
Strong Bases Design: Key Techniques and Stability Issues



Abstract
:1. Introduction
- Step 1:
- choosing the basicity center. That should be a negatively charged atom with a lone electron pair available for protonation. The trivalent nitrogen atom has been a common choice for more than a century; however, the generally weaker acidity of C–H bond compared to N–H bond makes carbene bases generally stronger. Nevertheless, a lot depends on further steps, making it possible to design strong bases with other types of basicity centers [16,17,18,19,20,21,22].
- Step 2:
- steric loading of the basicity center by groups with lone electron pairs, which destabilize B to a greater extent than BH+. That is implemented, for example, in various classes of “proton sponges” [23,24,25,26,27], Verkade bases [28,29], cyclic aminopyridines [30,31], rotaxane or catenane superbases [32,33,34], and adamanzanes [34,35,36].
- Step 3:
- Step 4:
- Step 5:
2. Results and Discussion
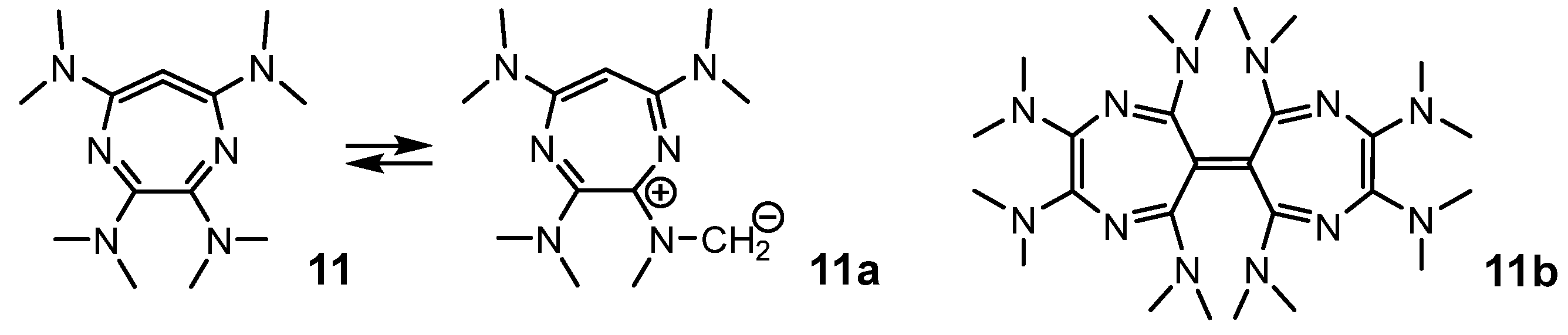
2.1. Flawed Design Examples
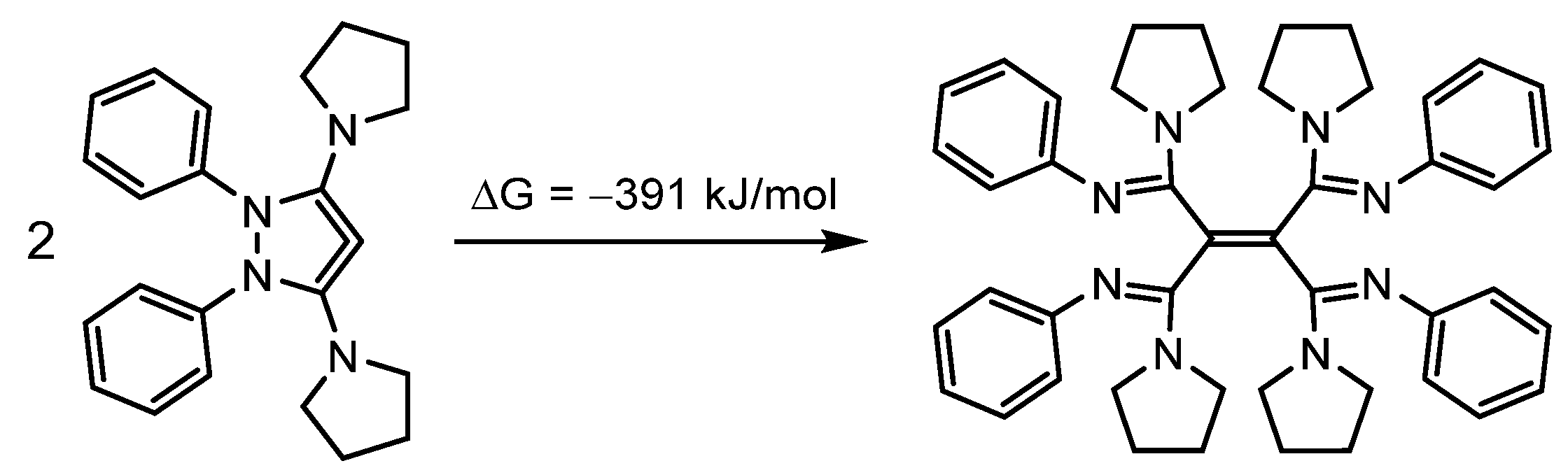
2.2. Successful Design Examples
- Step 6.
- Avoid the presence of groups in B that are either Brønsted or Lewis acidic, and also groups that tend to rearrange upon deprotonation.
- Step 7.
- Check that both B protonation and BH+ deprotonation are favored at the same sites; identify possible tautomers.
- Step 8.
- Perform a full conformational analysis of all tautomers of both B and BH+ in their ground and low-lying excited states to ensure their relaxation.
- Step 9.
- Check that neither B nor BH+ tends to dimerization or other rearrangements.
- Step 10.
- Look for as many degradation ways of B as possible, find the lowest-barrier one, and estimate the corresponding half-life under the conditions being proposed for practical usage, including the possible interaction with the solvent.
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schenck, M. Zur Kenntnis der methylierten Guanidine. Hoppe-Seyler’s Z. Physiol. Chem. 1912, 77, 328–393. [Google Scholar] [CrossRef]
- McKay, A.F.; Kreling, M.-E. Preparation and Chemistry of Δ8-hexahydro-1,4,8-pyrimidazole, Δ9-1,5,9-triazabicyclo(4.4.0)decene, and Δ9-1,4,9-triazabicyclo(5.3.0)decene. Can. J. Chem. 1957, 35, 1438–1445. [Google Scholar] [CrossRef]
- Issleib, K.; Lischewski, M. Dimethylamino-Iminophosphorane. Synth. Inorg. Met.-Org. Chem. 1973, 3, 255–266. [Google Scholar] [CrossRef]
- Schwesinger, R. Tricyclic 2,4-Diaminovinamidines—Readily Accessible, Very Strong CHN Bases. Angew. Chem. Int. Ed. Engl. 1987, 26, 1164–1165. [Google Scholar] [CrossRef]
- Schwesinger, R.; Mißfeldt, M.; Peters, K.; von Schnering, H.G. Novel, Very Strongly Basic, Pentacyclic “Proton Sponges” with Vinamidine Structure. Angew. Chem. Int. Ed. Engl. 1987, 26, 1165–1167. [Google Scholar] [CrossRef]
- Schwesinger, R.; Schlemper, H. Peralkylated Polyaminophosphazenes—Extremely Strong, Neutral Nitrogen Bases. Angew. Chem. Int. Ed. Engl. 1987, 26, 1167–1169. [Google Scholar] [CrossRef]
- Schwesinger, R.; Hasenfratz, C.; Schlemper, H.; Walz, L.; Peters, E.-M.; Peters, K.; von Schnering, H.G. How Strong and How Hindered Can Uncharged Phosphazene Bases Be? Angew. Chem. Int. Ed. Engl. 1993, 32, 1361–1363. [Google Scholar] [CrossRef]
- Schwesinger, R.; Schlemper, H.; Hasenfratz, C.; Willaredt, J.; Dambacher, T.; Breuer, T.; Ottaway, C.; Fletschinger, M.; Boele, J.; Fritz, H.; et al. Extremely Strong, Uncharged Auxiliary Bases; Monomeric and Polymer-Supported Polyaminophosphazenes (P2–P5). Liebigs Ann. 1996, 1996, 1055–1081. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Cao, Z.-Y.; Li, Q.-H.; Lin, G.-Q.; Zhou, J.; Tian, P. Activating Pronucleophiles with High pKa Values: Chiral Organo-Superbases. Angew. Chem. Int. Ed. 2020, 59, 8004–8014. [Google Scholar] [CrossRef]
- Puleo, T.R.; Sujansky, S.J.; Wright, S.E.; Bandar, J.S. Organic Superbases in Recent Synthetic Methodology Research. Chem. Eur. J. 2021, 27, 4216–4229. [Google Scholar] [CrossRef]
- Trofimov, B.A.; Schmidt, E.Y. Superbasis in Organic Synthesis. Chem. Probl. 2022, 20, 325–340. [Google Scholar] [CrossRef]
- Vazdar, K.; Margetić, D.; Kovačević, B.; Sundermeyer, J.; Leito, I.; Jahn, U. Design of Novel Uncharged Organic Superbases: Merging Basicity and Functionality. Acc. Chem. Res. 2021, 54, 3108–3123. [Google Scholar] [CrossRef] [PubMed]
- Pozharskii, A.F.; Ozeryanskii, V.A.; Filatova, E.A. Heterocyclic superbases: Retrospective and current trends. Chem. Heterocycl. Compd. 2012, 48, 200–219. [Google Scholar] [CrossRef]
- Ullrich, S.; Kovačević, B.; Koch, B.; Harms, K.; Sundermeyer, J. Design of non-ionic carbon superbases: Second generation carbodiphosphoranes. Chem. Sci. 2019, 10, 9483–9492. [Google Scholar] [CrossRef]
- Vermersch, F.; Yazdani, S.; Junor, G.P.; Grotjahn, D.B.; Jazzar, R.; Bertrand, G. Stable Singlet Carbenes as Organic Superbases. Angew. Chem. Int. Ed. 2021, 60, 27253–27257. [Google Scholar] [CrossRef] [PubMed]
- Mehlmann, P.; Mück-Lichtenfeld, C.; Tan, T.T.Y.; Dielmann, F. Tris(imidazolin-2-ylidenamino)phosphine: A crystalline phosphorus(III) superbase that splits carbon dioxide. Chem. Eur. J. 2017, 23, 5929–5933. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Kovačević, B.; Xie, X.; Sundermeyer, J. Phosphazenyl Phosphines: The Most Electron-Rich Uncharged Phosphorus Brønsted and Lewis Bases. Angew. Chem. Int. Ed. 2019, 58, 10335–10339. [Google Scholar] [CrossRef]
- Weitkamp, R.F.; Neumann, B.; Stammler, H.-G.; Hoge, B. Phosphorus-Containing Superbases: Recent Progress in the Chemistry of Electron-Abundant Phosphines and Phosphazenes. Chem. Eur. J. 2021, 27, 10807–10825. [Google Scholar] [CrossRef] [PubMed]
- Buß, F.; Röthel, M.B.; Werra, J.A.; Rotering, P.; Wilm, L.F.B.; Daniliuc, C.G.; Löwe, P.; Dielmann, F. Tris(tetramethylguanidinyl)phosphine: The Simplest Non-ionic Phosphorus Superbase and Strongly Donating Phosphine Ligand. Chem. Eur. J. 2022, 28, e202104021. [Google Scholar] [CrossRef]
- Lv, W.; Dai, Y.; Guo, R.; Su, Y.; Ruiz, D.A.; Liu, L.L.; Tung, C.-H.; Kong, L. Geometrically Constrained Organoboron Species as Lewis Superacids and Organic Superbases. Angew. Chem. Int. Ed. 2023, 62, e202308467. [Google Scholar] [CrossRef]
- Schwesinger, R.; Link, R.; Thiele, G.; Rotter, H.; Honert, D.; Limbach, H.-H.; Männle, F. Stable Phosphazenium Ions in Synthesis—An Easily Accessible, Extremely Reactive “Naked” Fluoride Salt. Angew. Chem. Int. Ed. Engl. 1991, 30, 1372–1375. [Google Scholar] [CrossRef]
- Schwesinger, R.; Link, R.; Wenzl, P.; Kossek, S. Anhydrous Phosphazenium Fluorides as Sources for Extremely Reactive Fluoride Ions in Solution. Chem. Eur. J. 2005, 12, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Kögel, J.F.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. A New Synthetic Pathway to the Second and Third Generation of Superbasic Bisphosphazene Proton Sponges: The Run for the Best Chelating Ligand for a Proton. J. Am. Chem. Soc. 2013, 135, 17768–17774. [Google Scholar] [CrossRef] [PubMed]
- Belding, L.; Dudding, T. Synthesis and Theoretical Investigation of a 1,8-Bis(bis(diisopropylamino)cyclopropeniminyl)naphthalene Proton Sponge Derivative. Chem. Eur. J. 2014, 20, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Kögel, J.F.; Xie, X.; Baal, E.; Gesevičius, D.; Oelkers, B.; Kovačević, B.; Sundermeyer, J. Superbasic Alkyl-Substituted Bisphosphazene Proton Sponges: Synthesis, Structural Features, Thermodynamic and Kinetic Basicity, Nucleophilicity and Coordination Chemistry. Chem. Eur. J. 2014, 20, 7670–7685. [Google Scholar] [CrossRef] [PubMed]
- Belding, L.; Stoyanov, P.; Dudding, T. Synthesis, Theoretical Analysis, and Experimental pKa Determination of a Fluorescent, Nonsymmetric, In–Out Proton Sponge. J. Org. Chem. 2016, 81, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Kögel, J.F.; Margetić, D.; Xie, X.; Finger, L.H.; Sundermeyer, J. A Phosphorus Bisylide: Exploring a New Class of Superbases with Two Interacting Carbon Atoms as Basicity Centers. Angew. Chem. Int. Ed. 2017, 56, 3090–3093. [Google Scholar] [CrossRef] [PubMed]
- Kisanga, P.B.; Verkade, J.G.; Schwesinger, R. pKa Measurements of P(RNCH2CH3)3N. J. Org. Chem. 2000, 65, 5431–5432. [Google Scholar] [CrossRef] [PubMed]
- Kisanga, P.B.; Verkade, J.G. Synthesis of new proazaphosphatranes and their application in organic synthesis. Tetrahedron 2001, 57, 467–475. [Google Scholar] [CrossRef]
- Uchida, N.; Taketoshi, A.; Kuwabara, J.; Yamamoto, T.; Inoue, Y.; Watanabe, Y.; Kanbara, T. Synthesis, Characterization, and Catalytic Reactivity of a Highly Basic Macrotricyclic Aminopyridine. Org. Lett. 2010, 12, 5242–5245. [Google Scholar] [CrossRef]
- Uchida, N.; Kuwabara, J.; Taketoshi, A.; Kanbara, T. Molecular Design of Organic Superbases, Azacalix[3](2,6)pyridines: Catalysts for 1,2- and 1,4-Additions. J. Org. Chem. 2012, 77, 10631–10637. [Google Scholar] [CrossRef] [PubMed]
- Power, M.J.; Morris, D.T.J.; Vitorica-Yrezabal, I.J.; Leigh, D.A. Compact Rotaxane Superbases. J. Am. Chem. Soc. 2023, 145, 8583–8599. [Google Scholar] [CrossRef] [PubMed]
- Capocasa, G.; Frateloreto, F.; Valentini, M.; Di Stefano, S. Molecular entanglement can strongly increase basicity. Commun. Chem. 2024, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- Chambron, J.-C.; Meyer, M. The ins and outs of proton complexation. Chem. Soc. Rev. 2009, 38, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Miyahara, Y.; Tanaka, Y.; Amimoto, K.; Akazawa, T.; Sakuragi, T.; Kobayashi, H.; Kubota, K.; Suenaga, M.; Koyama, H.; Inazu, T. The Proton Cryptate of Hexaethylenetetramine. Angew. Chem. Int. Ed. 1999, 38, 956–959. [Google Scholar] [CrossRef]
- Springborg, J. Adamanzanes–Bi- and tricyclic tetraamines and their coordination compounds. Dalton Trans. 2003, 2003, 1653–1665. [Google Scholar] [CrossRef]
- Vazdar, K.; Kunetskiy, R.; Saame, J.; Kaupmees, K.; Leito, I.; Jahn, U. Very Strong Organosuperbases Formed by Combining Imidazole and Guanidine Bases: Synthesis, Structure, and Basicity. Angew. Chem. Int. Ed. 2014, 53, 1435–1438. [Google Scholar] [CrossRef] [PubMed]
- Nasca, E.D.; Lambert, T.H. Higher-Order Cyclopropenimine Superbases: Direct Neutral Brønsted Base Catalyzed Michael Reactions with α-Aryl Esters. J. Am. Chem. Soc. 2015, 137, 10246–10253. [Google Scholar] [CrossRef]
- Kunetskiy, R.A.; Polyakova, S.M.; Vavřík, J.; Císařová, I.; Saame, J.; Nerut, E.R.; Koppel, I.; Koppel, I.A.; Kütt, A.; Leito, I.; et al. A New Class of Organosuperbases, N-Alkyl- and N-Aryl-1,3-dialkyl-4,5-dimethylimidazol-2-ylidene Amines: Synthesis, Structure, pKBH+ Measurements, and Properties. Chem. Eur. J. 2012, 18, 3621–3630. [Google Scholar] [CrossRef]
- Glasovac, Z.; Kovačević, B.; Meštrović, E.; Eckert-Maksić, M. Synthesis and properties of novel guanidine bases. N,N′,N″-Tris(3-dimethylaminopropyl)-guanidine. Tetrahedron Lett. 2005, 46, 8733–8736. [Google Scholar] [CrossRef]
- Ullrich, S.; Barić, D.; Xie, X.; Kovačević, B.; Sundermeyer, J. Basicity Enhancement by Multiple Intramolecular Hydrogen Bonding in Organic Superbase N,N′,N″,N‴-Tetrakis(3-(dimethylamino)propyl)triaminophosphazene. Org. Lett. 2019, 21, 9142–9146. [Google Scholar] [CrossRef] [PubMed]
- Maksić, Z.B.; Kovačević, B. Spatial and Electronic Structure of Highly Basic Organic Molecules: Cyclopropeneimines and Some Related Systems. J. Phys. Chem. A 1999, 103, 6678–6684. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Kovačević, B. Absolute Proton Affinity of Some Polyguanides. J. Org. Chem. 2000, 65, 3303–3309. [Google Scholar] [CrossRef] [PubMed]
- Maksić, Z.B.; Vianello, R. Quest for the Origin of Basicity: Initial vs Final State Effect in Neutral Nitrogen Bases. J. Phys. Chem. A 2002, 106, 419–430. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Glasovac, Z.; Despotović, I. Predicted high proton affinity of poly-2,5-dihydropyrrolimines—The aromatic domino effect. J. Phys. Org. Chem. 2002, 15, 499–508. [Google Scholar] [CrossRef]
- Kovačević, B.; Glasovac, Z.; Maksić, Z.B. The intramolecular hydrogen bond and intrinsic proton affinity of neutral organic molecules: N,N′,N″-tris(3-aminopropyl)guanidine and some related systems. J. Phys. Org. Chem. 2002, 15, 765–774. [Google Scholar] [CrossRef]
- Kovačević, B.; Maksić, Z.B.; Vianello, R.; Primorac, M. Computer aided design of organic superbases: The role of intramolecular hydrogen bonding. New J. Chem. 2002, 26, 1329–1334. [Google Scholar] [CrossRef]
- Bucher, G. DFT Calculations on a New Class of C3-Symmetric Organic Bases: Highly Basic Proton Sponges and Ligands for Very Small Metal Cations. Angew. Chem. Int. Ed. 2003, 42, 4039–4042. [Google Scholar] [CrossRef]
- Alder, R.W. Design of C2-Chiral Diamines that Are Computationally Predicted to Be a Million-fold More Basic than the Original Proton Sponges. J. Am. Chem. Soc. 2005, 127, 7924–7931. [Google Scholar] [CrossRef]
- Kovačević, B.; Maksić, Z.B. High basicity of phosphorus—Proton affinity of tris-(tetramethylguanidinyl)phosphine and tris-(hexamethyltriaminophosphazenyl)phosphine by DFT calculations. Chem. Commun. 2006, 42, 1524–1526. [Google Scholar] [CrossRef]
- Kovačević, B.; Maksić, Z.B. High basicity of tris-(tetramethylguanidinyl)-phosphine imide in the gas phase and acetonitrile—A DFT study. Tetrahedron Lett. 2006, 47, 2553–2555. [Google Scholar] [CrossRef]
- Despotović, I.; Maksić, Z.B.; Vianello, R. Engineering Neutral Organic Bases and Superbases by Computational DFT Methods—Carbonyl Polyenes. Eur. J. Org. Chem. 2006, 2006, 5505–5514. [Google Scholar] [CrossRef]
- Despotović, I.; Maksić, Z.B.; Vianello, R. Computational design of Brønsted neutral organic superbases—[3]iminoradialenes and quinonimines are important synthetic targets. New J. Chem. 2007, 31, 52–62. [Google Scholar] [CrossRef]
- Despotović, I.; Kovačević, B.; Maksić, Z.B. Pyridine and s-triazine as building blocks of nonionic organic superbases—A density functional theory B3LYP study. New J. Chem. 2007, 31, 447–457. [Google Scholar] [CrossRef]
- Despotović, I.; Maksić, Z.B.; Vianello, R. Design of Brønsted Neutral Organic Bases and Superbases by Computational DFT Methods: Cyclic and Polycyclic Quinones and [3]Carbonylradialenes. Eur. J. Org. Chem. 2007, 2007, 3402–3413. [Google Scholar] [CrossRef]
- Despotović, I.; Kovačević, B.; Maksić, Z.B. Hyperstrong Neutral Organic Bases: Phosphazeno Azacalix[3](2,6)pyridines. Org. Lett. 2007, 9, 4709–4712. [Google Scholar] [CrossRef]
- Margetić, D.; Trošelj, P.; Ishikawa, T.; Kumamoto, T. Design of New Scaffolds for Increased Superbasicity of Bisguanidine Proton Sponges. Bull. Chem. Soc. Jpn. 2010, 83, 1055–1057. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Peran, N. Polycyclic croissant-like organic compounds are powerful superbases in the gas phase and acetonitrile—A DFT study. Chem. Commun. 2011, 47, 1327–1329. [Google Scholar] [CrossRef]
- Lo, R.; Singh, A.; Kesharwani, M.K.; Ganguly, B. Rational design of a new class of polycyclic organic bases bearing two superbasic sites and their applications in the CO2 capture and activation process. Chem. Commun. 2012, 48, 5865–5867. [Google Scholar] [CrossRef]
- Biswas, A.K.; Lo, R.; Ganguly, B. First Principles Studies toward the Design of Silylene Superbases: A Density Functional Theory Study. J. Phys. Chem. A 2013, 117, 3109–3117. [Google Scholar] [CrossRef]
- Despotović, I.; Vianello, R. Engineering exceptionally strong oxygen superbases with 1,8-diazanaphthalene di-N-oxides. Chem. Commun. 2014, 50, 10941–10944. [Google Scholar] [CrossRef] [PubMed]
- Barić, D.; Dragičević, I.; Kovačević, B. Cyclopropenimine as a hydrogen bond acceptor—Towards the strongest non-phosphorus superbases. Tetrahedron 2014, 70, 8571–8576. [Google Scholar] [CrossRef]
- Leito, I.; Koppel, I.A.; Koppel, I.; Kaupmees, K.; Tshepelevitsh, S.; Saame, J. Basicity Limits of Neutral Organic Superbases. Angew. Chem. Int. Ed. 2015, 54, 9262–9265. [Google Scholar] [CrossRef] [PubMed]
- Barić, D.; Kovačević, B. Designing a next generation of proton sponges: Cyclopropeniminophosphazenes as the strongest pincer ligands. Tetrahedron Lett. 2016, 57, 442–445. [Google Scholar] [CrossRef]
- Barić, D.; Kovačević, B. Cyclopropenimine as pincer ligand and strong electron donor in proton sponges. J. Phys. Org. Chem. 2016, 29, 750–758. [Google Scholar] [CrossRef]
- Margetić, D.; Antol, I. A DFT study of endocyclic allenes: Unprecedentedly superbasic hydrocarbons. New J. Chem. 2016, 40, 8191–8193. [Google Scholar] [CrossRef]
- Biswas, A.K.; Ganguly, B. Revealing Germylene Compounds to Attain Superbasicity with Sigma Donor Substituents: A Density Functional Theory Study. Chem. Eur. J. 2017, 23, 2700–2705. [Google Scholar] [CrossRef] [PubMed]
- Tandarić, T.; Vianello, R. Design of Exceptionally Strong Organic Superbases Based on Aromatic Pnictogen Oxides: Computational DFT Analysis of the Oxygen Basicity in the Gas Phase and Acetonitrile Solution. J. Phys. Chem. A 2018, 122, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.K.; Si, M.K.; Ganguly, B. The effect of σ/π, σ and π donors on the basicity of silylene superbases: A density functional theory study. New J. Chem. 2018, 42, 11153–11159. [Google Scholar] [CrossRef]
- Saadat, K.; Shiri, A.; Kovačević, B. Substituted troponimines: When aromatization of the conjugate acid leads to very strong neutral organic superbases. New J. Chem. 2018, 42, 14568–14575. [Google Scholar] [CrossRef]
- Saeidian, H.; Barfinejad, E. Design of Exceptional Strong Organosuperbases Based on Iminophosphorane and Azaphosphiridine Derivatives: Harnessing Ring Strain and Aromaticity to Engineer Neutral Superbases. ChemistrySelect 2019, 4, 3088–3095. [Google Scholar] [CrossRef]
- Radić, N.; Maksić, Z.B. Carbon Atom as an Extremely Strong Nucleophilic and Electrophilic Center: Dendritic Allenes Are Powerful Organic Proton and Hydride Sponges. J. Org. Chem. 2019, 84, 2425–2438. [Google Scholar] [CrossRef] [PubMed]
- Barić, D. Utilizing the Azaazulene Scaffolds in the Design of New Organic Superbases. ACS Omega 2019, 4, 15197–15207. [Google Scholar] [CrossRef] [PubMed]
- Valadbeigi, Y.; Vianello, R. Is It Possible to Achieve Organic Superbases Beyond the Basicity Limit Using Tetrahedrane Scaffolds? ChemistrySelect 2020, 5, 5794–5798. [Google Scholar] [CrossRef]
- Saeidian, H.; Mirjafary, Z. Engineering non-ionic carbon super- and hyperbases by a computational DFT approach: Substituted allenes have unprecedented cation affinities. New J. Chem. 2020, 44, 12967–12977. [Google Scholar] [CrossRef]
- Saadat, K.; Shiri, A.; Kovačević, B. Step Forward to Stronger Neutral Organic Superbases: Fused Troponimines. J. Org. Chem. 2020, 85, 11375–11381. [Google Scholar] [CrossRef] [PubMed]
- Deljuie, F.; Rouhani, M.; Saeidian, H. Exceptional design of super/hyperbases based on spiro-alleneic structures in gas phase: A density functional theory study. J. Phys. Org. Chem. 2022, 35, e4423. [Google Scholar] [CrossRef]
- Koneshlou, T.; Rouhani, M.; Saeidian, H.; Aliabad, J.M. Super/hyperbasicity of novel diquinonimino derivatives of guanidine in gas phase. Chem. Phys. Lett. 2022, 804, 139915. [Google Scholar] [CrossRef]
- Jalezadeh, A.; Mirjafary, Z.; Rouhani, M.; Saeidian, H. Basicity of five-membered cyclic allenes: Proton and cation affinity evaluation using density functional theory calculations. Int. J. Mass Spectrom. 2022, 482, 116929. [Google Scholar] [CrossRef]
- Valadbeigi, Y.; Taheri, R. Superbasicity of imines with bicyclo[5.1.0]octa-1,3,5,7-tetraene scaffold due to electron delocalization in the conjugated acids. Comput. Theor. Chem. 2023, 1222, 114076. [Google Scholar] [CrossRef]
- Koneshlou, T.; Rouhani, M.; Saeidian, H.; Aliabad, J.M. Biguanide-dihydropyrimidine dual scaffolds with impressive basicities according to DFT calculations. Comput. Theor. Chem. 2023, 1225, 114178. [Google Scholar] [CrossRef]
- Saha, A.; Ganguly, B. Exploiting the (–C–H···C–) Interaction to Design Cage-Functionalized Organic Superbases and Hyperbases: A Computational Study. ACS Omega 2023, 8, 38546–38556. [Google Scholar] [CrossRef]
- Yarikordeh, S.; Rouhani, M.; Saeidian, H. Computationally design aspects of superbasic amidine-arsinine 1-oxide binary frameworks. Chem. Phys. Lett. 2023, 833, 140905. [Google Scholar] [CrossRef]
- Al-Husseini, S.; Rouhani, M.; Saeidian, H. A brief computational look at the basicity strength of some experimentally observed strained allenes. Chem. Phys. Lett. 2024, 842, 141221. [Google Scholar] [CrossRef]
- Kulsha, A.V.; Ragoyja, E.G.; Ivashkevich, O.A. Strong Bases Design: Predicted Limits of Basicity. J. Phys. Chem. A 2022, 126, 3642–3652. [Google Scholar] [CrossRef]
- Maksić, Z.B.; Kovačević, B.; Vianello, R. Advances in Determining the Absolute Proton Affinities of Neutral Organic Molecules in the Gas Phase and Their Interpretation: A Theoretical Account. Chem. Rev. 2012, 112, 5240–5270. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. Enhanced Basicity of Push–Pull Nitrogen Bases in the Gas Phase. Chem. Rev. 2016, 116, 13454–13511. [Google Scholar] [CrossRef]
- Raczyńska, E.D.; Gal, J.-F.; Maria, P.-C. Strong Bases and beyond: The Prominent Contribution of Neutral Push–Pull Organic Molecules towards Superbases in the Gas Phase. Int. J. Mol. Sci. 2024, 25, 5591. [Google Scholar] [CrossRef]
- Kaljurand, I.; Saame, J.; Rodima, T.; Koppel, I.; Koppel, I.A.; Kögel, J.F.; Sundermeyer, J.; Köhn, U.; Coles, M.P.; Leito, I. Experimental Basicities of Phosphazene, Guanidinophosphazene, and Proton Sponge Superbases in the Gas Phase and Solution. J. Phys. Chem. A 2016, 120, 2591–2604. [Google Scholar] [CrossRef]
- Lõkov, M.; Tshepelevitsh, S.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Conjugated Nitrogen Heterocycles in Different Media. Eur. J. Org. Chem. 2017, 2017, 4475–4489. [Google Scholar] [CrossRef]
- Glasovac, Z.; Eckert-Maksić, M.; Maksić, Z.B. Basicity of organic bases and superbases in acetonitrile by the polarized continuum model and DFT calculations. New J. Chem. 2009, 33, 588–597. [Google Scholar] [CrossRef]
- Saame, J.; Rodima, T.; Tshepelevitsh, S.; Kütt, A.; Kaljurand, I.; Haljasorg, T.; Koppel, I.A.; Leito, I. Experimental Basicities of Superbasic Phosphonium Ylides and Phosphazenes. J. Org. Chem. 2016, 81, 7349–7361. [Google Scholar] [CrossRef]
- Rossini, E.; Bochevarov, A.D.; Knapp, E.W. Empirical Conversion of pKa Values between Different Solvents and Interpretation of the Parameters: Application to Water, Acetonitrile, Dimethyl Sulfoxide, and Methanol. ACS Omega 2018, 3, 1653–1662. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- Glasovac, Z.; Kovačević, B. Modeling pKa of the Brønsted Bases as an Approach to the Gibbs Energy of the Proton in Acetonitrile. Int. J. Mol. Sci. 2022, 23, 10576. [Google Scholar] [CrossRef]
- Kulsha, A.V.; Ivashkevich, O.A. Quantum-chemical study of the stability of solvents with respect to strong organic bases. Dokl. Natl. Acad. Sci. Belarus 2023, 67, 380–387. [Google Scholar] [CrossRef]
- Ebel, H.F.; Schneider, R. Ionization of Benzylmagnesium Chloride. Angew. Chem. Int. Ed. Engl. 1965, 4, 878. [Google Scholar] [CrossRef]
- Clayden, J.; Yasin, S.A. Pathways for decomposition of THF by organolithiums: The role of HMPA. New J. Chem. 2002, 26, 191–192. [Google Scholar] [CrossRef]
- Gremmo, N.; Randles, J.E.B. Solvated electrons in hexamethylphosphoramide. Part 1.—Conductivity of solutions of alkali metals. J. Chem. Soc. Faraday Trans. 1 1974, 70, 1480–1487. [Google Scholar] [CrossRef]
- Lee, K.P.; Trochimowicz, H.J. Morphogenesis of Nasal Tumors in Rats Exposed to Hexamethylphosphoramide by Inhalation. Environ. Res. 1984, 33, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Harman, A.E.; Voigt, J.M.; Frame, S.R.; Bogdanffy, M.S. Mitogenic responses of rat nasal epithelium to hexamethylphosphoramide inhalation exposure. Mutat. Res. Fundam. Mol. Mech. Mutagen. 1997, 380, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Kulsha, A.V.; Ivashkevich, O.A. pH Indicators for Strong Molecular Bases: A Theoretical Approach. J. Phys. Chem. A 2024, 128, 4701–4704. [Google Scholar] [CrossRef] [PubMed]
- Bockman, T.M.; Kochi, J.K. Isolation and Oxidation-Reduction of Methylviologen Cation Radicals. Novel Disproportionation in Charge-Transfer Salts by X-ray Crystallography. J. Org. Chem. 1990, 55, 4127–4135. [Google Scholar] [CrossRef]
- Alder, R.W.; Blake, M.E.; Chaker, L.; Harvey, J.N.; Paolini, F.; Schütz, J. When and How Do Diaminocarbenes Dimerize? Angew. Chem. Int. Ed. 2004, 43, 5896–5911. [Google Scholar] [CrossRef] [PubMed]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A Stable Two-Coordinate Acyclic Silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [Google Scholar] [CrossRef]
- Lui, M.W.; Merten, C.; Ferguson, M.J.; McDonald, R.; Xu, Y.; Rivard, E. Contrasting Reactivities of Silicon and Germanium Complexes Supported by an N-Heterocyclic Guanidine Ligand. Inorg. Chem. 2015, 54, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.A.L.; Ghosh, A. The curious chemistry of carbones. Nat. Chem. 2023, 15, 1042. [Google Scholar] [CrossRef] [PubMed]
- Loh, Y.K.; Melaimi, M.; Munz, D.; Bertrand, G. An Air-Stable “Masked” Bis(imino)carbene: A Carbon-Based Dual Ambiphile. J. Am. Chem. Soc. 2023, 145, 2064–2069. [Google Scholar] [CrossRef]
- Lavallo, V.; Dyker, C.A.; Donnadieu, B.; Bertrand, G. Synthesis and Ligand Properties of Stable Five-Membered-Ring Allenes Containing Only Second-Row Elements. Angew. Chem. Int. Ed. 2008, 47, 5411–5414. [Google Scholar] [CrossRef]
- Melaimi, M.; Parameswaran, P.; Donnadieu, B.; Frenking, G.; Bertrand, G. Synthesis and Ligand Properties of a Persistent, All-Carbon Four-Membered-Ring Allene. Angew. Chem. Int. Ed. 2009, 48, 4792–4795. [Google Scholar] [CrossRef]
- Ariai, J.; Ziegler, M.; Würtele, C.; Gellrich, U. An N-Heterocyclic Quinodimethane: A Strong Organic Lewis Base Exhibiting Diradical Reactivity. Angew. Chem. Int. Ed. 2024, 63, e202316720. [Google Scholar] [CrossRef]
- Erdmann, P.; Leitner, J.; Schwarz, J.; Greb, L. An Extensive Set of Accurate Fluoride Ion Affinities for p-Block Element Lewis Acids and Basic Design Principles for Strong Fluoride Ion Acceptors. ChemPhysChem 2020, 21, 987–994. [Google Scholar] [CrossRef]
- Dempsey, S.H.; Kass, S.R. Liberating the Anion: Evaluating Weakly Coordinating Cations. J. Org. Chem. 2022, 87, 15466–15482. [Google Scholar] [CrossRef]
- Brown, S.J.; Clark, J.H. Tetraphenylfluorophosphorane. J. Chem. Soc. Chem. Commun. 1983, 19, 1256–1257. [Google Scholar] [CrossRef]
- Brüggeller, P. Five-coordinate platinum(II) hydrides containing 1,1,4,7,10,10-hexaphenyl-1,4,7,10-tetraphosphadecane as a tetradentate monometallic ligand. Inorg. Chem. 1990, 29, 1742–1750. [Google Scholar] [CrossRef]
- King, R.B.; Cloyd, J.C., Jr. Poly(tertiary phosphines and arsines). X. Synthesis of methylated poly(tertiary phosphines). J. Am. Chem. Soc. 1975, 97, 53–60. [Google Scholar] [CrossRef]
- Bampos, N.; Field, L.D.; Messerle, B.A.; Smernik, R.J. Synthesis of new tetradentate oligophosphine ligands. Inorg. Chem. 1993, 32, 4084–4088. [Google Scholar] [CrossRef]
- Qi, X.-J.; Liu, L.; Fu, Y.; Guo, Q.-X. Ab Initio Calculations of pKa Values of Transition-Metal Hydrides in Acetonitrile. Organometallics 2006, 25, 5879–5886. [Google Scholar] [CrossRef]
- Morris, R.H. Brønsted–Lowry Acid Strength of Metal Hydride and Dihydrogen Complexes. Chem. Rev. 2016, 116, 8588–8654. [Google Scholar] [CrossRef]
- Schwesinger, R. Starke ungeladene Stickstoffbasen. Nachr. Chem. Tech. Lab. 1990, 38, 1214–1226. [Google Scholar] [CrossRef]
- Curtis, C.J.; Miedaner, A.; Ellis, W.W.; DuBois, D.L. Measurement of the Hydride Donor Abilities of [HM(diphosphine)2]+ Complexes (M = Ni, Pt) by Heterolytic Activation of Hydrogen. J. Am. Chem. Soc. 2002, 124, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Drews, T.; Rusch, D.; Seidel, S.; Willemsen, S.; Seppelt, K. Systematic Reactions of [Pt(PF3)4]. Chem. Eur. J. 2008, 14, 4280–4286. [Google Scholar] [CrossRef] [PubMed]
- Ozari, Y.; Jagur-Grodzinski, J. Donor strength of N-substituted phosphoramides. J. Chem. Soc. Chem. Commun. 1974, 10, 295–296. [Google Scholar] [CrossRef]
- Liakos, D.G.; Sparta, M.; Kesharwani, M.K.; Martin, J.M.L.; Neese, F. Exploring the Accuracy Limits of Local Pair Natural Orbital Coupled-Cluster Theory. J. Chem. Theory Comput. 2015, 11, 1525–1539. [Google Scholar] [CrossRef]
- Liakos, D.G.; Neese, F. Is It Possible to Obtain Coupled Cluster Quality Energies at near Density Functional Theory Cost? Domain-Based Local Pair Natural Orbital Coupled Cluster vs Modern Density Functional Theory. J. Chem. Theory Comput. 2015, 11, 4054–4063. [Google Scholar] [CrossRef]
- Riplinger, C.; Pinski, P.; Becker, U.; Valeev, E.F.; Neese, F. Sparse maps—A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. J. Chem. Phys. 2016, 144, 024109. [Google Scholar] [CrossRef]
- Saitow, M.; Becker, U.; Riplinger, C.; Valeev, E.F.; Neese, F. A new near-linear scaling, efficient and accurate, open-shell domain-based local pair natural orbital coupled cluster singles and doubles theory. J. Chem. Phys. 2017, 146, 164105. [Google Scholar] [CrossRef]
- Guo, Y.; Riplinger, C.; Becker, U.; Liakos, D.G.; Minenkov, Y.; Cavallo, L.; Neese, F. Communication: An improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD(T)]. J. Chem. Phys. 2018, 148, 011101. [Google Scholar] [CrossRef]
- Mallick, S.; Roy, B.; Kumar, P. A comparison of DLPNO-CCSD(T) and CCSD(T) method for the determination of the energetics of hydrogen atom transfer reactions. Comput. Theor. Chem. 2020, 1187, 112934. [Google Scholar] [CrossRef]
- Sandler, I.; Chen, J.; Taylor, M.; Sharma, S.; Ho, J. Accuracy of DLPNO-CCSD(T): Effect of Basis Set and System Size. J. Phys. Chem. A 2021, 125, 1553–1563. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard III, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Mahajan, G.R.; Kumbharkhane, A.C. Dielectric relaxation study of hexamethylphosphoramide—1,4-dioxane mixtures using time domain reflectometry (TDR) technique. Phys. Chem. Liq. 2012, 50, 513–522. [Google Scholar] [CrossRef]
- Simon, S.; Duran, M.; Dannenberg, J.J. How does basis set superposition error change the potential surfaces for hydrogen-bonded dimers? J. Chem. Phys. 1996, 105, 11024–11031. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Weigend, F.; Köhn, A.; Hättig, C. Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations. J. Chem. Phys. 2002, 116, 3175–3183. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Metz, B.; Stoll, H.; Dolg, M. Small-core multiconfiguration-Dirac–Hartree–Fock-adjusted pseudopotentials for post-d main group elements: Application to PbH and PbO. J. Chem. Phys. 2000, 113, 2563–2569. [Google Scholar] [CrossRef]
- Stoychev, G.L.; Auer, A.A.; Neese, F. Automatic Generation of Auxiliary Basis Sets. J. Chem. Theory Comput. 2017, 13, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Figgen, D.; Peterson, K.A.; Dolg, M.; Stoll, H. Energy-consistent pseudopotentials and correlation consistent basis sets for the 5d elements Hf–Pt. J. Chem. Phys. 2009, 130, 164108. [Google Scholar] [CrossRef] [PubMed]
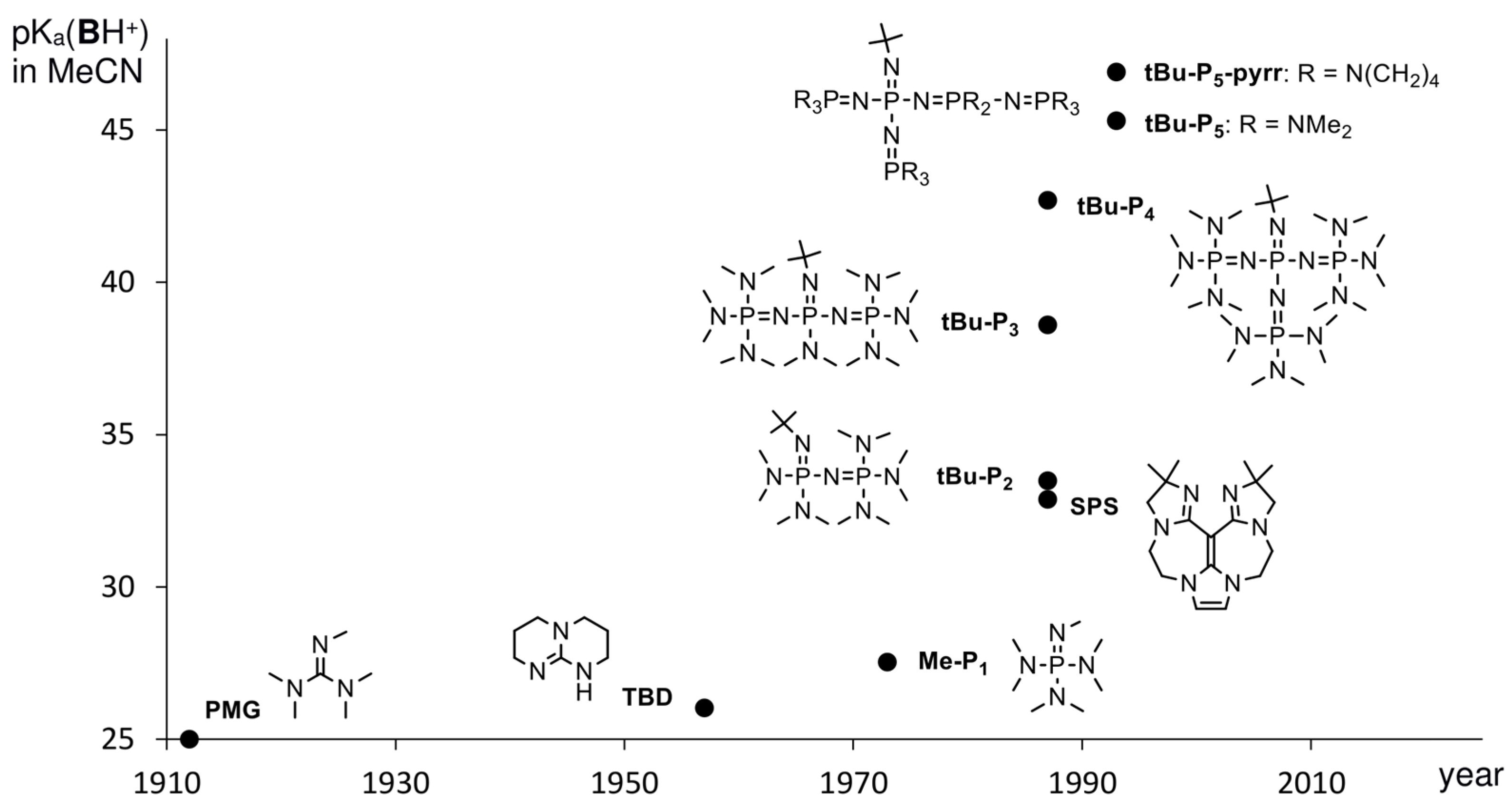
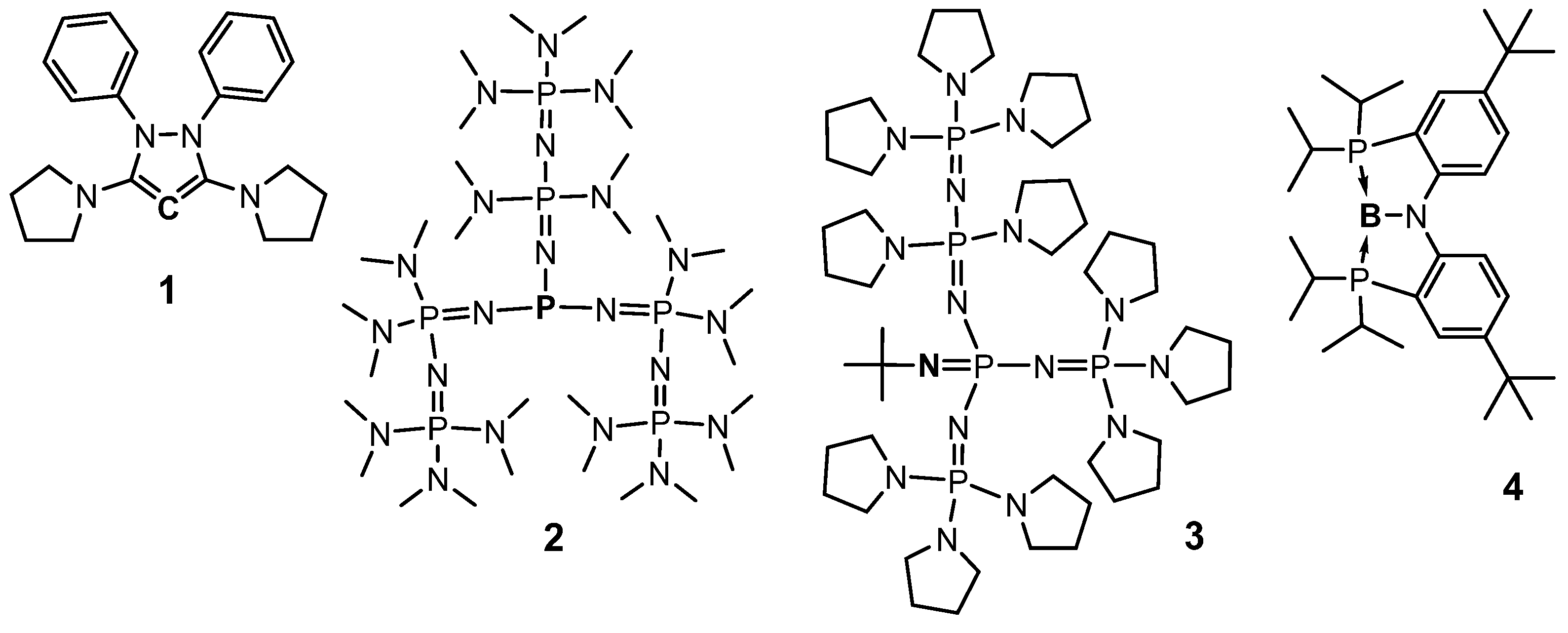
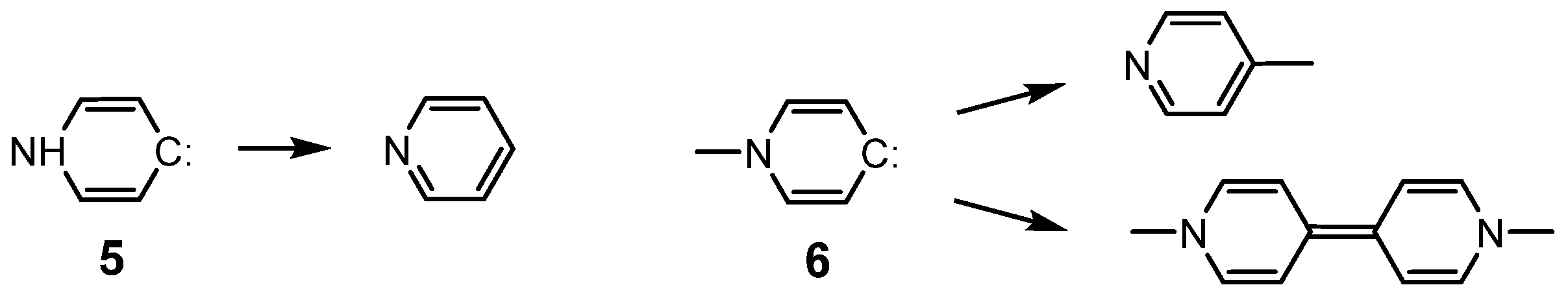
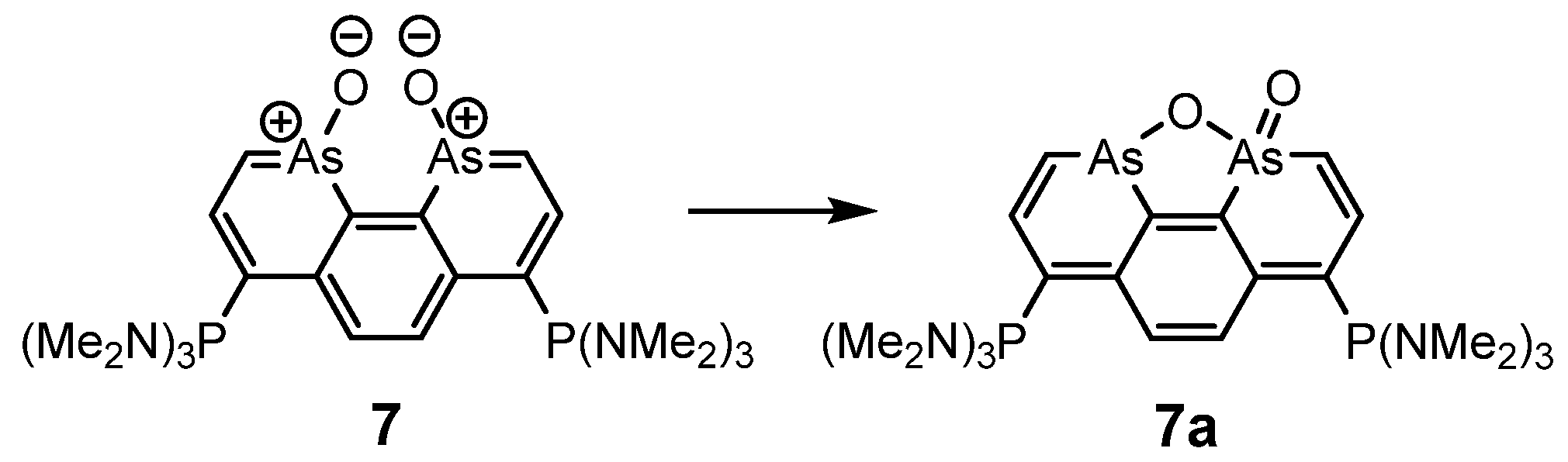






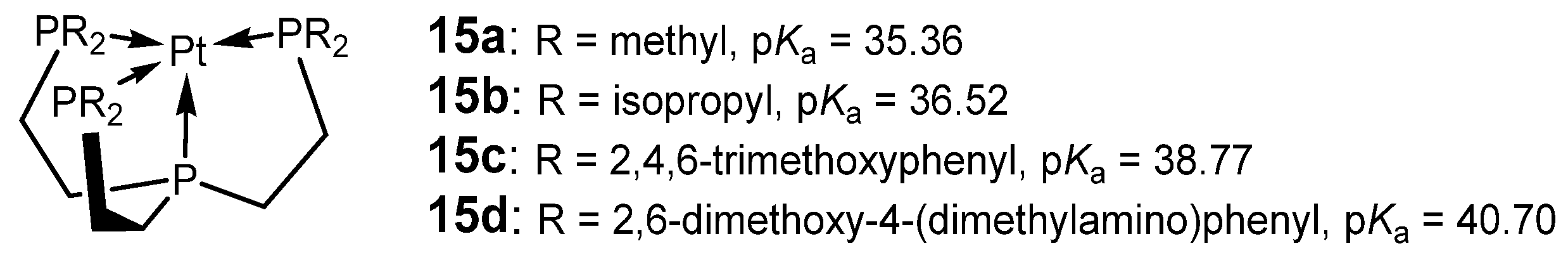

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base B | pKa(BH+) |
|---|---|
| pyridine | 1.94 |
| Et3N | 8.88 |
| TBD | 17.01 |
| SPS | 25.78 |
| tBu-P4 | 35.65 |
| carbene 1 | 39.28 |
| phosphine 2 | 40.59 |
| azene 3 | 40.82 |
| borylene 4 | 40.87 |
| Protonation Degree | 14a | 14b |
|---|---|---|
| neutral base | 53.02 | 54.67 |
| monoprotonated | 49.04 | 52.24 |
| diprotonated | 10.35 | 13.01 |
| triprotonated | 5.03 | 9.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulsha, A.V.; Ivashkevich, O.A.; Lyakhov, D.A.; Michels, D. Strong Bases Design: Key Techniques and Stability Issues. Int. J. Mol. Sci. 2024, 25, 8716. https://doi.org/10.3390/ijms25168716
Kulsha AV, Ivashkevich OA, Lyakhov DA, Michels D. Strong Bases Design: Key Techniques and Stability Issues. International Journal of Molecular Sciences. 2024; 25(16):8716. https://doi.org/10.3390/ijms25168716
Chicago/Turabian StyleKulsha, Andrey V., Oleg A. Ivashkevich, Dmitry A. Lyakhov, and Dominik Michels. 2024. "Strong Bases Design: Key Techniques and Stability Issues" International Journal of Molecular Sciences 25, no. 16: 8716. https://doi.org/10.3390/ijms25168716
APA StyleKulsha, A. V., Ivashkevich, O. A., Lyakhov, D. A., & Michels, D. (2024). Strong Bases Design: Key Techniques and Stability Issues. International Journal of Molecular Sciences, 25(16), 8716. https://doi.org/10.3390/ijms25168716