Bleeding Outcomes. Ann. Pharmacother. 2021, 55, 711–722. [Google Scholar] [CrossRef]
- Nopp, S.; Königsbrügge, O.; Schmaldienst, S.; Klauser-Braun, R.; Lorenz, M.; Pabinger, I.; Säemann, M.; Ay, C. Growth Differentiation Factor-15 Predicts Major Bleeding, Major Adverse Cardiac Events and Mortality in Patients with End-Stage Kidney Disease on Haemodialysis: Findings from the VIVALDI Study. Nephrol. Dial. Transplant. 2023, 38, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, E.G.; Georgatzakou, H.T.; Fortis, S.P.; Tsante, K.A.; Tsantes, A.G.; Nomikou, E.G.; Kapota, A.I.; Petras, D.I.; Venetikou, M.S.; Papageorgiou, E.G.; et al. Coagulation Abnormalities in Renal Pathology of Chronic Kidney Disease: The Interplay between Blood Cells and Soluble Factors. Biomolecules 2021, 11, 1309. [Google Scholar] [CrossRef] [PubMed]
- Abdelmaguid, A.; Roberts, L.N.; Tugores, L.; Joslin, J.R.; Hunt, B.J.; Parmar, K.; Nebres, D.; Naga, S.S.; Khalil, E.S.; Bramham, K. Evaluation of Novel Coagulation and Platelet Function Assays in Patients with Chronic Kidney Disease. J. Thromb. Haemost. 2022, 20, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Ocak, G.; Lijfering, W.M.; Verduijn, M.; Dekker, F.W.; Rosendaal, F.R.; Cannegieter, S.C.; Vossen, C.Y. Risk of Venous Thrombosis in Patients with Chronic Kidney Disease: Identification of High-Risk Groups. J. Thromb. Haemost. 2013, 11, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, K.; Hurják, B.; Katona, É.; Becs, G.; Balla, J.; Muszbek, L. Terminal Phase Components of the Clotting Cascade in Patients with End-Stage Renal Disease Undergoing Hemodiafiltration or Hemodialysis Treatment. Int. J. Mol. Sci. 2020, 21, 8426. [Google Scholar] [CrossRef]
- Khalid, A.; White, D.; Sharp, M.; Duff, E.; MacDonald, S.; Fry, A.; Thomas, W. Investigation of Platelet Function in Patients with Chronic Kidney Disease Stages IV-V. Int. J. Lab. Hematol. 2021, 43, 1606–1611. [Google Scholar] [CrossRef]
- Martins, S.R.; Alves, L.V.; Cardoso, C.N.; Silva, L.G.; Nunes, F.F.C.; de Lucas Júnior, F.d.M.; Silva, A.C.S.; Dusse, L.M.S.; Alpoim, P.N.; Mota, A.P.L. Cell-Derived Microparticles and von Willebrand Factor in Brazilian Renal Transplant Recipients. Nephrology 2019, 24, 1304–1312. [Google Scholar] [CrossRef]
- Amabile, N.; Guérin, A.P.; Leroyer, A.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating Endothelial Microparticles Are Associated with Vascular Dysfunction in Patients with End-Stage Renal Failure. J. Am. Soc. Nephrol. 2005, 16, 3381–3388. [Google Scholar] [CrossRef]
- Mörtberg, J.; Salzinger, B.; Lundwall, K.; Edfors, R.; Jacobson, S.H.; Wallén, H.N.; Jernberg, T.; Baron, T.; Erlinge, D.; Andell, P.; et al. Prognostic Importance of Biomarkers Associated with Haemostatic, Vascular and Endothelial Disturbances in Acute Coronary Syndrome Patients in Relation to Kidney Function. Int. J. Cardiol. 2023, 373, 64–71. [Google Scholar] [CrossRef]
- Figuer, A.; Alique, M.; Valera, G.; Serroukh, N.; Ceprían, N.; De Sequera, P.; Morales, E.; Carracedo, J.; Ramírez, R.; Bodega, G. New Mechanisms Involved in the Development of Cardiovascular Disease in Chronic Kidney Disease. Nefrol. Engl. Ed. 2023, 43, 63–80. [Google Scholar] [CrossRef]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic Serum and Solutes Increase Post–Vascular Interventional Thrombotic Risk Through Altered Stability of Smooth Muscle Cell Tissue Factor. Circulation 2013, 127, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Fryc, J.; Naumnik, B. Thrombolome and Its Emerging Role in Chronic Kidney Diseases. Toxins 2021, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Nosseir, N.M.; Tawfik, M.S.; Bakheet, O.H.; El-Maghraby, D.F. Procoagulant FVIII and Anticoagulant Protein C in Renal Failure Patients on Hemodialysis. Egypt. J. Radiat. Sci. Appl. 2022, 35, 13–18. [Google Scholar] [CrossRef]
- Caruana, J.; Riva, N.; Vella, K.; Davenport, A.; Gatt, A. Navigating through the Haemostatic Paradox in Kidney Failure: A Practical Overview. Br. J. Haematol. 2023, 202, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Mallamaci, F.; Zoccali, C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef]
- Baaten, C.C.F.M.J.; Schröer, J.R.; Floege, J.; Marx, N.; Jankowski, J.; Berger, M.; Noels, H. Platelet Abnormalities in CKD and Their Implications for Antiplatelet Therapy. Clin. J. Am. Soc. Nephrol. 2022, 17, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T. Decreased Plasma Fibrinolytic Potential As a Risk for Venous and Arterial Thrombosis. Semin. Thromb. Hemost. 2016, 43, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Wang, C.; Xiong, J.; Zhao, J.; Yang, K. Activated Platelets, the Booster of Chronic Kidney Disease and Cardiovascular Complications. Kidney Dis. 2022, 8, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Finsterbusch, M.; Norman, M.U.; Hall, P.; Kitching, A.R.; Hickey, M.J. Platelet Retention in Inflamed Glomeruli Occurs via Selective Prolongation of Interactions with Immune Cells. Kidney Int. 2019, 95, 363–374. [Google Scholar] [CrossRef]
- Nishi, T.; Ariyoshi, N.; Nakayama, T.; Fujimoto, Y.; Sugimoto, K.; Wakabayashi, S.; Hanaoka, H.; Kobayashi, Y. Impact of Chronic Kidney Disease on Platelet Inhibition of Clopidogrel and Prasugrel in Japanese Patients. J. Cardiol. 2017, 69, 752–755. [Google Scholar] [CrossRef]
- Binder, V.; Chrúscicka-Smaga, B.; Bergum, B.; Jaisson, S.; Gillery, P.; Sivertsen, J.; Hervig, T.; Kaminska, M.; Tilvawala, R.; Nemmara, V.V.; et al. Carbamylation of Integrin αIIbβ3: The Mechanistic Link to Platelet Dysfunction in ESKD. J. Am. Soc. Nephrol. 2022, 33, 1841–1856. [Google Scholar] [CrossRef] [PubMed]
- Luciano, R.L.; Moeckel, G.W. Update on the Native Kidney Biopsy: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 73, 404–415. [Google Scholar] [CrossRef]
- Lim, C.C.; Tan, R.Y.; Choo, J.C.J.; Tan, H.Z.; Mok, I.; Chin, Y.M.; Tan, C.S. Estimation of Risk for Major Bleeding in Native Kidney Biopsies in Patients with Multiple Risk Factors. Int. Urol. Nephrol. 2022, 54, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Baaten, C.C.F.M.J.; ten Cate, H.; van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet Populations and Priming in Hematological Diseases. Blood Rev. 2017, 31, 389–399. [Google Scholar] [CrossRef]
- Nair, A.B.; Parker, R.I. Hemostatic Testing in Critically Ill Infants and Children. Front. Pediatr. 2021, 8, 606643. [Google Scholar] [CrossRef] [PubMed]
- Mörtberg, J.; Lundwall, K.; Mobarrez, F.; Wallén, H.; Jacobson, S.H.; Spaak, J. Increased Concentrations of Platelet- and Endothelial-Derived Microparticles in Patients with Myocardial Infarction and Reduced Renal Function—A Descriptive Study. BMC Nephrol. 2019, 20, 71. [Google Scholar] [CrossRef]
- Fonseca, F.; Ballerini, A.P.; Izar, M.C.; Kato, J.; Ferreira, C.E.; Fonzar, W.; do Amaral, J.; Rezende, P.; Machado-Santelli, G.; França, C. Advanced Chronic Kidney Disease Is Associated with Higher Serum Concentration of Monocyte Microparticles. Life Sci. 2020, 260, 118295. [Google Scholar] [CrossRef] [PubMed]
- De Laval, P.; Mobarrez, F.; Almquist, T.; Vassil, L.; Fellström, B.; Soveri, I. Acute Effects of Haemodialysis on Circulating Microparticles. Clin. Kidney J. 2019, 12, 456–462. [Google Scholar] [CrossRef]
- Lau, Y.C.; Xiong, Q.; Blann, A.D.; Lip, G.Y.H. Relationship between Renal Function and Circulating Microparticles, Soluble P-Selectin and E-Selectin Levels in Atrial Fibrillation. J. Thromb. Thrombolysis 2017, 43, 18–23. [Google Scholar] [CrossRef]
- Freyssinet, J.-M.; Toti, F. Formation of Procoagulant Microparticles and Properties. Thromb. Res. 2010, 125, S46–S48. [Google Scholar] [CrossRef]
- McMillan, R.; Skiadopoulos, L.; Hoppensteadt, D.; Guler, N.; Bansal, V.; Parasuraman, R.; Fareed, J. Biomarkers of Endothelial, Renal, and Platelet Dysfunction in Stage 5 Chronic Kidney Disease Hemodialysis Patients with Heart Failure. Clin. Appl. Thromb. 2018, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Yaker, L.; Kamel, S.; Ausseil, J.; Boullier, A. Effects of Chronic Kidney Disease and Uremic Toxins on Extracellular Vesicle Biology. Toxins 2020, 12, 811. [Google Scholar] [CrossRef] [PubMed]
- Van Bladel, E.R.; De Jager, R.L.; Walter, D.; Cornelissen, L.; Gaillard, C.A.; Boven, L.A.; Roest, M.; Fijnheer, R. Platelets of Patients with Chronic Kidney Disease Demonstrate Deficient Platelet Reactivity In Vitro. BMC Nephrol. 2012, 13, 127. [Google Scholar] [CrossRef]
- Almquist, T.; Mobarrez, F.; Jacobson, S.H.; Wallén, H.; Hjemdahl, P. Effects of Lipid-Lowering Treatment on Circulating Microparticles in Patients with Diabetes Mellitus and Chronic Kidney Disease. Nephrol. Dial. Transplant. 2016, 31, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, M.; Xiao, F.; Abujrad, H.; Al-Rewashdy, Y.; Tang, V.A.; Langlois, M.A.; Sorisky, A.; Ooi, T.C.; Burger, D. Effect of Hemodialysis on Extracellular Vesicles and Circulating Submicron Particles. BMC Nephrol. 2019, 20, 294. [Google Scholar] [CrossRef]
- Van Der Vorm, L.N.; Visser, R.; Huskens, D.; Veninga, A.; Adams, D.L.; Remijn, J.A.; Hemker, H.C.; Rensma, P.L.; Van Horssen, R.; De Laat, B. Circulating Active von Willebrand Factor Levels Are Increased in Chronic Kidney Disease and End-Stage Renal Disease. Clin. Kidney J. 2019, 13, 72–74. [Google Scholar] [CrossRef]
- Wattanakit, K.; Cushman, M. Chronic Kidney Disease and Venous Thromboembolism: Epidemiology and Mechanisms. Curr. Opin. Pulm. Med. 2009, 15, 408–412. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Fried, L.F.; Crump, C.; Bleyer, A.J.; Manolio, T.A.; Tracy, R.P.; Furberg, C.D.; Psaty, B.M. Elevations of Inflammatory and Procoagulant Biomarkers in Elderly Persons with Renal Insufficiency. Circulation 2003, 107, 87–92. [Google Scholar] [CrossRef]
- Adam, S.S.; Key, N.S.; Greenberg, C.S. D-Dimer Antigen: Current Concepts and Future Prospects. Blood 2009, 113, 2878–2887. [Google Scholar] [CrossRef]
- Yu, G.; Jiang, Y.; Xu, Z.; Cheng, J.; Li, H.; Li, X.; Chen, J. Plasma D-Dimer as a Potential Predictor of Progression in IgA Nephropathy: A Cohort Study. Ren. Fail. 2023, 45, 2251587. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, S.; Ge, X.; Shang, D.; Xie, Q.; Hao, C.; Zhu, T. Plasma Fibrinogen: A Driver of Left Ventricular Remodeling in Patients Undergoing Peritoneal Dialysis and Its Related Risk Factors. Ren. Fail. 2023, 45, 2255679. [Google Scholar] [CrossRef] [PubMed]
- Lorentz, C.U.; Tucker, E.I.; Verbout, N.G.; Shatzel, J.J.; Olson, S.R.; Markway, B.D.; Wallisch, M.; Ralle, M.; Hinds, M.T.; McCarty, O.J.T.; et al. The Contact Activation Inhibitor AB023 in Heparin-Free Hemodialysis: Results of a Randomized Phase 2 Clinical Trial. Blood 2021, 138, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Feng, S.; Wu, L. Glomerular Endothelium-Inspired Anticoagulant Surface Coating on Polyethersulfone Hemodialysis Membrane. Mater. Chem. Phys. 2023, 297, 127364. [Google Scholar] [CrossRef]
- Ji, H.; Li, Y.; Su, B.; Zhao, W.; Kizhakkedathu, J.N.; Zhao, C. Advances in Enhancing Hemocompatibility of Hemodialysis Hollow-Fiber Membranes. Adv. Fiber Mater. 2023, 5, 1198–1240. [Google Scholar] [CrossRef] [PubMed]
- Abdelrasoul, A.; Westphalen, H.; Kalugin, D.; Doan, H.; Shoker, A. In Situ Synchrotron Quantitative Analysis of Competitive Adsorption Tendency of Human Serum Protein to Different Clinical Hemodialysis Membranes and Assessment of Potential Impacts. Biomed. Eng. Adv. 2023, 6, 100104. [Google Scholar] [CrossRef]
- Abdelrasoul, A.; Shoker, A. Induced Hemocompatibility of Polyethersulfone (PES) Hemodialysis Membrane Using Polyvinylpyrrolidone: Investigation on Human Serum Fibrinogen Adsorption and Inflammatory Biomarkers Released. Chem. Eng. Res. Des. 2022, 177, 615–624. [Google Scholar] [CrossRef]
- van Eck van der Sluijs, A.; Abrahams, A.C.; Rookmaaker, M.B.; Verhaar, M.C.; Bos, W.J.W.; Blankestijn, P.J.; Dekker, F.W.; van Diepen, M.; Ocak, G. Bleeding Risk of Haemodialysis and Peritoneal Dialysis Patients. Nephrol. Dial. Transplant. 2021, 36, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Danielle, F.M.E.E.H.; Lionel, E.; Elvire, D.T.; Nasser, A.; Josiane, N.B.V.; Patrice, H.M.; Eveline, N.D. Evaluation of Bleeding Risk by Hemostatic Parameters in Hemodialysis at the Douala General Hospital: Comparison between Patients on Hemodialysis before 3 Months and after 12 Months. Open J. Nephrol. 2023, 13, 31–38. [Google Scholar] [CrossRef]
- Livio, M.; Marchesi, D.; Remuzzi, G.; Gotti, E.; Mecca, G.; De Gaetano, G. Uraemic Bleeding: Role of Anaemia and Beneficial Effect of Red Cell Transfusions. Lancet 1982, 320, 1013–1015. [Google Scholar] [CrossRef]
- Lunkes, G.I.; Lunkes, D.S.; Leal, D.; Araújo, M.D.C.; Corrêa, M.; Becker, L.; Rosa, C.S.D.; Morsch, V.M.; Schetinger, M.R.C. Effect of High Glucose Levels in Human Platelet NTPDase and 5′-Nucleotidase Activities. Diabetes Res. Clin. Pract. 2008, 81, 351–357. [Google Scholar] [CrossRef]
- Hirata, K.; Shikata, K.; Matsuda, M.; Akiyama, K.; Sugimoto, H.; Kushiro, M.; Makino, H. Increased Expression of Selectins in Kidneys of Patients with Diabetic Nephropathy. Diabetologia 1998, 41, 185–192. [Google Scholar] [CrossRef]
- Speer, T.; Ridker, P.M.; von Eckardstein, A.; Schunk, S.J.; Fliser, D. Lipoproteins in Chronic Kidney Disease: From Bench to Bedside. Eur. Heart J. 2021, 42, 2170–2185. [Google Scholar] [CrossRef]
- Reis, A.; Rudnitskaya, A.; Chariyavilaskul, P.; Dhaun, N.; Melville, V.; Goddard, J.; Webb, D.J.; Pitt, A.R.; Spickett, C.M. Top-down Lipidomics of Low Density Lipoprotein Reveal Altered Lipid Profiles in Advanced Chronic Kidney Disease. J. Lipid Res. 2015, 56, 413–422. [Google Scholar] [CrossRef]
- Barbagelata, L.; Masson, W.; Corral, P.; Lavalle-Cobo, A.; Nogueira, J.P.; Rosa Diez, G. Relationship between Lipoprotein(a) Levels, Cardiovascular Outcomes and Death in Patients with Chronic Kidney Disease: A Systematic Review of Prospective Studies. J. Nephrol. 2023, 36, 1549–1559. [Google Scholar] [CrossRef]
- Lu, H.Y.; Liao, K.M. Increased Risk of Deep Vein Thrombosis in End-Stage Renal Disease Patients. BMC Nephrol. 2018, 19, 204. [Google Scholar] [CrossRef]
- Yang, M.; Kholmukhamedov, A. Platelet Reactivity in Dyslipidemia: Atherothrombotic Signaling and Therapeutic Implications. Rev. Cardiovasc. Med. 2021, 22, 67–81. [Google Scholar] [CrossRef]
- Collet, J.-P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef]
- Mann, J.F.E.; Joseph, P.; Gao, P.; Pais, P.; Tyrwhitt, J.; Xavier, D.; Dans, T.; Jaramillo, P.L.; Gamra, H.; Yusuf, S. Effects of Aspirin on Cardiovascular Outcomes in Patients with Chronic Kidney Disease. Kidney Int. 2023, 103, 403–410. [Google Scholar] [CrossRef]
- Oh, Y.J.; Kim, A.J.; Ro, H.; Chang, J.H.; Lee, H.H.; Chung, W.; Hyun, Y.Y.; Lee, J.; Kim, Y.H.; Han, S.H.; et al. Low-Dose Aspirin Was Associated with an Increased Risk of Cardiovascular Events in Patients with Chronic Kidney Disease Patients and Low Bodyweight: Results from KNOW-CKD Study. Sci. Rep. 2021, 11, 6691. [Google Scholar] [CrossRef] [PubMed]
- Haim-Pinhas, H.; Yoskovitz, G.; Lishner, M.; Pereg, D.; Kitay-Cohen, Y.; Topaz, G.; Sela, Y.; Wand, O.; Rozenberg, I.; Benchetrit, S.; et al. Effect of Aspirin on Primary Prevention of Cardiovascular Disease and Mortality among Patients with Chronic Kidney Disease. Sci. Rep. 2022, 12, 17788. [Google Scholar] [CrossRef]
- Su, X.; Yan, B.; Wang, L.; Lv, J.; Cheng, H.; Chen, Y. Effect of Antiplatelet Therapy on Cardiovascular and Kidney Outcomes in Patients with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. BMC Nephrol. 2019, 20, 309. [Google Scholar] [CrossRef]
- Jain, N.; Corken, A.; Arthur, J.M.; Ware, J.; Arulprakash, N.; Dai, J.; Phadnis, M.A.; Davis, O.; Rahmatallah, Y.; Mehta, J.L.; et al. Ticagrelor Inhibits Platelet Aggregation and Reduces Inflammatory Burden More than Clopidogrel in Patients with Stages 4 or 5 Chronic Kidney Disease. Vasc. Pharmacol. 2023, 148, 107143. [Google Scholar] [CrossRef]
- Yu, Y.; Pan, D.; Bai, R.; Luo, J.; Tan, Y.; Duan, W.; Shi, D. P2y12 Inhibitor Monotherapy after 1–3 Months Dual Antiplatelet Therapy in Patients with Coronary Artery Disease and Chronic Kidney Disease Undergoing Percutaneous Coronary Intervention: A Meta-Analysis of Randomized Controlled Trials. Front. Cardiovasc. Med. 2023, 10, 1197161. [Google Scholar] [CrossRef]
- Kim, C.; Choi, D.W.; Lee, S.J.; Suh, Y.; Hong, S.J.; Ahn, C.M.; Kim, J.S.; Kim, B.K.; Ko, Y.G.; Choi, D.; et al. Benefit and Risk of Prolonged Dual Antiplatelet Therapy after Drug-Eluting Stent Implantation in Patients with Chronic Kidney Disease: A Nationwide Cohort Study. Atherosclerosis 2022, 352, 69–75. [Google Scholar] [CrossRef]
- Hwang, D.; Park, K.W.; Lee, J.M.; Rhee, T.M.; Hong, M.K.; Jang, Y.; Valgimigli, M.; Colombo, A.; Gilard, M.; Palmerini, T.; et al. Efficacy and Safety of Dual Antiplatelet Therapy after Coronary Stenting in Patients with Chronic Kidney Disease. Am. Heart J. 2018, 197, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Porlán, M.V.; Tello-Montoliu, A.; López-García, C.; Gil-Pérez, P.; Quintana-Giner, M.; López-Gálvez, R.; Rivera-Caravaca, J.M.; Marín, F.; Figal, D.P. Impact of Renal Function on Ticagrelor-Induced Antiplatelet Effects in Coronary Artery Disease Patients. IJC Heart Vasc. 2023, 46, 101195. [Google Scholar] [CrossRef]
- Mohsen, M.; Zhang, T.; Battistella, M. Anticoagulation in CKD: Trials and Tribulations. Kidney Med. 2023, 5, 100686. [Google Scholar] [CrossRef] [PubMed]
- Zagoridis, K.; Karatisidis, L.; Mprotsis, T.; Pentidou, A.; Bezirgianidou, Z.; Misidou, C.; Spanoudakis, E. Apixaban Reduces the Risk of Major and Clinically Relevant Non-Major Bleeding Compared to Warfarin in Patients with End Stage Renal Disease; a Systematic Review and Meta-Analysis of Ten Studies. Thromb. Res. 2023, 231, 17–24. [Google Scholar] [CrossRef]
- Catella, J.; Bertoletti, L.; Mismetti, P.; Ollier, E.; Samperiz, A.; Soler, S.; Suriñach, J.M.; Mahé, I.; Lorente, M.A.; Braester, A.; et al. Severe Renal Impairment and Risk of Bleeding during Anticoagulation for Venous Thromboembolism. J. Thromb. Haemost. 2020, 18, 1728–1737. [Google Scholar] [CrossRef]
- Welander, F.; Renlund, H.; Dimény, E.; Holmberg, H.; Själander, A. Warfarin Treatment Quality and Outcomes in Patients with Non-Valvular Atrial Fibrillation and CKD G3-G5D. Thromb. Res. 2023, 229, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Steffel, J.; Collins, R.; Antz, M.; Cornu, P.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; Rowell, N.; et al. 2021 European Heart Rhythm Association Practical Guide on the Use of Non-Vitamin K Antagonist Oral Anticoagulants in Patients with Atrial Fibrillation. EP Eur. 2021, 23, 1612–1676. [Google Scholar] [CrossRef] [PubMed]
- Sin, C.-F.; Wong, K.-P.; Wong, H.-M.; Siu, C.-W.; Yap, D.Y.H. Plasma Rivaroxaban Level in Patients with Early Stages of Chronic Kidney Disease—Relationships with Renal Function and Clinical Events. Front. Pharmacol. 2022, 13, 888660. [Google Scholar] [CrossRef]
- Jain, N.; Reilly, R.F. Clinical Pharmacology of Oral Anticoagulants in Patients with Kidney Disease. Clin. J. Am. Soc. Nephrol. 2019, 14, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tirucherai, G.; Marbury, T.C.; Wang, J.; Chang, M.; Zhang, D.; Song, Y.; Pursley, J.; Boyd, R.A.; Frost, C. Pharmacokinetics, Pharmacodynamics, and Safety of Apixaban in Subjects with End-stage Renal Disease on Hemodialysis. J. Clin. Pharmacol. 2016, 56, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Burlacu, A.; Genovesi, S.; Ortiz, A.; Combe, C.; Basile, C.; Schneditz, D.; Van Der Sande, F.; Popa, G.T.; Morosanu, C.; Covic, A. Pros and Cons of Antithrombotic Therapy in End-Stage Kidney Disease: A 2019 Update. Nephrol. Dial. Transplant. 2019, 34, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Skripka, A.; Sychev, D.; Bochkov, P.; Shevchenko, R.; Krupenin, P.; Kogay, V.; Listratov, A.; Krainyaya, A.; Gurinovich, O.; Sokolova, A.; et al. Factors Affecting Trough Plasma Dabigatran Concentrations in Patients with Atrial Fibrillation and Chronic Kidney Disease. High Blood Press. Cardiovasc. Prev. 2020, 27, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Parada Barcia, J.A.; Raposeiras Roubin, S.; Abu-Assi, E.; Erquicia, P.D.; Lizancos Castro, A.; Lestón, L.R.; Míguez, J.O.; González Bermúdez, I.; Íñiguez-Romo, A. Comparison of Stroke and Bleeding Risk Profile in Patients with Atrial Fibrillation and Chronic Kidney Disease. Am. J. Cardiol. 2023, 196, 31–37. [Google Scholar] [CrossRef] [PubMed]
- de Vriese, A.S.; Caluwé, R.; van der Meersch, H.; de Boeck, K.; de Bacquer, D. Safety and Efficacy of Vitamin K Antagonists versus Rivaroxaban in Hemodialysis Patients with Atrial Fibrillation: A Multicenter Randomized Controlled Trial. J. Am. Soc. Nephrol. 2021, 32, 1474–1483. [Google Scholar] [CrossRef]
- Siontis, K.C.; Zhang, X.; Eckard, A.; Bhave, N.; Schaubel, D.E.; He, K.; Tilea, A.; Stack, A.G.; Balkrishnan, R.; Yao, X.; et al. Outcomes Associated with Apixaban Use in Patients with End-Stage Kidney Disease and Atrial Fibrillation in the United States. Circulation 2018, 138, 1519–1529. [Google Scholar] [CrossRef]
- Pokorney, S. RENAL-AF Trial: Apixaban Similar to Warfarin; Medicom Medical Publishers: Baarn, The Netherlands, 2019. [Google Scholar]
- Fordyce, C.B.; Hellkamp, A.S.; Lokhnygina, Y.; Lindner, S.M.; Piccini, J.P.; Becker, R.C.; Berkowitz, S.D.; Breithardt, G.; Fox, K.A.A.; Mahaffey, K.W.; et al. On-Treatment Outcomes in Patients with Worsening Renal Function with Rivaroxaban Compared with Warfarin. Circulation 2016, 134, 37–47. [Google Scholar] [CrossRef]
- Kao, T.; Chen, Z.; Lin, Y. Anticoagulation for Patients with Concomitant Atrial Fibrillation and End-Stage Renal Disease: A Systematic Review and Network Meta-Analysis. J. Am. Heart Assoc. 2024, 13, e034176. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.S.; Giugliano, R.P.; Rangaswami, J. Anticoagulation-related Nephropathy. J. Thromb. Haemost. 2016, 14, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Nadasdy, T.; Rovin, B.H.; Satoskar, A.A.; Nadasdy, G.M.; Wu, H.M.; Bhatt, U.Y.; Hebert, L.A. Warfarin-Related Nephropathy Occurs in Patients with and without Chronic Kidney Disease and Is Associated with an Increased Mortality Rate. Kidney Int. 2011, 80, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tangri, N.; Gersh Bernard, J.; Sangaralingham Lindsey, R.; Shah Nilay, D.; Nath Karl, A.; Noseworthy Peter, A. Renal Outcomes in Anticoagulated Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2017, 70, 2621–2632. [Google Scholar] [CrossRef]
- Trevisan, M.; Hjemdahl, P.; Clase, C.M.; de Jong, Y.; Evans, M.; Bellocco, R.; Fu, E.L.; Carrero, J.J. Cardiorenal Outcomes among Patients with Atrial Fibrillation Treated with Oral Anticoagulants. Am. J. Kidney Dis. 2023, 81, 307–317.e1. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Ou, S.-M.; Chu, Y.-C.; Lin, Y.-P.; Tsai, M.-T.; Tarng, D.-C. Antithrombotic Therapy for Chronic Kidney Disease Patients with Concomitant Atrial Fibrillation and Coronary Artery Disease. Front. Cardiovasc. Med. 2021, 8, 751359. [Google Scholar] [CrossRef] [PubMed]
- Kato, C.; Oakes, M.; Kim, M.; Desai, A.; Olson, S.R.; Raghunathan, V.; Shatzel, J.J. Anticoagulation strategies in extracorporeal circulatory devices in adult populations. Eur. J. Haematol. 2021, 106, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, M.; Piscitani, L.; Di Liberato, L.; Sirolli, V. Biocompatibility of Surface-Modified Membranes for Chronic Hemodialysis Therapy. Biomedicines 2022, 10, 844. [Google Scholar] [CrossRef]
- Xu, L.; Ji, H.; Zhong, R.; Cheng, S.; Dang, G.; Xu, T.; Zhao, W.; Zhao, C. Antioxidative Hydrogel-Embedded Polyethersulfone Membrane with Improved Hemocompatibility to Alleviate Oxidative Stress. J. Membr. Sci. 2023, 684, 121866. [Google Scholar] [CrossRef]
- Zaman, S.U.; Rafiq, S.; Ali, A.; Mehdi, M.S.; Arshad, A.; Rehman, S.U.; Muhammad, N.; Irfan, M.; Khurram, M.S.; Zaman, M.K.U.; et al. Recent Advancement Challenges with Synthesis of Biocompatible Hemodialysis Membranes. Chemosphere 2022, 307, 135626. [Google Scholar] [CrossRef]
- Zhi, L.; Li, S.; He, X.; Feng, Y.; Cheng, C.; Li, S.; Sun, S.; Zhao, C. In-Situ Modified Polyethersulfone Oxygenation Membrane with Improved Hemocompatibility and Gas Transfer Efficiency. J. Membr. Sci. 2023, 667, 121162. [Google Scholar] [CrossRef]
- Voigt, M.; Gebert, M.; Haug, U.; Hulko, M.; Storr, M.; Boschetti-de-Fierro, A.; Beck, W.; Krause, B. Retention of Beneficial Molecules and Coagulation Factors during Haemodialysis and Haemodiafiltration. Sci. Rep. 2019, 9, 6370. [Google Scholar] [CrossRef]
- Hou, X.; Huang, L.; Zhang, H.; Xin, Q.; Li, H.; Ye, H.; Zhang, Y. Adsorption Resin/Polyethersulfone Membrane Used for Plasma Separation and Middle Molecular Toxins Adsorption. J. Ind. Eng. Chem. 2023, 123, 447–458. [Google Scholar] [CrossRef]
- Laville, S.M.; Gras-Champel, V.; Hamroun, A.; Moragny, J.; Lambert, O.; Metzger, M.; Jacquelinet, C.; Combe, C.; Fouque, D.; Laville, M.; et al. Kidney Function Decline and Serious Adverse Drug Reactions in Patients with CKD. Am. J. Kidney Dis. 2024, 83, 601–614.e1. [Google Scholar] [CrossRef]
- Leblond, F.A.; Petrucci, M.; Dubé, P.; Bernier, G.; Bonnardeaux, A.; Pichette, V. Downregulation of Intestinal Cytochrome P450 in Chronic Renal Failure. J. Am. Soc. Nephrol. 2002, 13, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Naud, J.; Michaud, J.; Boisvert, C.; Desbiens, K.; Leblond, F.A.; Mitchell, A.; Jones, C.; Bonnardeaux, A.; Pichette, V. Down-Regulation of Intestinal Drug Transporters in Chronic Renal Failure in Rats. J. Pharmacol. Exp. Ther. 2007, 320, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Dobesh, P.P.; Oestreich, J.H. Ticagrelor: Pharmacokinetics, Pharmacodynamics, Clinical Efficacy, and Safety. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2014, 34, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Mullangi, R.; Srinivas, N.R. Clopidogrel: Review of Bioanalytical Methods, Pharmacokinetics/Pharmacodynamics, and Update on Recent Trends in Drug–Drug Interaction Studies. Biomed. Chromatogr. 2009, 23, 26–41. [Google Scholar] [CrossRef]
- Pereira, N.L.; Cresci, S.; Angiolillo, D.J.; Batchelor, W.; Capers, Q.; Cavallari, L.H.; Leifer, D.; Luzum, J.A.; Roden, D.M.; Stellos, K.; et al. CYP2C19 Genetic Testing for Oral P2Y12 Inhibitor Therapy: A Scientific Statement from the American Heart Association. Circulation 2024, 149, e1–e22. [Google Scholar] [CrossRef]
- Mathew, R.O.; Sidhu, M.S.; Rihal, C.S.; Lennon, R.; El-Hajjar, M.; Yager, N.; Lyubarova, R.; Abdul-Nour, K.; Weitz, S.; O’Cochlain, D.F.; et al. Safety and Efficacy of CYP2C19 Genotype-Guided Escalation of P2Y12 Inhibitor Therapy After Percutaneous Coronary Intervention in Chronic Kidney Disease: A Post Hoc Analysis of the TAILOR-PCI Study. Cardiovasc. Drugs Ther. 2024, 38, 447–457. [Google Scholar] [CrossRef]
- Thomas, C.D.; Franchi, F.; Rossi, J.S.; Keeley, E.C.; Anderson, R.D.; Beitelshees, A.L.; Duarte, J.D.; Ortega-Paz, L.; Gong, Y.; Kerensky, R.A.; et al. Effectiveness of Clopidogrel vs Alternative P2Y12 Inhibitors Based on the ABCD-GENE Score. J. Am. Coll. Cardiol. 2024, 83, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, F.; Gargiulo, G.; Paolillo, R.; Ferrone, M.; Cimino, S.; Giugliano, G.; Schiattarella, G.G.; Verde, N.; Stabile, E.; Perrino, C.; et al. Impact of Chronic Kidney Disease on Platelet Aggregation in Patients with Acute Coronary Syndrome. J. Cardiovasc. Med. 2020, 21, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Mayer, K.; Bernlochner, I.; Braun, S.; Schulz, S.; Orban, M.; Morath, T.; Cala, L.; Hoppmann, P.; Schunkert, H.; Laugwitz, K.-L.; et al. Aspirin Treatment and Outcomes after Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2014, 64, 863–871. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane | Uremic Toxin Removal (%) | BSA Retention (%) | Hemolysis Ratio (%) | PRT (s) | aPTT (s) | References |
|---|---|---|---|---|---|---|
| CA/PEG/PVA | 93 | 96 | 3.2 | 300 | - | Azhar et al. [18] |
| PES/TA/α-LA | 96 | 99.7 | 0.3 | 297 | 80 | Wei et al. [83] |
| PES/G/E-TA | 54 | 93 | 0 | - | 45 | Xu et al. [130] |
| NZ/PCNF | 59 | - | <2 | - | - | Yang et al. [16] |
| PES/AR | - | >90 | 0.1 | - | - | Hou et al. [134] |
| PES/PVP/PAA | - | - | 0 | >10 | >60 | Zhi et al. [132] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, Z.; Sirolli, V.; Bonomini, M.; Gallina, S.; Renda, G. Hallmarks for Thrombotic and Hemorrhagic Risks in Chronic Kidney Disease Patients. Int. J. Mol. Sci. 2024, 25, 8705. https://doi.org/10.3390/ijms25168705
Saeed Z, Sirolli V, Bonomini M, Gallina S, Renda G. Hallmarks for Thrombotic and Hemorrhagic Risks in Chronic Kidney Disease Patients. International Journal of Molecular Sciences. 2024; 25(16):8705. https://doi.org/10.3390/ijms25168705
Chicago/Turabian StyleSaeed, Zeeba, Vittorio Sirolli, Mario Bonomini, Sabina Gallina, and Giulia Renda. 2024. "Hallmarks for Thrombotic and Hemorrhagic Risks in Chronic Kidney Disease Patients" International Journal of Molecular Sciences 25, no. 16: 8705. https://doi.org/10.3390/ijms25168705