
Structural Characterization of Heat Shock Protein 90β and Molecular Interactions with Geldanamycin and Ritonavir: A Computational Study

Abstract
:1. Introduction
2. Results
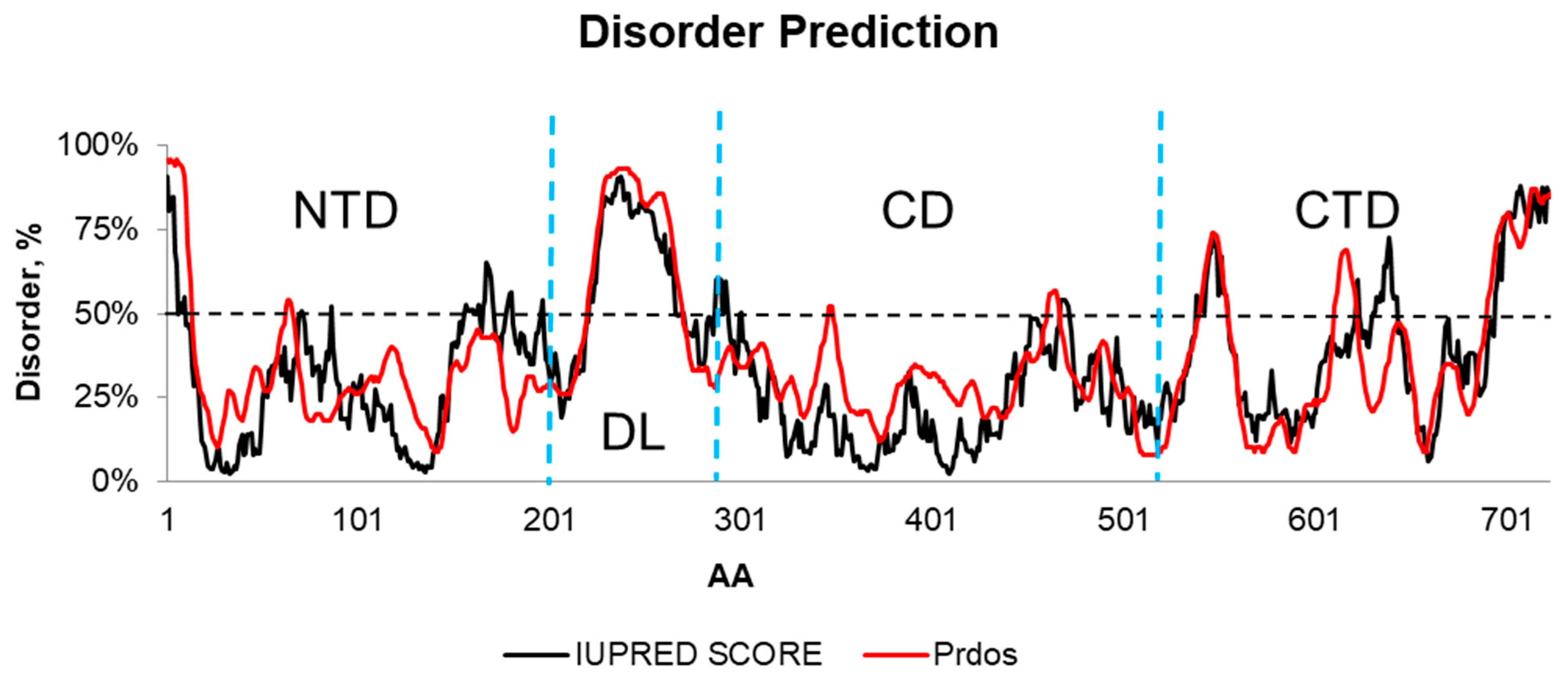
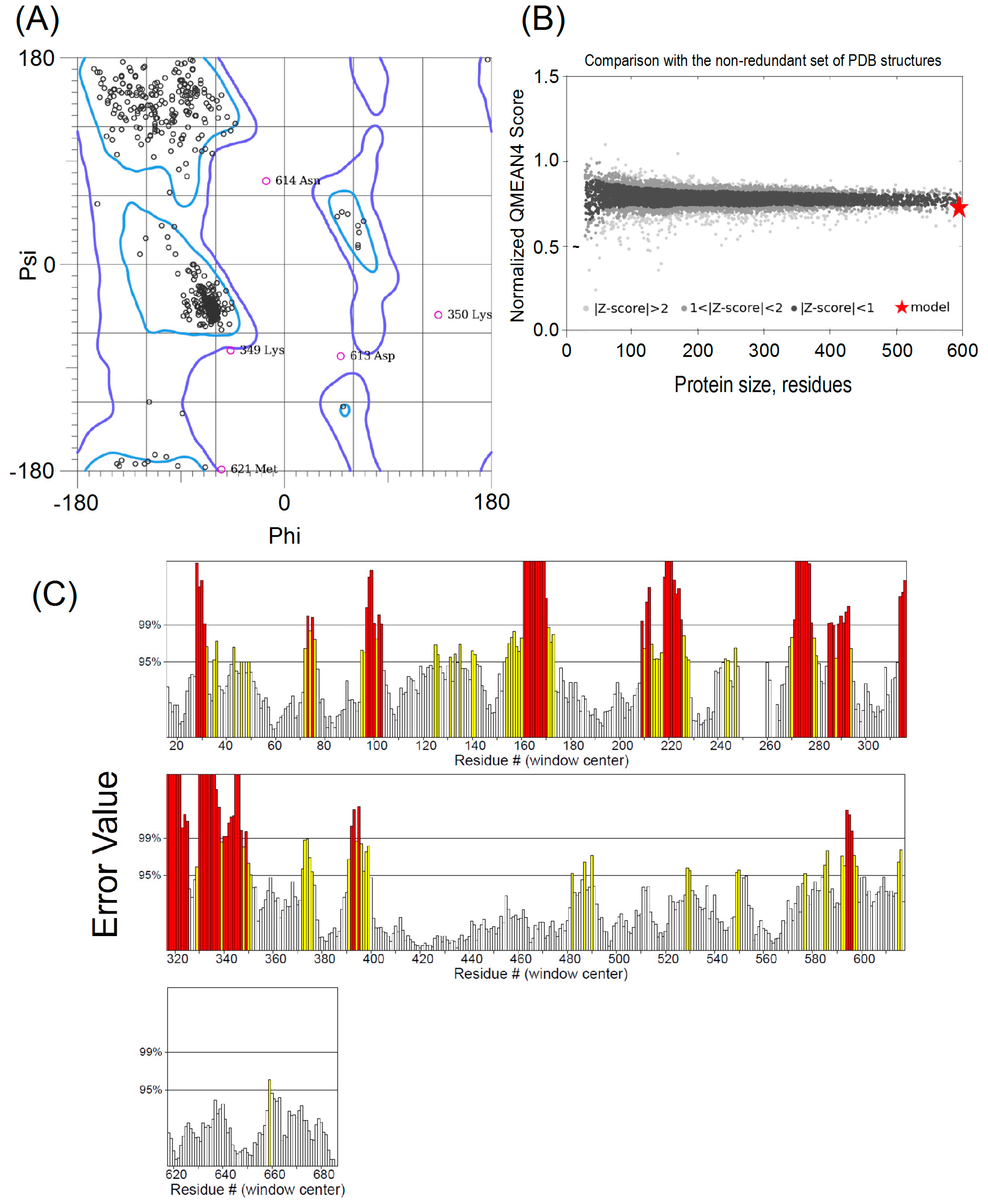
2.1. Comparative Modeling
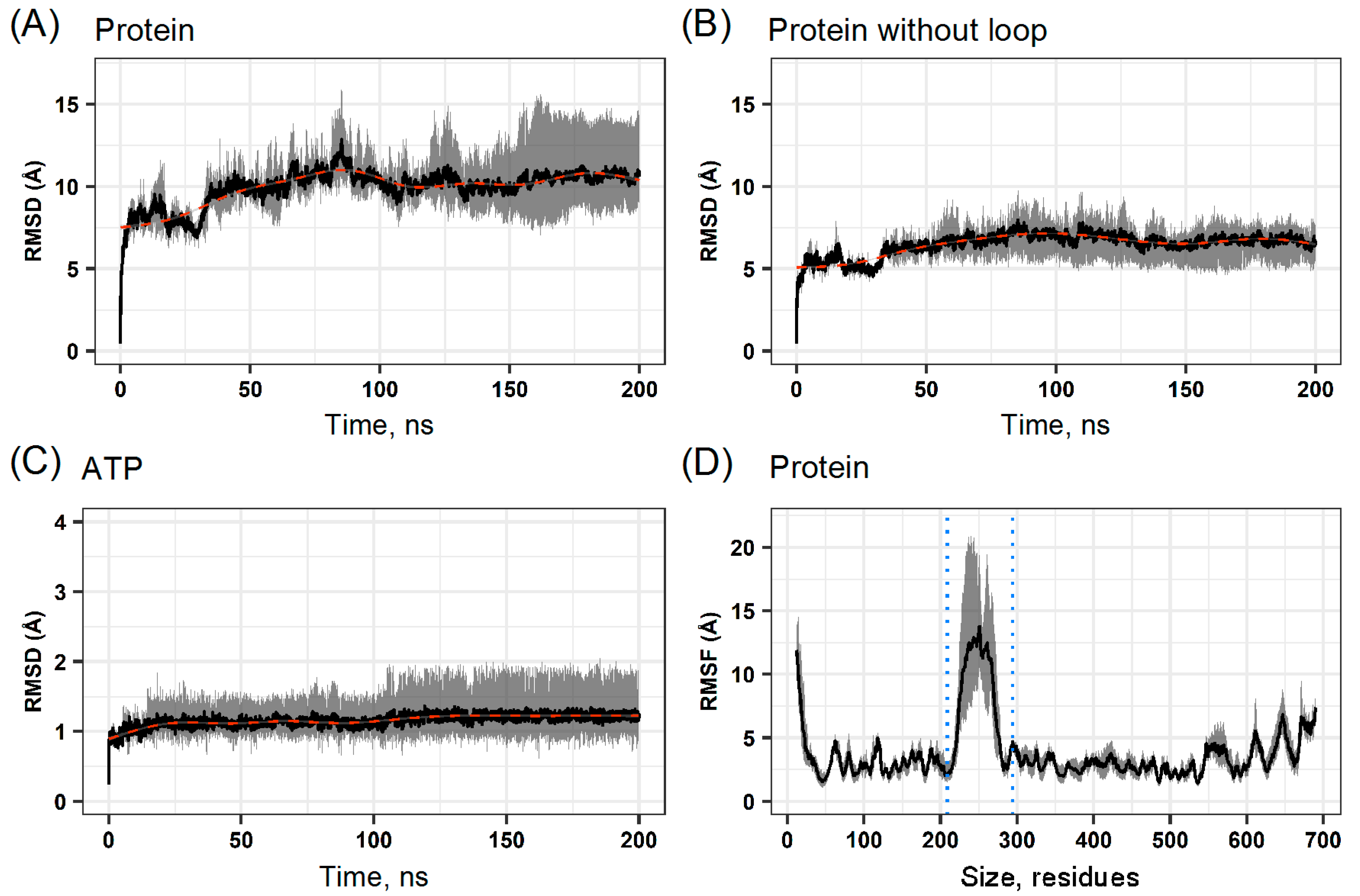
2.2. Molecular Dynamics of the Hsp90β Model
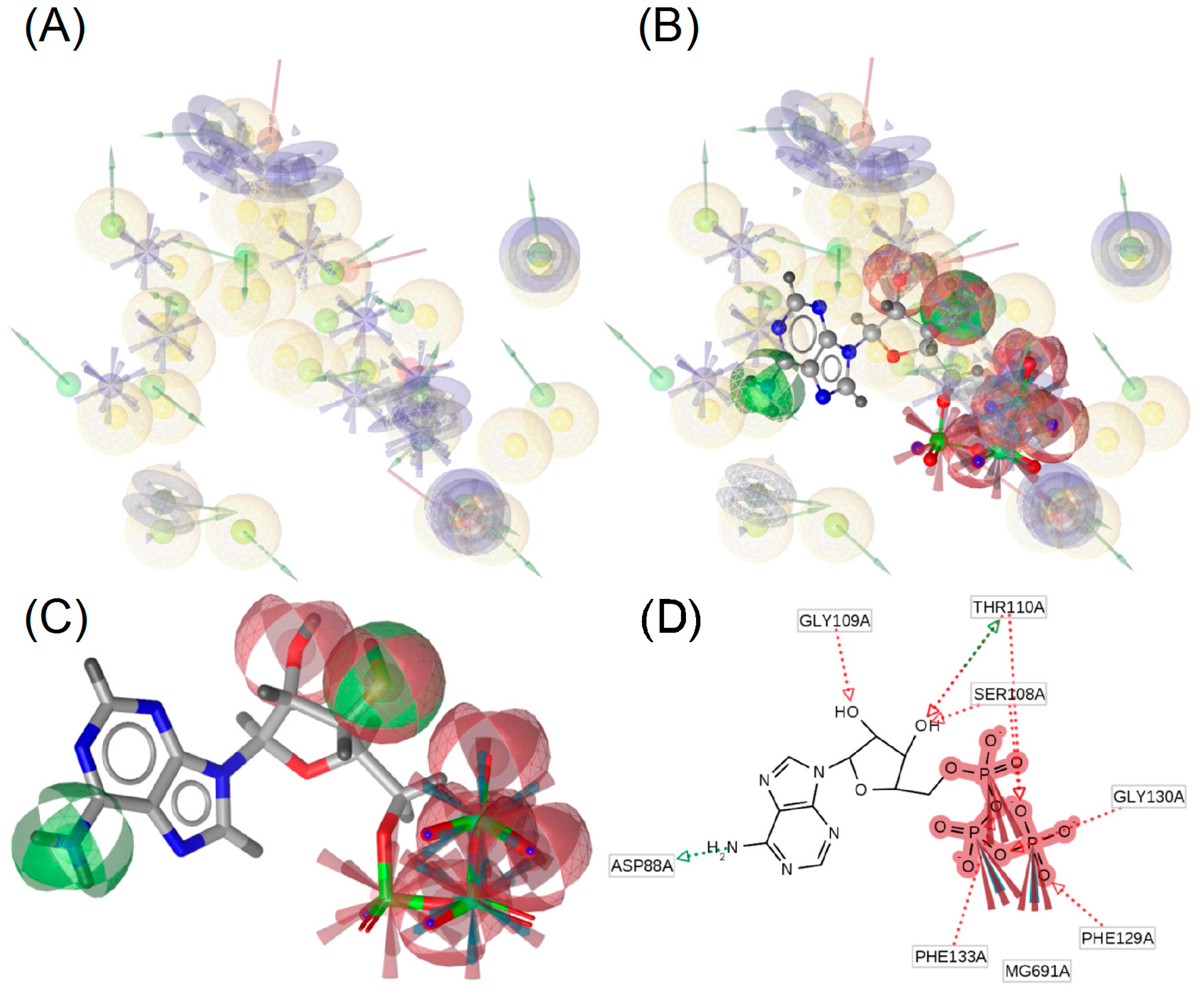
2.3. Molecular Docking and Ligand Interactions
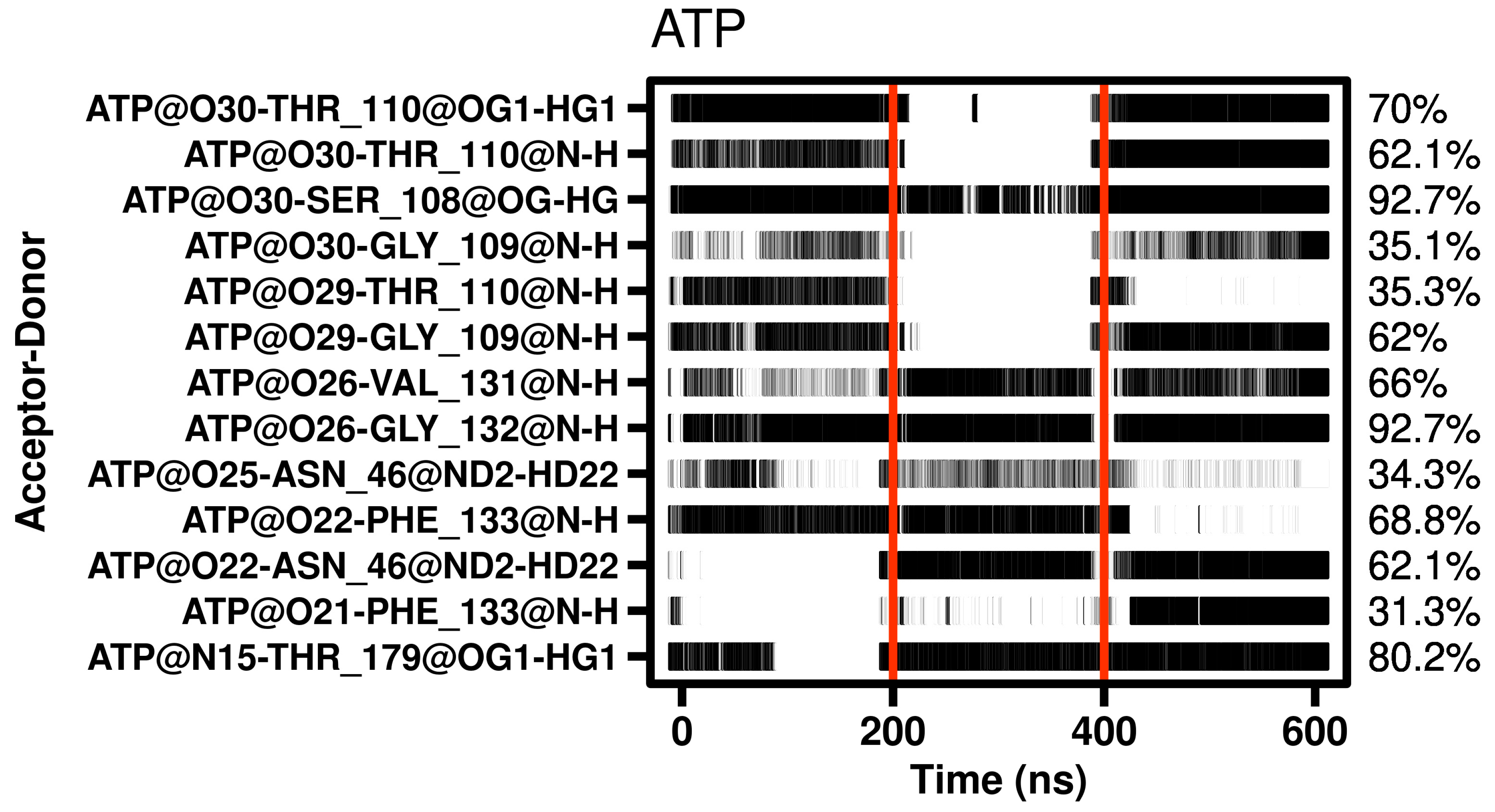
2.4. Molecular Dynamics of Geldanamycin and Ritonavir
2.5. Free Energy
3. Discussion
4. Methodology
4.1. Molecular Modeling and Quality Assessment
4.2. Ligand Parameterization and Molecular Docking
4.3. Interaction Modeling
4.4. Molecular Dynamics
4.5. Free Energy Calculation Using MMPBSA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Diniz, C.S.G.; Pellini, A.C.G.; Ribeiro, A.G.; Tedardi, M.V.; de Miranda, M.J.; Touso, M.M.; Baquero, O.S.; dos Santos, P.C.; Chiaravalloti-Neto, F. Breast Cancer Mortality and Associated Factors in São Paulo State, Brazil: An Ecological Analysis. BMJ Open 2017, 7, e016395. [Google Scholar] [CrossRef] [PubMed]
- Tilli, T.M.; Carels, N.; Tuszynski, J.A.; Pasdar, M. Validation of a Network-Based Strategy for the Optimization of Combinatorial Target Selection in Breast Cancer Therapy: siRNA Knockdown of Network Targets in MDA-MB-231 Cells as an in Vitro Model for Inhibition of Tumor Development. Oncotarget 2016, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Korsching, E.; Packeisen, J.; Liedtke, C.; Hungermann, D.; Wülfing, P.; van Diest, P.J.; Brandt, B.; Boecker, W.; Buerger, H. The Origin of Vimentin Expression in Invasive Breast Cancer: Epithelial–Mesenchymal Transition, Myoepithelial Histogenesis or Histogenesis from Progenitor Cells with Bilinear Differentiation Potential? J. Pathol. 2005, 206, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-L.; Eriksson, S.; Chang, Z.-F. Regulation and Functional Contribution of Thymidine Kinase 1 in Repair of DNA Damage*. J. Biol. Chem. 2010, 285, 27327–27335. [Google Scholar] [CrossRef] [PubMed]
- Alegre, M.M.; Robison, R.A.; O’Neill, K.L. Thymidine Kinase 1 Upregulation Is an Early Event in Breast Tumor Formation. J. Oncol. 2012, 2012, e575647. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, V.C.; Agarwal, V.; ELFadl, D.; Fox, J.N.; McManus, P.L.; Mahapatra, T.K.; Kneeshaw, P.J.; Drew, P.J.; Lind, M.J.; Cawkwell, L. Pilot and Feasibility Study: Comparative Proteomic Analysis by 2-DE MALDI TOF/TOF MS Reveals 14-3-3 Proteins as Putative Biomarkers of Response to Neoadjuvant Chemotherapy in ER-Positive Breast Cancer. J. Proteom. 2012, 75, 2745–2752. [Google Scholar] [CrossRef]
- Bergamaschi, A.; Frasor, J.; Borgen, K.; Stanculescu, A.; Johnson, P.; Rowland, K.; Wiley, E.L.; Katzenellenbogen, B.S. 14-3-3ζ as a Predictor of Early Time to Recurrence and Distant Metastasis in Hormone Receptor-Positive and -Negative Breast Cancers. Breast Cancer Res. Treat. 2013, 137, 689–696. [Google Scholar] [CrossRef]
- Boudreau, A.; Tanner, K.; Wang, D.; Geyer, F.C.; Reis-Filho, J.S.; Bissell, M.J. 14-3-3σ Stabilizes a Complex of Soluble Actin and Intermediate Filament to Enable Breast Tumor Invasion. Proc. Natl. Acad. Sci. USA 2013, 110, E3937–E3944. [Google Scholar] [CrossRef]
- Ge, J.; Normant, E.; Porter, J.R.; Ali, J.A.; Dembski, M.S.; Gao, Y.; Georges, A.T.; Grenier, L.; Pak, R.H.; Patterson, J.; et al. Design, Synthesis, and Biological Evaluation of Hydroquinone Derivatives of 17-Amino-17-Demethoxygeldanamycin as Potent, Water-Soluble Inhibitors of Hsp90. J. Med. Chem. 2006, 49, 4606–4615. [Google Scholar] [CrossRef]
- Kren, B.T.; Unger, G.M.; Abedin, M.J.; Vogel, R.I.; Henzler, C.M.; Ahmed, K.; Trembley, J.H. Preclinical Evaluation of Cyclin Dependent Kinase 11 and Casein Kinase 2 Survival Kinases as RNA Interference Targets for Triple Negative Breast Cancer Therapy. Breast Cancer Res. 2015, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.; Chou, P.-C.; Velazquez-Torres, G.; Samudio, I.; Parreno, K.; Huang, Y.; Tseng, C.; Vu, T.; Gully, C.; Su, C.-H.; et al. The Cell Cycle Regulator 14-3-3σ Opposes and Reverses Cancer Metabolic Reprogramming. Nat. Commun. 2015, 6, 7530. [Google Scholar] [CrossRef] [PubMed]
- Carels, N.; Tilli, T.; Tuszynski, J.A. A Computational Strategy to Select Optimized Protein Targets for Drug Development toward the Control of Cancer Diseases. PLoS ONE 2015, 10, e0115054. [Google Scholar] [CrossRef] [PubMed]
- Srirangam, A.; Mitra, R.; Wang, M.; Gorski, J.C.; Badve, S.; Baldridge, L.; Hamilton, J.; Kishimoto, H.; Hawes, J.; Li, L.; et al. Effects of HIV Protease Inhibitor Ritonavir on Akt-Regulated Cell Proliferation in Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1883–1896. [Google Scholar] [CrossRef]
- Ou, J.-R.; Tan, M.-S.; Xie, A.-M.; Yu, J.-T.; Tan, L. Heat Shock Protein 90 in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 796869. [Google Scholar] [CrossRef] [PubMed]
- Haase, M.; Fitze, G. HSP90AB1: Helping the Good and the Bad. Gene 2016, 575, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat Shock Protein 90 Inhibitors: An Update on Achievements, Challenges, and Future Directions. J. Med. Chem. 2020, 63, 1798–1822. [Google Scholar] [CrossRef] [PubMed]
- Murata, S.; Chiba, T.; Tanaka, K. CHIP: A Quality-Control E3 Ligase Collaborating with Molecular Chaperones. Proteasome Regualtion Cell Funct. 2003, 35, 572–578. [Google Scholar] [CrossRef]
- Gorska, M.; Popowska, U.; Sielicka-Dudzin, A.; Kuban-Jankowska, A.; Sawczuk, W.; Knap, N.; Cicero, G.; Wozniak, F. Geldanamycin and Its Derivatives as Hsp90 Inhibitors. FBL 2012, 17, 2269–2277. [Google Scholar] [CrossRef]
- Sato, A. The Human Immunodeficiency Virus Protease Inhibitor Ritonavir Is Potentially Active against Urological Malignancies. OncoTargets Ther. 2015, 8, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Moawad, E.Y. Identifying the Optimal Dose of Ritonavir in the Treatment of Malignancies. Metab. Brain Dis. 2014, 29, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, F.I.; Poulos, L.T. Ritonavir Analogues as a Probe for Deciphering the Cytochrome P450 3A4 Inhibitory Mechanism. Curr. Top. Med. Chem. 2014, 14, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- El-Rayes, B.F.; Ali, S.; Heilbrun, L.K.; Lababidi, S.; Bouwman, D.; Visscher, D.; Philip, P.A. Cytochrome P450 and Glutathione Transferase Expression in Human Breast Cancer. Clin. Cancer Res. 2003, 9, 1705. [Google Scholar] [PubMed]
- van Eijk, M.; Boosman, R.J.; Schinkel, A.H.; Huitema, A.D.R.; Beijnen, J.H. Cytochrome P450 3A4, 3A5, and 2C8 Expression in Breast, Prostate, Lung, Endometrial, and Ovarian Tumors: Relevance for Resistance to Taxanes. Cancer Chemother. Pharmacol. 2019, 84, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Verba, K.A.; Wang, R.Y.-R.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic Structure of Hsp90-Cdc37-Cdk4 Reveals That Hsp90 Traps and Stabilizes an Unfolded Kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- McGuffin, L.J.; Aldowsari, F.M.F.; Alharbi, S.M.A.; Adiyaman, R. ModFOLD8: Accurate Global and Local Quality Estimates for 3D Protein Models. Nucleic Acids Res. 2021, 49, W425–W430. [Google Scholar] [CrossRef]
- Benkert, P.; Tosatto, S.C.E.; Schomburg, D. QMEAN: A Comprehensive Scoring Function for Model Quality Assessment. Proteins Struct. Funct. Bioinform. 2008, 71, 261–277. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 Chaperone Machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Dernovšek, J.; Tomašič, T. Following the Design Path of Isoform-Selective Hsp90 Inhibitors: Small Differences, Great Opportunities. Pharmacol. Ther. 2023, 245, 108396. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.R.; Silva, J.R.A.; De Tássia Carvalho Cardoso, É.; Silva, E.O.; Lameira, J.; Do Nascimento, J.L.; Do Socorro Barros Brasil, D.; Alves, C.N. Combined Kinetic Studies and Computational Analysis on Kojic Acid Analogs as Tyrosinase Inhibitors. Molecules 2014, 19, 9591–9605. [Google Scholar] [CrossRef]
- Ahmed, F.; Soomro, A.M.; Chethikkattuveli Salih, A.R.; Samantasinghar, A.; Asif, A.; Kang, I.S.; Choi, K.H. A Comprehensive Review of Artificial Intelligence and Network Based Approaches to Drug Repurposing in Covid-19. Biomed. Pharmacother. 2022, 153, 113350. [Google Scholar] [CrossRef]
- Ahmed, F.; Lee, J.W.; Samantasinghar, A.; Kim, Y.S.; Kim, K.H.; Kang, I.S.; Memon, F.H.; Lim, J.H.; Choi, K.H. SperoPredictor: An Integrated Machine Learning and Molecular Docking-Based Drug Repurposing Framework With Use Case of COVID-19. Front. Public Health 2022, 10, 902123. [Google Scholar] [CrossRef]
- Wlodawer, A.; Minor, W.; Dauter, Z.; Jaskolski, M. Protein Crystallography for Aspiring Crystallographers or How to Avoid Pitfalls and Traps in Macromolecular Structure Determination. FEBS J. 2013, 280, 5705–5736. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.V.; Tayara, H.; Chong, K.T. Recent Studies of Artificial Intelligence on In Silico Drug Distribution Prediction. Int. J. Mol. Sci. 2023, 24, 1815. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Y.; Zhang, C.; Zheng, W.; Zhang, G.; Zhang, Y. Progressive Assembly of Multi-Domain Protein Structures from Cryo-EM Density Maps. Nat. Comput. Sci. 2022, 2, 265–275. [Google Scholar] [CrossRef]
- Chang, Y.; Hawkins, B.A.; Du, J.J.; Groundwater, P.W.; Hibbs, D.E.; Lai, F. A Guide to In Silico Drug Design. Pharmaceutics 2022, 15, 49. [Google Scholar] [CrossRef]
- Subramaniam, S.; Mehrotra, M.; Gupta, D. Virtual High Throughput Screening (vHTS)—A Perspective. Bioinformation 2008, 3, 14–17. [Google Scholar] [CrossRef]
- Mushebenge, A.G.-A.; Ugbaja, S.C.; Mbatha, N.A.; Khan, R.B.; Kumalo, H.M. Assessing the Potential Contribution of In Silico Studies in Discovering Drug Candidates That Interact with Various SARS-CoV-2 Receptors. Int. J. Mol. Sci. 2023, 24, 15518. [Google Scholar] [CrossRef]
- Gennari, L.; Merlotti, D.; Paola, V.D.; Nuti, R. Raloxifene in Breast Cancer Prevention. Expert Opin. Drug Saf. 2008, 7, 259–270. [Google Scholar] [CrossRef]
- Patel, V.; Liaw, B.; Oh, W. The Role of Ketoconazole in Current Prostate Cancer Care. Nat. Rev. Urol. 2018, 15, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, Q.; Zhang, S.; Liu, H.; Zhao, B.; Du, B.; Wang, W.; Lin, P.; Zhang, Z.; Zhong, Y.; et al. Digoxin Enhances the Anticancer Effect on Non-Small Cell Lung Cancer While Reducing the Cardiotoxicity of Adriamycin. Front. Pharmacol. 2020, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Visan, A.I.; Negut, I. Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life 2024, 14, 233. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 Chaperone Machinery*. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, J.; Guiu, X.A.; Outwin, E.A.; Schellenberger, P.; Roumeliotis, T.I.; Choudhary, J.S.; Pearl, L.H. HSP90-CDC37-PP5 Forms a Structural Platform for Kinase Dephosphorylation. Nat. Commun. 2022, 13, 7343. [Google Scholar] [CrossRef]
- Conforte, A.J.; Tuszynski, J.A.; da Silva, F.A.B.; Carels, N. Signaling Complexity Measured by Shannon Entropy and Its Application in Personalized Medicine. Front. Genet. 2019, 10, 930. [Google Scholar] [CrossRef]
- Pires, J.G.; da Silva, G.F.; Weyssow, T.; Conforte, A.J.; Pagnoncelli, D.; da Silva, F.A.B.; Carels, N. Galaxy and MEAN Stack to Create a User-Friendly Workflow for the Rational Optimization of Cancer Chemotherapy. Front. Genet. 2021, 12, 624259. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal Structure of an Hsp90–Nucleotide–P23/Sba1 Closed Chaperone Complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef]
- Jahn, M.; Rehn, A.; Pelz, B.; Hellenkamp, B.; Richter, K.; Rief, M.; Buchner, J.; Hugel, T. The Charged Linker of the Molecular Chaperone Hsp90 Modulates Domain Contacts and Biological Function. Proc. Natl. Acad. Sci. USA 2014, 111, 17881–17886. [Google Scholar] [CrossRef]
- Hainzl, O.; Lapina, M.C.; Buchner, J.; Richter, K. The Charged Linker Region Is an Important Regulator of Hsp90 Function*. J. Biol. Chem. 2009, 284, 22559–22567. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Mollapour, M.; Graf, C.; Lee, C.-T.; Scroggins, B.T.; Xu, W.; Haslerova, L.; Hessling, M.; Konstantinova, A.A.; Trepel, J.B.; et al. Hsp90 Charged-Linker Truncation Reverses the Functional Consequences of Weakened Hydrophobic Contacts in the N Domain. Nat. Struct. Mol. Biol. 2009, 16, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C. Mechanisms of Hsp90 Regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Won, J.; Kryshtafovych, A.; Seok, C. Assessment of Protein Model Structure Accuracy Estimation in CASP14: Old and New Challenges. Proteins Struct. Funct. Bioinform. 2021, 89, 1940–1948. [Google Scholar] [CrossRef]
- Pereira, J.; Simpkin, A.J.; Hartmann, M.D.; Rigden, D.J.; Keegan, R.M.; Lupas, A.N. High-Accuracy Protein Structure Prediction in CASP14. Proteins Struct. Funct. Bioinform. 2021, 89, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- Dutta Gupta, S.; Bommaka, M.K.; Banerjee, A. Inhibiting Protein-Protein Interactions of Hsp90 as a Novel Approach for Targeting Cancer. Eur. J. Med. Chem. 2019, 178, 48–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, C.; Chen, W.; Xu, Y.; Shi, Y.; Wen, Y.; Zhang, N. A Dynamic View of ATP-Coupled Functioning Cycle of Hsp90 N-Terminal Domain. Sci. Rep. 2015, 5, 9542. [Google Scholar] [CrossRef]
- Chadli, A.; Bouhouche, I.; Sullivan, W.; Stensgard, B.; McMahon, N.; Catelli, M.G.; Toft, D.O. Dimerization and N-Terminal Domain Proximity Underlie the Function of the Molecular Chaperone Heat Shock Protein 90. Proc. Natl. Acad. Sci. USA 2000, 97, 12524. [Google Scholar] [CrossRef] [PubMed]
- Salsbury, F.R. Molecular Dynamics Simulations of Protein Dynamics and Their Relevance to Drug Discovery. Endocr. Metab. Dis. Technol.-Importance Protein Dyn. 2010, 10, 738–744. [Google Scholar] [CrossRef]
- Sakkiah, S.; Thangapandian, S.; John, S.; Lee, K.W. Pharmacophore Based Virtual Screening, Molecular Docking Studies to Design Potent Heat Shock Protein 90 Inhibitors. Eur. J. Med. Chem. 2011, 46, 2937–2947. [Google Scholar] [CrossRef]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef]
- Piippo, N.; Korhonen, E.; Hytti, M.; Skottman, H.; Kinnunen, K.; Josifovska, N.; Petrovski, G.; Kaarniranta, K.; Kauppinen, A. Hsp90 Inhibition as a Means to Inhibit Activation of the NLRP3 Inflammasome. Sci. Rep. 2018, 8, 6720. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.; Workman, P. Inhibitors of the HSP90 Molecular Chaperone: Current Status. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2006; Volume 95, pp. 323–348. ISBN 0065-230X. [Google Scholar]
- Neckers, L.; Workman, P. Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, K.C.; Atreya, H.S.; Harsha Raj, M.; Salimath, B.P.; Aparna, H.S. A Pyrrole-Based Natural Small Molecule Mitigates HSP90 Expression in MDA-MB-231 Cells and Inhibits Tumor Angiogenesis in Mice by Inactivating HSF-1. Cell Stress Chaperones 2017, 22, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.; Pettit, S.N.; Magnolo, S.K.; Sanvoisin, J.; Chen, W.; Wood, S.P.; Freeman, L.D.; Pengelly, R.J.; Hughes, D.E. Fragment Screening Using Capillary Electrophoresis (CEfrag) for Hit Identification of Heat Shock Protein 90 ATPase Inhibitors. J. Biomol. Screen. 2012, 17, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Gatti, G.; Di Biagio, A.; Casazza, R.; De Pascalis, C.; Bassetti, M.; Cruciani, M.; Vella, S.; Bassetti, D. The Relationship between Ritonavir Plasma Levels and Side-Effects: Implications for Therapeutic Drug Monitoring. AIDS 1999, 13, 2083–2089. [Google Scholar] [CrossRef]
- Arodola, O.A.; Soliman, M.E. Could the FDA-Approved Anti-HIV PR Inhibitors Be Promising Anticancer Agents? An Answer from Enhanced Docking Approach and Molecular Dynamics Analyses. Drug Des. Devel. Ther. 2015, 9, 6055–6065. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Atkins, J.D.; Boateng, S.Y.; Sorensen, T.; McGuffin, L.J. Disorder Prediction Methods, Their Applicability to Different Protein Targets and Their Usefulness for Guiding Experimental Studies. Int. J. Mol. Sci. 2015, 16, 19040–19054. [Google Scholar] [CrossRef]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of Disordered Protein Regions from Amino Acid Sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef]
- Erdős, G.; Dosztányi, Z. Analyzing Protein Disorder with IUPred2A. Curr. Protoc. Bioinform. 2020, 70, e99. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y. How Significant Is a Protein Structure Similarity with TM-Score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Tejero, R.; Montelione, G.T. Evaluating Protein Structures Determined by Structural Genomics Consortia. Proteins Struct. Funct. Bioinform. 2007, 66, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. Sect. D 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of Protein Structures: Patterns of Nonbonded Atomic Interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Zhang, Y. Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization. Biophys. J. 2011, 101, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson–Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- DesJarlais, R.L.; Sheridan, R.P.; Seibel, G.L.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Using Shape Complementarity as an Initial Screen in Designing Ligands for a Receptor Binding Site of Known Three-Dimensional Structure. J. Med. Chem. 1988, 31, 722–729. [Google Scholar] [CrossRef]
- Biovia, D.S. Discovery Studio Modeling Environment; Release 2021; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Showalter, S.A.; Brüschweiler, R. Validation of Molecular Dynamics Simulations of Biomolecules Using NMR Spin Relaxation as Benchmarks: Application to the AMBER99SB Force Field. J. Chem. Theory Comput. 2007, 3, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Swain, S.S.; Paidesetty, S.K.; Dehury, B.; Sahoo, J.; Vedithi, S.C.; Mahapatra, N.; Hussain, T.; Padhy, R.N. Molecular Docking and Simulation Study for Synthesis of Alternative Dapsone Derivative as a Newer Antileprosy Drug in Multidrug Therapy. J. Cell. Biochem. 2018, 119, 9838–9852. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Cerutti, D.S.; Cisneros, G.A.; Cruzeiro, V.W.D.; Forouzesh, N.; Giese, T.J.; Götz, A.W.; Gohlke, H.; et al. Amber 24; University of California: San Francisco, CA, USA, 2023. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | ΔH, Average | −TΔS, Average | ΔG | Experimental |
|---|---|---|---|---|
| ATP | −30.43 ± 2.30 | 16.69 ± 0.12 | −13.73 ± 3.92 | −9.07 |
| GDM | −20.98 ± 2.90 | 10.62 ± 3.39 | −10.35 ± 7.15 | −8.39 |
| RIT | −16.81 ± 2.62 | 10.16 ± 1.83 | −6.66 ± 8.47 | −7.24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, C.R.; Antunes, D.; Caffarena, E.; Carels, N. Structural Characterization of Heat Shock Protein 90β and Molecular Interactions with Geldanamycin and Ritonavir: A Computational Study. Int. J. Mol. Sci. 2024, 25, 8782. https://doi.org/10.3390/ijms25168782
Lima CR, Antunes D, Caffarena E, Carels N. Structural Characterization of Heat Shock Protein 90β and Molecular Interactions with Geldanamycin and Ritonavir: A Computational Study. International Journal of Molecular Sciences. 2024; 25(16):8782. https://doi.org/10.3390/ijms25168782
Chicago/Turabian StyleLima, Carlyle Ribeiro, Deborah Antunes, Ernesto Caffarena, and Nicolas Carels. 2024. "Structural Characterization of Heat Shock Protein 90β and Molecular Interactions with Geldanamycin and Ritonavir: A Computational Study" International Journal of Molecular Sciences 25, no. 16: 8782. https://doi.org/10.3390/ijms25168782