
Embryonic Lethal Phenotyping to Identify Candidate Genes Related with Birth Defects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
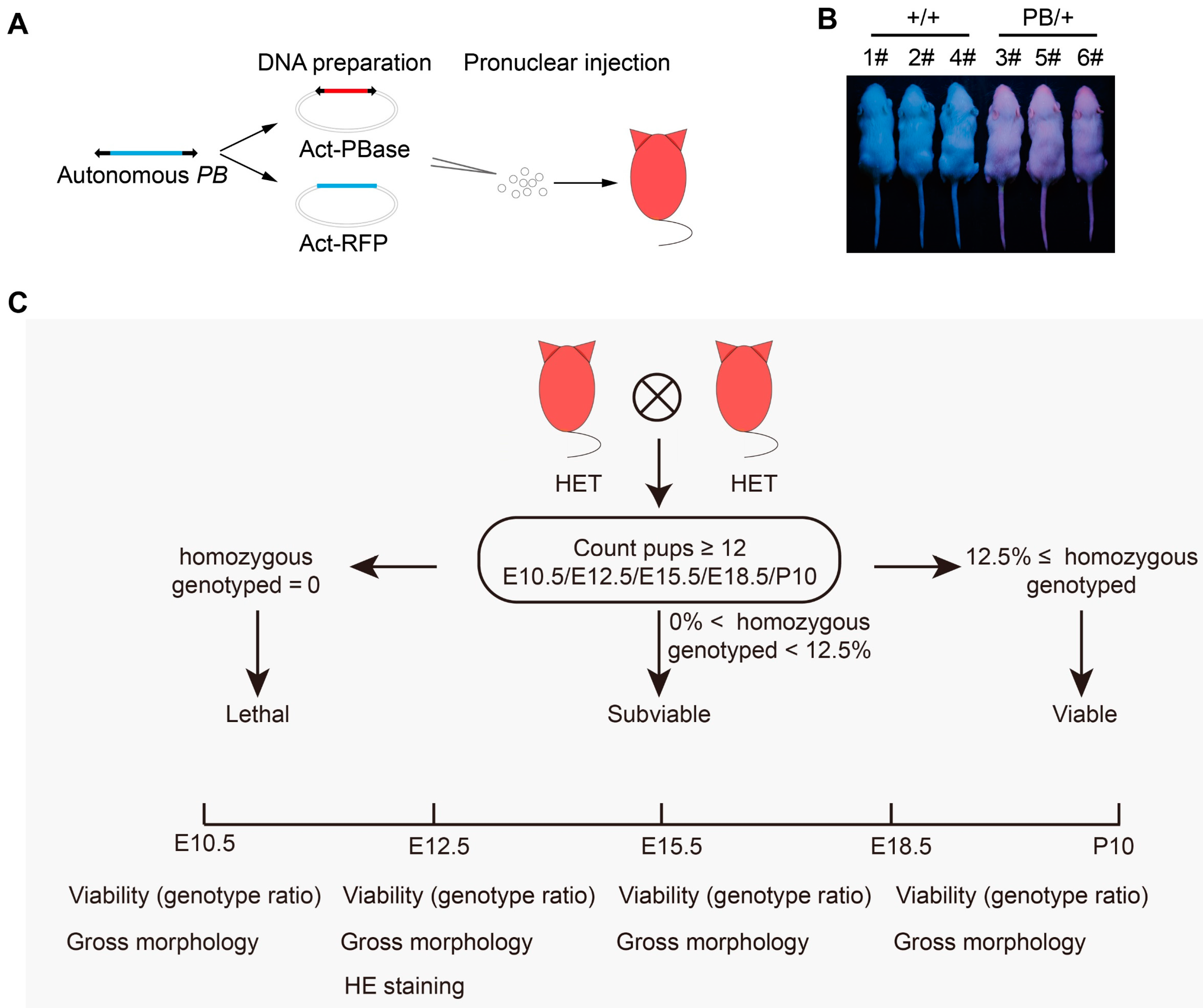
2.1. The Phenotyping Pipeline and Identification of Lethal Phenotypes in Mice
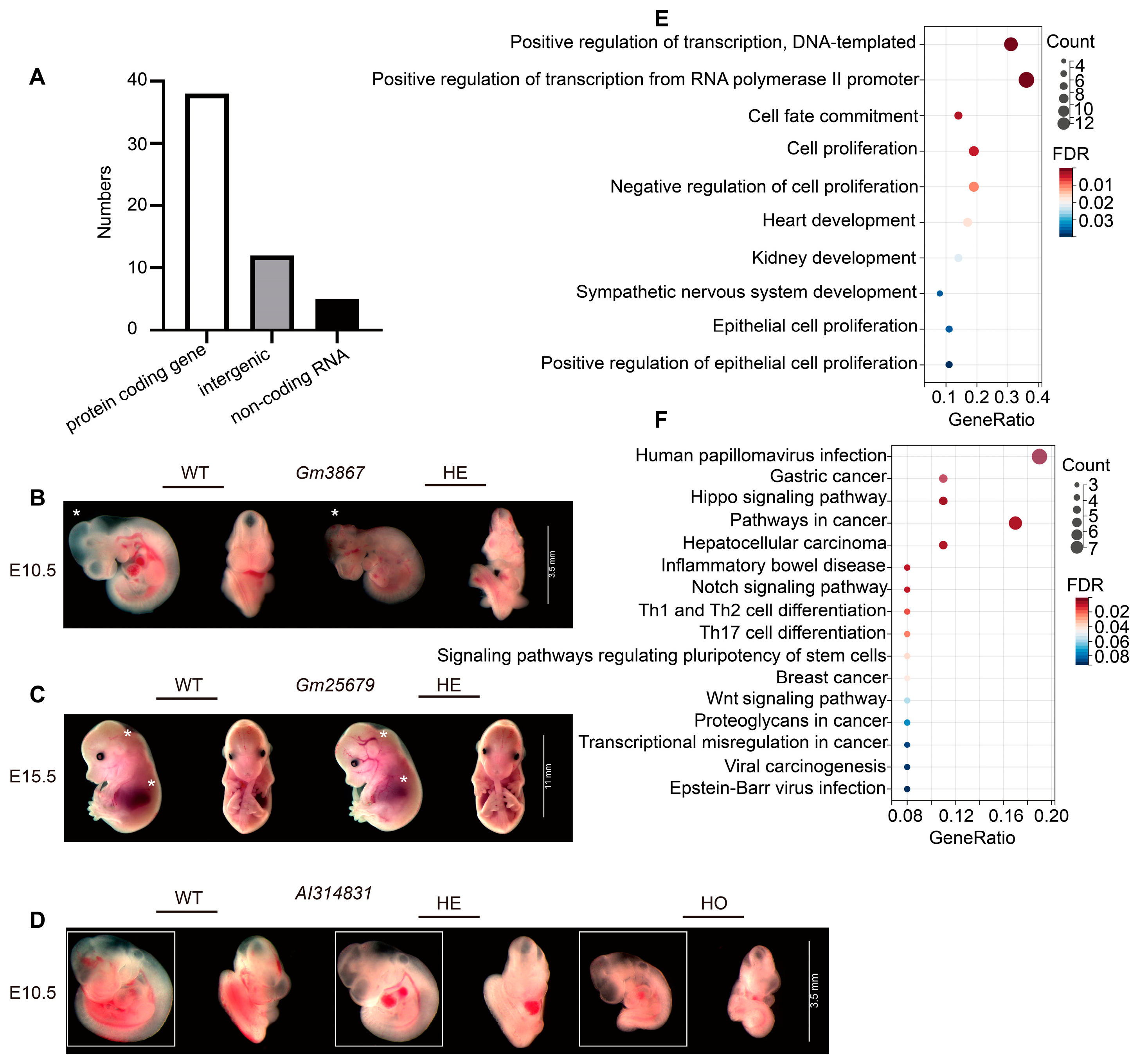
2.2. Functional Enrichment Analysis of Embryonic Lethal Gene
2.3. Body Weight and Composition Analysis of Embryonic Lethal Gene Suggest Smad2 as a Metabolic Regulator
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Gene List Analysis
4.3. Measurement of Body Composition and Body Length
4.4. Metabolic Rate and Physical Activity
4.5. Morphological Observation of Embryos
4.6. Histological Analysis
4.7. Quantitative Real-Time PCR Analysis
4.8. Bioinformatic Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calcagni, G.; Pugnaloni, F.; Digilio, M.C.; Unolt, M.; Putotto, C.; Niceta, M.; Baban, A.; Piceci Sparascio, F.; Drago, F.; De Luca, A.; et al. Cardiac Defects and Genetic Syndromes: Old Uncertainties and New Insights. Genes 2021, 12, 1047. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Gleeson, J.G. Closing in on Mechanisms of Open Neural Tube Defects. Trends Neurosci. 2020, 43, 519–532. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Announcement: World Birth Defects Day—March 3, 2017. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 195. [CrossRef] [PubMed]
- Wright, N.J.; Anderson, J.E.; Ozgediz, D.; Farmer, D.L.; Banu, T. Addressing paediatric surgical care on World Birth Defects Day. Lancet 2018, 391, 1019. [Google Scholar] [CrossRef]
- Bhandari, J.; Thada, P.K. Neural Tube Disorders; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Kang, L.; Cao, G.; Jing, W.; Liu, J.; Liu, M. Global, regional, and national incidence and mortality of congenital birth defects from 1990 to 2019. Eur. J. Pediatr. 2023, 182, 1781–1792. [Google Scholar] [CrossRef]
- Lee, K.S.; Choi, Y.J.; Cho, J.; Lee, H.; Lee, H.; Park, S.J.; Park, J.S.; Hong, Y.C. Environmental and Genetic Risk Factors of Congenital Anomalies: An Umbrella Review of Systematic Reviews and Meta-Analyses. J. Korean Med. Sci. 2021, 36, e183. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.C.H.D. Global, regional, and national burden of congenital heart disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Child Adolesc. Health 2020, 4, 185–200. [Google Scholar] [CrossRef]
- Mouse Genome Sequencing, C.; Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef]
- Adams, D.; Baldock, R.; Bhattacharya, S.; Copp, A.J.; Dickinson, M.; Greene, N.D.; Henkelman, M.; Justice, M.; Mohun, T.; Murray, S.A.; et al. Bloomsbury report on mouse embryo phenotyping: Recommendations from the IMPC workshop on embryonic lethal screening. Dis. Models Mech. 2013, 6, 571–579. [Google Scholar] [CrossRef]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef]
- Ding, S.; Wu, X.; Li, G.; Han, M.; Zhuang, Y.; Xu, T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005, 122, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Wilkinson, M.N.; Chen, X.; Sankararaman, S.; Mayhew, D.; Mitra, R.D. An optimized, broadly applicable piggyBac transposon induction system. Nucleic Acids Res. 2017, 45, e55. [Google Scholar] [CrossRef]
- Tian, J.; Tong, D.; Li, Z.; Wang, E.; Yu, Y.; Lv, H.; Hu, Z.; Sun, F.; Wang, G.; He, M.; et al. Mage transposon: A novel gene delivery system for mammalian cells. Nucleic Acids Res. 2024, 52, 2724–2739. [Google Scholar] [CrossRef] [PubMed]
- Collier, L.S.; Carlson, C.M.; Ravimohan, S.; Dupuy, A.J.; Largaespada, D.A. Cancer gene discovery in solid tumours using transposon-based somatic mutagenesis in the mouse. Nature 2005, 436, 272–276. [Google Scholar] [CrossRef]
- Weber, J.; de la Rosa, J.; Grove, C.S.; Schick, M.; Rad, L.; Baranov, O.; Strong, A.; Pfaus, A.; Friedrich, M.J.; Engleitner, T.; et al. PiggyBac transposon tools for recessive screening identify B-cell lymphoma drivers in mice. Nat. Commun. 2019, 10, 1415. [Google Scholar] [CrossRef]
- Chang, H.; Pan, Y.; Landrette, S.; Ding, S.; Yang, D.; Liu, L.; Tian, L.; Chai, H.; Li, P.; Li, D.M.; et al. Efficient genome-wide first-generation phenotypic screening system in mice using the piggyBac transposon. Proc. Natl. Acad. Sci. USA 2019, 116, 18507–18516. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.V.; Jin, K.; Liu, Y.; Yang, W.; Xie, X.; Ye, L.; Wang, L.; Zhu, L.; Ding, S.; Su, Y.; et al. PBmice: An integrated database system of piggyBac (PB) insertional mutations and their characterizations in mice. Nucleic Acids Res. 2008, 36, D729–D734. [Google Scholar] [CrossRef]
- Cui, J.; Ding, Y.; Chen, S.; Zhu, X.; Wu, Y.; Zhang, M.; Zhao, Y.; Li, T.R.; Sun, L.V.; Zhao, S.; et al. Disruption of Gpr45 causes reduced hypothalamic POMC expression and obesity. J. Clin. Investig. 2016, 126, 3192–3206. [Google Scholar] [CrossRef]
- Perez-Garcia, V.; Fineberg, E.; Wilson, R.; Murray, A.; Mazzeo, C.I.; Tudor, C.; Sienerth, A.; White, J.K.; Tuck, E.; Ryder, E.J.; et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 2018, 555, 463–468. [Google Scholar] [CrossRef]
- Jonusiene, V.; Sasnauskiene, A. Notch and Endometrial Cancer. Adv. Exp. Med. Biol. 2021, 1287, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ming, T.; Tang, S.; Ren, S.; Yang, H.; Liu, M.; Tao, Q.; Xu, H. Wnt signaling in colorectal cancer: Pathogenic role and therapeutic target. Mol. Cancer 2022, 21, 144. [Google Scholar] [CrossRef] [PubMed]
- Tepekoy, F.; Akkoyunlu, G.; Demir, R. The role of Wnt signaling members in the uterus and embryo during pre-implantation and implantation. J. Assist. Reprod. Genet. 2015, 32, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Paonessa, M.; Borini, A.; Coticchio, G. Genetic causes of preimplantation embryo developmental failure. Mol. Reprod. Dev. 2021, 88, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Cuman, C.; Menkhorst, E.; Winship, A.; Van Sinderen, M.; Osianlis, T.; Rombauts, L.J.; Dimitriadis, E. Fetal-maternal communication: The role of Notch signalling in embryo implantation. Reproduction 2014, 147, R75–R86. [Google Scholar] [CrossRef]
- Massimiani, M.; Lacconi, V.; La Civita, F.; Ticconi, C.; Rago, R.; Campagnolo, L. Molecular Signaling Regulating Endometrium-Blastocyst Crosstalk. Int. J. Mol. Sci. 2019, 21, 23. [Google Scholar] [CrossRef]
- Dietrich, B.; Haider, S.; Meinhardt, G.; Pollheimer, J.; Knofler, M. WNT and NOTCH signaling in human trophoblast development and differentiation. Cell. Mol. Life Sci. 2022, 79, 292. [Google Scholar] [CrossRef]
- Duhl, D.M.; Stevens, M.E.; Vrieling, H.; Saxon, P.J.; Miller, M.W.; Epstein, C.J.; Barsh, G.S. Pleiotropic effects of the mouse lethal yellow (Ay) mutation explained by deletion of a maternally expressed gene and the simultaneous production of agouti fusion RNAs. Development 1994, 120, 1695–1708. [Google Scholar] [CrossRef] [PubMed]
- Perks, K.L.; Ferreira, N.; Richman, T.R.; Ermer, J.A.; Kuznetsova, I.; Shearwood, A.J.; Lee, R.G.; Viola, H.M.; Johnstone, V.P.A.; Matthews, V.; et al. Adult-onset obesity is triggered by impaired mitochondrial gene expression. Sci. Adv. 2017, 3, e1700677. [Google Scholar] [CrossRef]
- Roest, P.A.; Molin, D.G.; Schalkwijk, C.G.; van Iperen, L.; Wentzel, P.; Eriksson, U.J.; Gittenberger-de Groot, A.C. Specific local cardiovascular changes of Nepsilon-(carboxymethyl)lysine, vascular endothelial growth factor, and Smad2 in the developing embryos coincide with maternal diabetes-induced congenital heart defects. Diabetes 2009, 58, 1222–1228. [Google Scholar] [CrossRef]
- Wu, J.X.; Shi, M.; Gong, B.M.; Ji, B.W.; Hu, C.C.; Wang, G.C.; Lei, L.; Tang, C.; Sun, L.V.; Wu, X.H.; et al. An miRNA-mRNA integrative analysis in human placentas and mice: Role of the Smad2/miR-155-5p axis in the development of fetal growth restriction. Front. Bioeng. Biotechnol. 2023, 11, 1159805. [Google Scholar] [CrossRef]
- Georgi, B.; Voight, B.F.; Bucan, M. From mouse to human: Evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet. 2013, 9, e1003484. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, J.E.; Zhu, A.; Robertson, D.L.; Hentges, K.E. Defining the role of essential genes in human disease. PLoS ONE 2011, 6, e27368. [Google Scholar] [CrossRef] [PubMed]
- Alcolea, M.P.; Colom, B.; Llado, I.; Gianotti, M.; Garcia-Palmer, F.J. Mitochondrial transcription factor A (TFAM) is increased in rat embryo during placentation and associated with mitochondrial differentiation. Cell. Physiol. Biochem. 2006, 17, 79–88. [Google Scholar] [CrossRef]
- Salazar-Petres, E.; Pereira-Carvalho, D.; Lopez-Tello, J.; Sferruzzi-Perri, A.N. Placental structure, function, and mitochondrial phenotype relate to fetal size in each fetal sex in micedagger. Biol. Reprod. 2022, 106, 1292–1311. [Google Scholar] [CrossRef] [PubMed]
- Dunker, N.; Krieglstein, K. Targeted mutations of transforming growth factor-beta genes reveal important roles in mouse development and adult homeostasis. Eur. J. Biochem. 2000, 267, 6982–6988. [Google Scholar] [CrossRef]
- Green, R.M.; Feng, W.; Phang, T.; Fish, J.L.; Li, H.; Spritz, R.A.; Marcucio, R.S.; Hooper, J.; Jamniczky, H.; Hallgrimsson, B.; et al. Tfap2a-dependent changes in mouse facial morphology result in clefting that can be ameliorated by a reduction in Fgf8 gene dosage. Dis. Models Mech. 2015, 8, 31–43. [Google Scholar] [CrossRef]
- Szumska, D.; Pieles, G.; Essalmani, R.; Bilski, M.; Mesnard, D.; Kaur, K.; Franklyn, A.; El Omari, K.; Jefferis, J.; Bentham, J.; et al. VACTERL/caudal regression/Currarino syndrome-like malformations in mice with mutation in the proprotein convertase Pcsk5. Genes Dev. 2008, 22, 1465–1477. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, M.; Mi, D.; Lu, T.; Wang, B.; Dong, H.; Zhong, S.; Chen, Y.; Sun, L.; Zhou, X.; et al. Mouse and human share conserved transcriptional programs for interneuron development. Science 2021, 374, eabj6641. [Google Scholar] [CrossRef]
- Nemeth, K.; Bayraktar, R.; Ferracin, M.; Calin, G.A. Non-coding RNAs in disease: From mechanisms to therapeutics. Nat. Rev. Genet. 2024, 25, 211–232. [Google Scholar] [CrossRef]
- Kerr, A.; Baxter, R.C. Noncoding RNA actions through IGFs and IGF binding proteins in cancer. Oncogene 2022, 41, 3385–3393. [Google Scholar] [CrossRef] [PubMed]
- Grafanaki, K.; Grammatikakis, I.; Ghosh, A.; Gopalan, V.; Olgun, G.; Liu, H.; Kyriakopoulos, G.C.; Skeparnias, I.; Georgiou, S.; Stathopoulos, C.; et al. Noncoding RNA circuitry in melanoma onset, plasticity, and therapeutic response. Pharmacol. Ther. 2023, 248, 108466. [Google Scholar] [CrossRef] [PubMed]
- Bella, F.; Campo, S. Long non-coding RNAs and their involvement in bipolar disorders. Gene 2021, 796–797, 145803. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef] [PubMed]
- Muntean, I.; Toganel, R.; Benedek, T. Genetics of Congenital Heart Disease: Past and Present. Biochem. Genet. 2017, 55, 105–123. [Google Scholar] [CrossRef]
- Cheng, Z.; Shang, Y.; Xu, X.; Dong, Z.; Zhang, Y.; Du, Z.; Lu, X.; Zhang, T. Presenilin 1 mutation likely contributes to U1 small nuclear RNA dysregulation and Alzheimer's disease-like symptoms. Neurobiol. Aging 2021, 100, 1–10. [Google Scholar] [CrossRef]
- Fasolo, F.; Jin, H.; Winski, G.; Chernogubova, E.; Pauli, J.; Winter, H.; Li, D.Y.; Glukha, N.; Bauer, S.; Metschl, S.; et al. Long Noncoding RNA MIAT Controls Advanced Atherosclerotic Lesion Formation and Plaque Destabilization. Circulation 2021, 144, 1567–1583. [Google Scholar] [CrossRef]
- Tan, Y.T.; Lin, J.F.; Li, T.; Li, J.J.; Xu, R.H.; Ju, H.Q. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun. 2021, 41, 109–120. [Google Scholar] [CrossRef]
- Ma, H.; Hu, T.; Tao, W.; Tong, J.; Han, Z.; Herndler-Brandstetter, D.; Wei, Z.; Liu, R.; Zhou, T.; Liu, Q.; et al. A lncRNA from an inflammatory bowel disease risk locus maintains intestinal host-commensal homeostasis. Cell Res. 2023, 33, 372–388. [Google Scholar] [CrossRef]
- Brown, S.D.M.; Hancock, J.M. The mouse genome. Vertebr. Genomes 2006, 2, 33–45. [Google Scholar] [CrossRef]
- Shen, Y.; Yue, F.; McCleary, D.F.; Ye, Z.; Edsall, L.; Kuan, S.; Wagner, U.; Dixon, J.; Lee, L.; Lobanenkov, V.V.; et al. A map of the cis-regulatory sequences in the mouse genome. Nature 2012, 488, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Oudelaar, A.M.; Higgs, D.R. The relationship between genome structure and function. Nat. Rev. Genet. 2021, 22, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Zhang, B.H.; Wu, B.Y.; Xiao, H.T.; Li, Z.H.; Li, R.Y.; Xu, X.W.; Li, T. Signaling pathways in obesity: Mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 298. [Google Scholar] [CrossRef] [PubMed]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.F.; Nguyen, H.T.; Veitia, R.A. Causes and effects of haploinsufficiency. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Irudayam, M.J.; Al Abdallah, Q.; Jones, T.L.; Mims, T.S.; Puchowicz, M.A.; Pierre, J.F.; Brown, C.W. SMAD2 and SMAD3 differentially regulate adiposity and the growth of subcutaneous white adipose tissue. FASEB J. 2021, 35, e22018. [Google Scholar] [CrossRef]
- Ding, S.; Xu, T.; Wu, X. Generation of genetically engineered mice by the piggyBac transposon system. Methods Mol. Biol. 2014, 1194, 171–185. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, B.; Gong, B.; Zheng, Y.; Sun, L.; Wu, X. Embryonic Lethal Phenotyping to Identify Candidate Genes Related with Birth Defects. Int. J. Mol. Sci. 2024, 25, 8788. https://doi.org/10.3390/ijms25168788
Yan B, Gong B, Zheng Y, Sun L, Wu X. Embryonic Lethal Phenotyping to Identify Candidate Genes Related with Birth Defects. International Journal of Molecular Sciences. 2024; 25(16):8788. https://doi.org/10.3390/ijms25168788
Chicago/Turabian StyleYan, Bing, Baoming Gong, Yufang Zheng, Lei Sun, and Xiaohui Wu. 2024. "Embryonic Lethal Phenotyping to Identify Candidate Genes Related with Birth Defects" International Journal of Molecular Sciences 25, no. 16: 8788. https://doi.org/10.3390/ijms25168788