
Regulation of TIR-1/SARM-1 by miR-71 Protects Dopaminergic Neurons in a C. elegans Model of LRRK2-Induced Parkinson’s Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
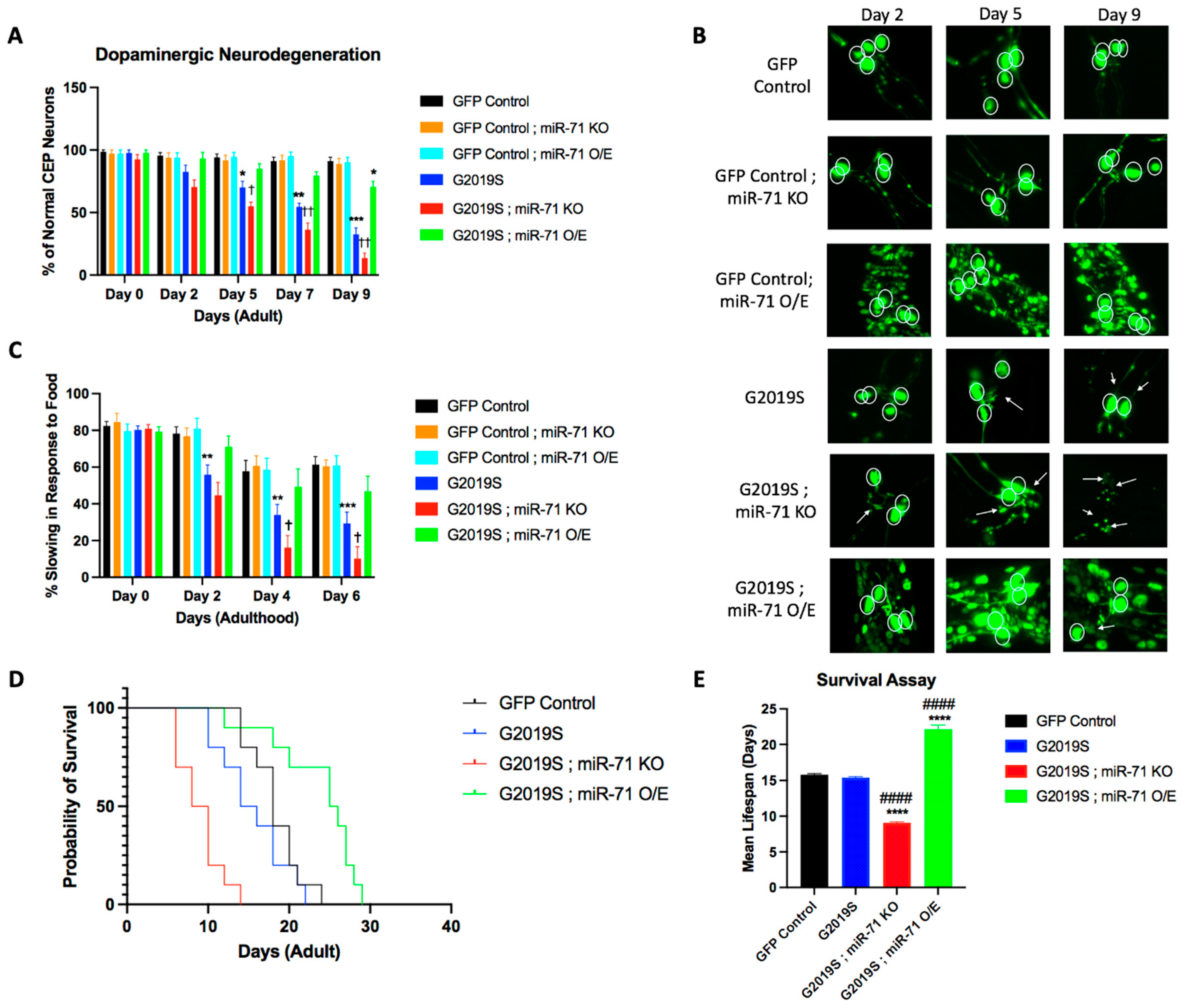
2.1. miR-71 Rescues Loss of Dopaminergic Neurons
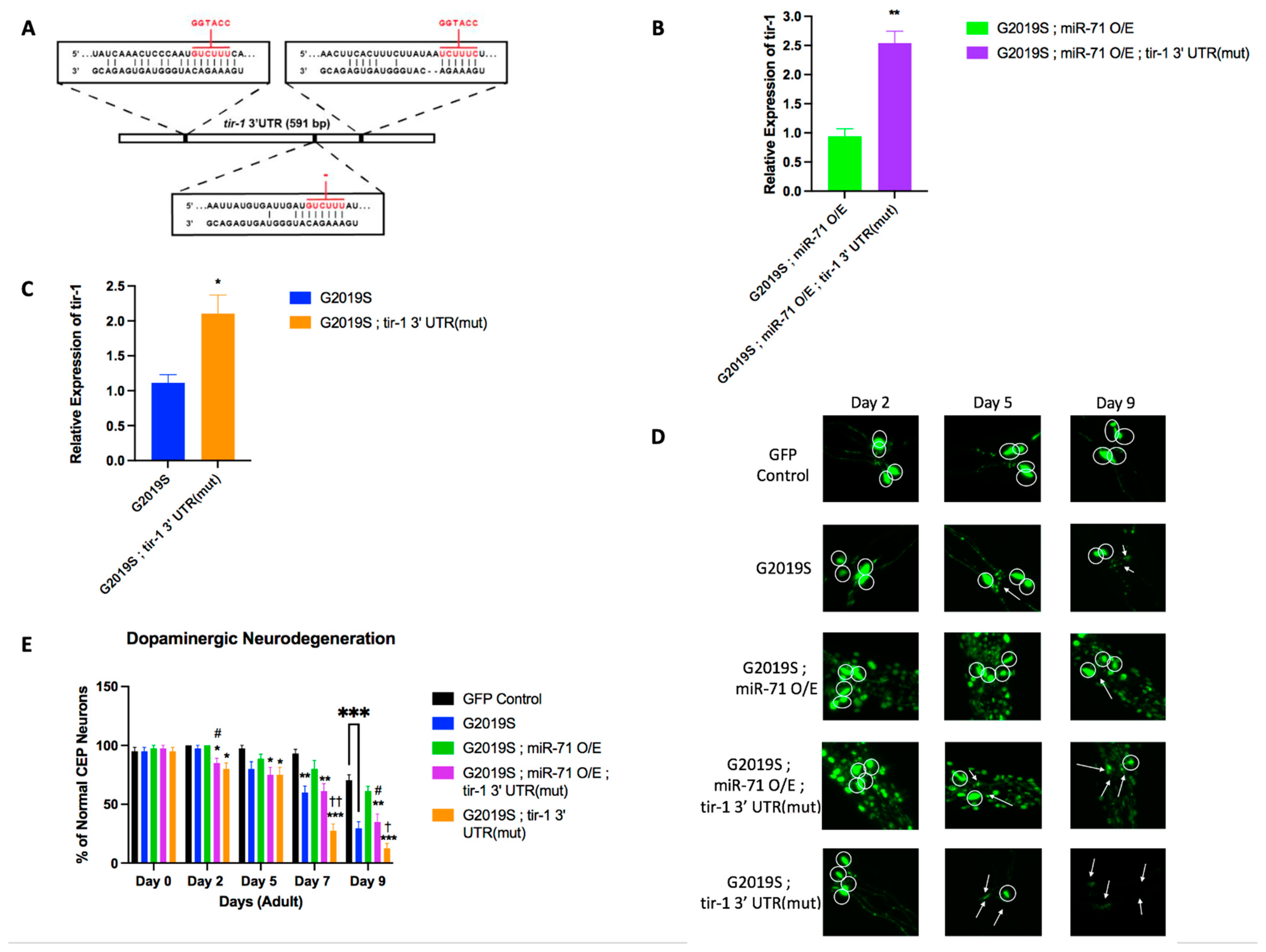
2.2. Loss of tir-1 Reverses Effects of miR-71 Knockout
2.3. miR-71 Regulates tir-1 to Rescue Dopaminergic Neurons
3. Discussion
4. Materials and Methods
4.1. Worm Culturing, Strains, and General Maintenance
4.2. Basal Slowing Assay
4.3. Dopaminergic Neurodegeneration Assay
4.4. qRT-PCR
4.5. Survival Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zafar, S.; Yaddanapudi, S.S. Parkinson Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018; ISBN 978-0-9944381-6-4. [Google Scholar]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef]
- Tokars, V.; Chen, C.; Parisiadou, L. Closing the Structure-to-Function Gap for LRRK2. Trends Biochem. Sci. 2022, 47, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Giaime, E.; Tong, Y.; Wagner, L.K.; Yuan, Y.; Huang, G.; Shen, J. Age-Dependent Dopaminergic Neurodegeneration and Impairment of the Autophagy-Lysosomal Pathway in LRRK-Deficient Mice. Neuron 2017, 96, 796–807.e6. [Google Scholar] [CrossRef]
- Rui, Q.; Ni, H.; Li, D.; Gao, R.; Chen, G. The Role of LRRK2 in Neurodegeneration of Parkinson Disease. Curr. Neuropharmacol. 2018, 16, 1348–1357. [Google Scholar] [CrossRef]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic Etiology of Parkinson Disease Associated with Mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 Genes: A Mutation Update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic Risk Factors in Parkinson’s Disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hamamichi, S.; Dae Lee, B.; Yang, D.; Ray, A.; Caldwell, G.A.; Caldwell, K.A.; Dawson, T.M.; Smith, W.W.; Dawson, V.L. Inhibitors of LRRK2 Kinase Attenuate Neurodegeneration and Parkinson-like Phenotypes in Caenorhabditis Elegans and Drosophila Parkinson’s Disease Models. Hum. Mol. Genet. 2011, 20, 3933–3942. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Khoury, R.E.; Wang, W.; Byrd, T.A.; Pehek, E.A.; Thacker, C.; Zhu, X.; Smith, M.A.; Wilson-Delfosse, A.L.; Chen, S.G. LRRK2-Mediated Neurodegeneration and Dysfunction of Dopaminergic Neurons in a Caenorhabditis Elegans Model of Parkinson’s Disease. Neurobiol. Dis. 2010, 40, 73–81. [Google Scholar] [CrossRef]
- Senchuk, M.M.; Van Raamsdonk, J.M.; Moore, D.J. Multiple Genetic Pathways Regulating Lifespan Extension Are Neuroprotective in a G2019S LRRK2 Nematode Model of Parkinson’s Disease. Neurobiol. Dis. 2021, 151, 105267. [Google Scholar] [CrossRef]
- Islam, M.S.; Moore, D.J. Mechanisms of LRRK2-Dependent Neurodegeneration: Role of Enzymatic Activity and Protein Aggregation. Biochem. Soc. Trans. 2017, 45, 163–172. [Google Scholar] [CrossRef]
- de Lencastre, A.; Pincus, Z.; Zhou, K.; Kato, M.; Lee, S.S.; Slack, F.J. MicroRNAs Both Promote and Antagonize Longevity in C. elegans. Curr. Biol. CB 2010, 20, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Boulias, K.; Horvitz, H.R. The C. Elegans MicroRNA Mir-71 Acts in Neurons to Promote Germline-Mediated Longevity through Regulation of DAF-16/FOXO. Cell Metab. 2012, 15, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Finger, F.; Ottens, F.; Springhorn, A.; Drexel, T.; Proksch, L.; Metz, S.; Cochella, L.; Hoppe, T. Olfaction Regulates Organismal Proteostasis and Longevity via MicroRNA-Dependent Signaling. Nat. Metab. 2019, 1, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-W.; Chang, C.; Chuang, C.-F. The MicroRNA Mir-71 Inhibits Calcium Signaling by Targeting the TIR-1/Sarm1 Adaptor Protein to Control Stochastic L/R Neuronal Asymmetry in C. Elegans. PLoS Genet. 2012, 8, e1002864. [Google Scholar] [CrossRef] [PubMed]
- Peterson, N.D.; Icso, J.D.; Salisbury, J.E.; Rodríguez, T.; Thompson, P.R.; Pukkila-Worley, R. Pathogen Infection and Cholesterol Deficiency Activate the C. Elegans P38 Immune Pathway through a TIR-1/SARM1 Phase Transition. eLife 2022, 11, e74206. [Google Scholar] [CrossRef] [PubMed]
- Horsefield, S.; Burdett, H.; Zhang, X.; Manik, M.K.; Shi, Y.; Chen, J.; Qi, T.; Gilley, J.; Lai, J.-S.; Rank, M.X.; et al. NAD+ Cleavage Activity by Animal and Plant TIR Domains in Cell Death Pathways. Science 2019, 365, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Loring, H.S.; Czech, V.L.; Icso, J.D.; O’Connor, L.; Parelkar, S.S.; Byrne, A.B.; Thompson, P.R. A Phase Transition Enhances the Catalytic Activity of SARM1, an NAD+ Glycohydrolase Involved in Neurodegeneration. eLife 2021, 10, e66694. [Google Scholar] [CrossRef]
- Czech, V.L.; O’Connor, L.C.; Philippon, B.; Norman, E.; Byrne, A.B. TIR-1/SARM1 Inhibits Axon Regeneration and Promotes Axon Degeneration. eLife 2023, 12, e80856. [Google Scholar] [CrossRef] [PubMed]
- Krauss, R.; Bosanac, T.; Devraj, R.; Engber, T.; Hughes, R.O. Axons Matter: The Promise of Treating Neurodegenerative Disorders by Targeting SARM1-Mediated Axonal Degeneration. Trends Pharmacol. Sci. 2020, 41, 281–293. [Google Scholar] [CrossRef]
- Sawin, E.R.; Ranganathan, R.; Horvitz, H.R. C. elegans Locomotory Rate Is Modulated by the Environment through a Dopaminergic Pathway and by Experience through a Serotonergic Pathway. Neuron 2000, 26, 619–631. [Google Scholar] [CrossRef]
- Zhang, X.; Zabinsky, R.; Teng, Y.; Cui, M.; Han, M. MicroRNAs Play Critical Roles in the Survival and Recovery of Caenorhabditis Elegans from Starvation-Induced L1 Diapause. Proc. Natl. Acad. Sci. USA 2011, 108, 17997–18002. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, D.; Brennan, R.; de Lencastre, A. Conservation and Targets of MiR-71: A Systematic Review and Meta-Analysis. Non-Coding RNA 2023, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Majoros, W.H.; Lekprasert, P.; Mukherjee, N.; Skalsky, R.L.; Corcoran, D.L.; Cullen, B.R.; Ohler, U. MicroRNA Target Site Identification by Integrating Sequence and Binding Information. Nat. Methods 2013, 10, 630–633. [Google Scholar] [CrossRef] [PubMed]
- Vérièpe, J.; Fossouo, L.; Parker, J.A. Neurodegeneration in C. Elegans Models of ALS Requires TIR-1/Sarm1 Immune Pathway Activation in Neurons. Nat. Commun. 2015, 6, 7319. [Google Scholar] [CrossRef]
- Yao, X.-Y.; Guan, L.-N.; Chen, Q.; Ren, C. LRRK2 G2019S and Parkinson’s Disease: Insight from Neuroinflammation. Postgrad. Med. J. 2023, 100, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Couillault, C.; Pujol, N.; Reboul, J.; Sabatier, L.; Guichou, J.-F.; Kohara, Y.; Ewbank, J.J. TLR-Independent Control of Innate Immunity in Caenorhabditis Elegans by the TIR Domain Adaptor Protein TIR-1, an Ortholog of Human SARM. Nat. Immunol. 2004, 5, 488–494. [Google Scholar] [CrossRef]
- Lezi, E.; Zhou, T.; Koh, S.; Chuang, M.; Sharma, R.; Pujol, N.; Chisholm, A.D.; Eroglu, C.; Matsunami, H.; Yan, D. An Antimicrobial Peptide and Its Neuronal Receptor Regulate Dendrite Degeneration in Aging and Infection. Neuron 2018, 97, 125–138.e5. [Google Scholar] [CrossRef]
- Hopkins, E.L.; Gu, W.; Kobe, B.; Coleman, M.P. A Novel NAD Signaling Mechanism in Axon Degeneration and Its Relationship to Innate Immunity. Front. Mol. Biosci. 2021, 8, 703532. [Google Scholar] [CrossRef] [PubMed]
- Figley, M.D.; DiAntonio, A. The SARM1 Axon Degeneration Pathway: Control of the NAD+ Metabolome Regulates Axon Survival in Health and Disease. Curr. Opin. Neurobiol. 2020, 63, 59–66. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, L.; Huang, Y. Differential Expression of Peripheral Circulating MicroRNA-146a Between Patients with Atherosclerotic Vulnerable Plaque and Stable Plaque. Int. Heart. J. 2023, 64, 847–855. [Google Scholar] [CrossRef]
- Wu, Y. Circ_0044516 Enriches the Level of SARM1 as a MiR-330-5p Sponge to Regulate Cell Malignant Behaviors and Tumorigenesis of Prostate Cancer. Biochem. Genet. 2022, 60, 1346–1361. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Hou, L.; Liu, T.; Jiao, W.; Ma, Q.; Fang, Z.; Zhang, S.; Song, D.; Liu, J.; Gao, X.; et al. LncRNA OGFRP1 Functions as a CeRNA to Promote the Progression of Prostate Cancer by Regulating SARM1 Level via MiR-124-3p. Aging 2020, 12, 8880–8892. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, Z.; Kim, E.; Henstridge, C.M.; Pena Altamira, E.; Hunt, C.K.; Burchill, E.; Callaghan, I.; Loreto, A.; Brown-Wright, H.; et al. Sarm1 Deletion Suppresses TDP-43-Linked Motor Neuron Degeneration and Cortical Spine Loss. Acta Neuropathol. Commun. 2019, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Bratkowski, M.; Burdett, T.C.; Danao, J.; Wang, X.; Mathur, P.; Gu, W.; Beckstead, J.A.; Talreja, S.; Yang, Y.-S.; Danko, G.; et al. Uncompetitive, Adduct-Forming SARM1 Inhibitors Are Neuroprotective in Preclinical Models of Nerve Injury and Disease. Neuron 2022, 110, 3711–3726.e16. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kerry, P.S.; Nanson, J.D.; Bosanac, T.; Sasaki, Y.; Krauss, R.; Saikot, F.K.; Adams, S.E.; Mosaiab, T.; Masic, V.; et al. Structural Basis of SARM1 Activation, Substrate Recognition, and Inhibition by Small Molecules. Mol. Cell 2022, 82, 1643–1659.e10. [Google Scholar] [CrossRef] [PubMed]
- Stiernagle, T. Maintenance of C. Elegans. WormBook 2006, 12, 203–205. [Google Scholar] [CrossRef]
- Caenorhabditis Genetics Center (CGC)—College of Biological Sciences. Available online: https://cgc.umn.edu/ (accessed on 23 August 2023).
- Fay, D.S. Classical Genetic Methods. WormBook 2013, 1, 1–58. [Google Scholar] [CrossRef]
- Ray, A.; Martinez, B.A.; Berkowitz, L.A.; Caldwell, G.A.; Caldwell, K.A. Mitochondrial Dysfunction, Oxidative Stress, and Neurodegeneration Elicited by a Bacterial Metabolite in a C. Elegans Parkinson’s Model. Cell Death Dis. 2014, 5, e984. [Google Scholar] [CrossRef]
- Cooper, J.F.; Dues, D.J.; Spielbauer, K.K.; Machiela, E.; Senchuk, M.M.; Van Raamsdonk, J.M. Delaying Aging Is Neuroprotective in Parkinson’s Disease: A Genetic Analysis in C. Elegans Models. NPJ Park. Dis. 2015, 1, 15022. [Google Scholar] [CrossRef]
- Saha, S.; Ash, P.E.A.; Gowda, V.; Liu, L.; Shirihai, O.; Wolozin, B. Mutations in LRRK2 Potentiate Age-Related Impairment of Autophagic Flux. Mol. Neurodegener. 2015, 10, 26. [Google Scholar] [CrossRef]
- Kuwahara, T.; Inoue, K.; D’Agati, V.D.; Fujimoto, T.; Eguchi, T.; Saha, S.; Wolozin, B.; Iwatsubo, T.; Abeliovich, A. LRRK2 and RAB7L1 Coordinately Regulate Axonal Morphology and Lysosome Integrity in Diverse Cellular Contexts. Sci. Rep. 2016, 6, 29945. [Google Scholar] [CrossRef] [PubMed]
- Porta-de-la-Riva, M.; Fontrodona, L.; Villanueva, A.; Cerón, J. Basic Caenorhabditis Elegans Methods: Synchronization and Observation. J. Vis. Exp. JoVE 2012, 64, 4019. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naidoo, D.; de Lencastre, A. Regulation of TIR-1/SARM-1 by miR-71 Protects Dopaminergic Neurons in a C. elegans Model of LRRK2-Induced Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 8795. https://doi.org/10.3390/ijms25168795
Naidoo D, de Lencastre A. Regulation of TIR-1/SARM-1 by miR-71 Protects Dopaminergic Neurons in a C. elegans Model of LRRK2-Induced Parkinson’s Disease. International Journal of Molecular Sciences. 2024; 25(16):8795. https://doi.org/10.3390/ijms25168795
Chicago/Turabian StyleNaidoo, Devin, and Alexandre de Lencastre. 2024. "Regulation of TIR-1/SARM-1 by miR-71 Protects Dopaminergic Neurons in a C. elegans Model of LRRK2-Induced Parkinson’s Disease" International Journal of Molecular Sciences 25, no. 16: 8795. https://doi.org/10.3390/ijms25168795
APA StyleNaidoo, D., & de Lencastre, A. (2024). Regulation of TIR-1/SARM-1 by miR-71 Protects Dopaminergic Neurons in a C. elegans Model of LRRK2-Induced Parkinson’s Disease. International Journal of Molecular Sciences, 25(16), 8795. https://doi.org/10.3390/ijms25168795