
Is Exon Skipping a Viable Therapeutic Approach for Vascular Ehlers–Danlos Syndrome with Mutations in COL3A1 Exon 10 or 15?

 , , ,
, , ,
Abstract
1. Introduction
2. Results
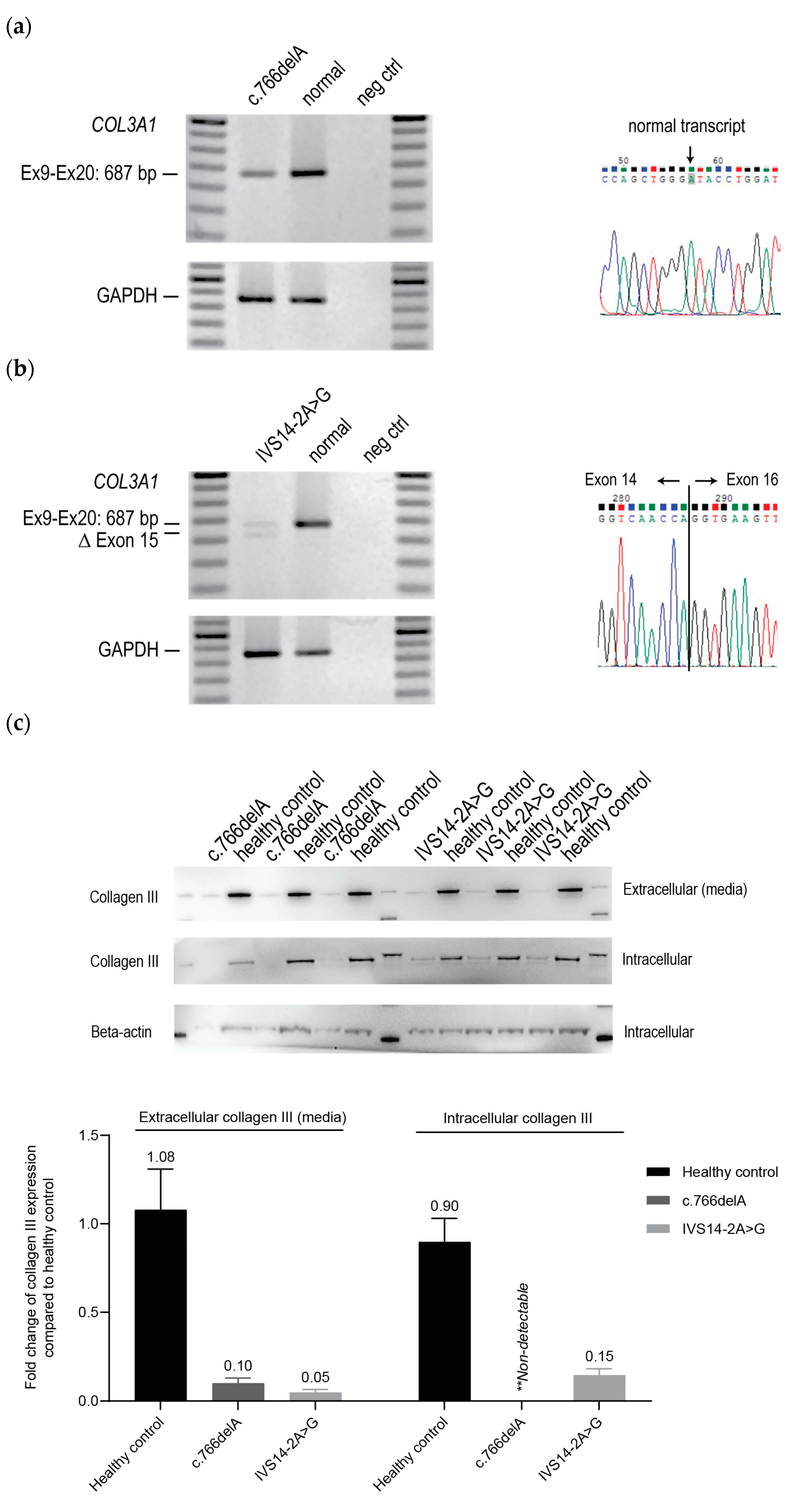
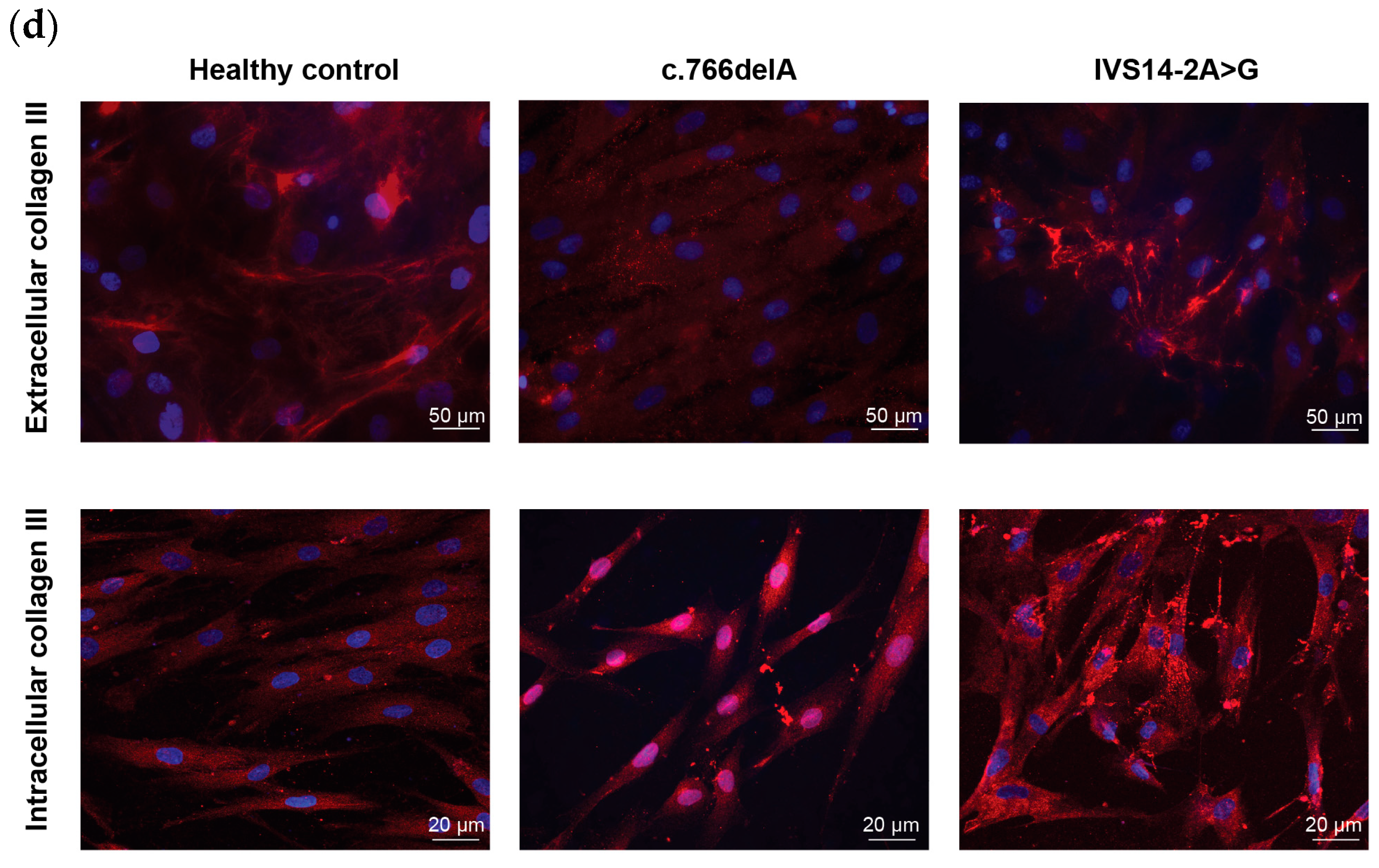
2.1. Cellular Phenotypes of vEDS Patient Fibroblasts Harbouring COL3A1 Mutations
2.2. ASO-Mediated Exon Skipping to Reframe COL3A1 Transcripts and Assessment of Collagen III Expression in vEDS Patient-Derived Fibroblasts Carrying COL3A1 c.766delA
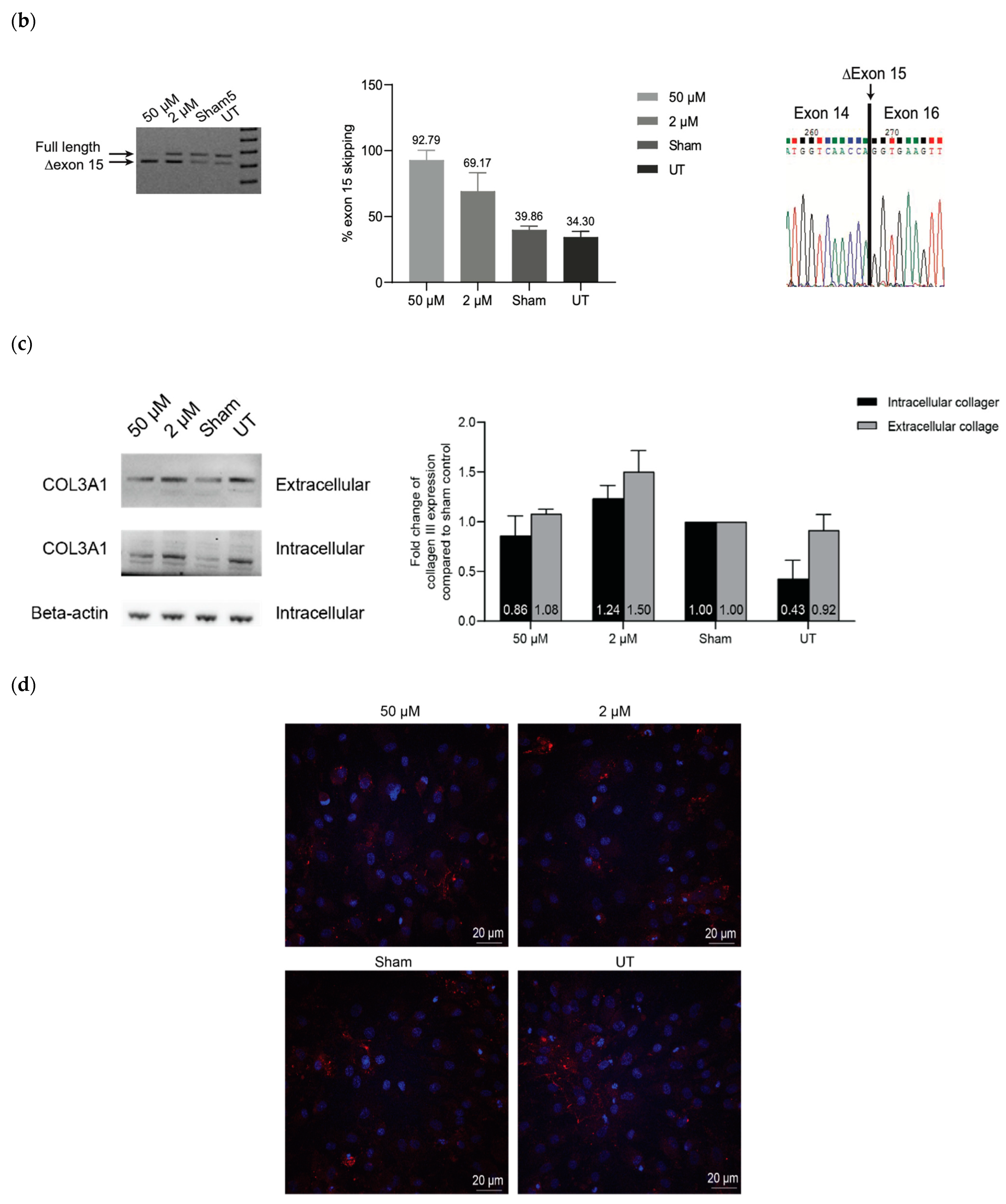
2.3. ASO-Mediated Exon Skipping to Reframe COL3A1 Transcripts and Assessment of Collagen III Expression in vEDS Patient-Derived Fibroblasts Carrying COL3A1 IVS14-2A>G
3. Discussion
4. Materials and Methods
4.1. Antisense Oligonucleotide Design and Synthesis
4.2. Cell Culture and Transfection
4.3. RNA Harvest and RT-PCR
4.4. Protein Harvest and Western Blot
4.4.1. Cell Lysate Preparation
4.4.2. Spent Media Preparation
4.4.3. Western Blot
4.5. Fibroblast-Derived Extracellular Matrix
4.6. Immunofluorescence Staining
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boot-Handford, R.P.; Tuckwell, D.S.; Plumb, D.A.; Rock, C.F.; Poulsom, R. A novel and highly conserved collagen (pro(alpha)1(XXVII)) with a unique expression pattern and unusual molecular characteristics establishes a new clade within the vertebrate fibrillar collagen family. J. Biol. Chem. 2003, 278, 31067–31077. [Google Scholar] [CrossRef] [PubMed]
- Exposito, J.-Y.; Cluzel, C.; Garrone, R.; Lethias, C. Evolution of collagens. Anat. Rec. 2002, 268, 302–316. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Laub, F.; Zhou, P.; Hahn, R.A.; Tanaka, S.; Burgeson, R.E.; Gerecke, D.R.; Ramirez, F.; Gordon, M.K. Collagen XXIV, a vertebrate fibrillar collagen with structural features of invertebrate collagens: Selective expression in developing cornea and bone. J. Biol. Chem. 2003, 278, 43236–43244. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.M.; Corrado, M.; Missero, C.; Byers, P.H. Identification, characterization and expression analysis of a new fibrillar collagen gene, COL27A1. Matrix Biol. 2003, 22, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Cheung, D.T.; Benya, P.D.; Perelman, N.; DiCesare, P.E.; Nimni, M.E. A highly specific and quantitative method for determining type III/I collagen ratios in tissues. Matrix 1990, 10, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Ramshaw, J.A.; Shah, N.K.; Brodsky, B. Gly-X-Y tripeptide frequencies in collagen: A context for host-guest triple-helical peptides. J. Struct. Biol. 1998, 122, 86–91. [Google Scholar] [CrossRef]
- Epstein, D.; Elias-Bishko, S.; Hershko, A. Requirement for protein synthesis in the regulation of protein breakdown in cultured hepatoma cells. Biochemistry 1975, 14, 5199–5204. [Google Scholar] [CrossRef]
- van Kuppevelt, T.H.; Veerkamp, J.H.; Timmermans, J.A. Immunoquantification of type I, III, IV and V collagen in small samples of human lung parenchyma. Int. J. Biochem. Cell Biol. 1995, 27, 775–782. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sumiyoshi, H.; Nakagami, K.; Tahara, E. Distribution of collagen types I, III, and V in fibrotic and neoplastic human liver. Acta Pathol. Jpn. 1984, 34, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Hurst, P.R.; Gibbs, R.D.; Clark, D.E.; Myers, D.B. Temporal changes to uterine collagen types I, III and V in relation to early pregnancy in the rat. Reprod. Fertil. Dev. 1994, 6, 669–677. [Google Scholar] [CrossRef] [PubMed]
- McCullagh, K.A.; Balian, G. Collagen characterisation and cell transformation in human atherosclerosis. Nature 1975, 258, 73–75. [Google Scholar] [CrossRef]
- Kaplan, E.P.; Richier, J.C.; Howard, P.S.; Ewalt, D.H.; Lin, V.K. Type III collagen messenger RNA is modulated in non-compliant human bladder tissue. J. Urol. 1997, 157, 2366–2369. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.T. Cardiac interstitium in health and disease: The fibrillar collagen network. J. Am. Coll. Cardiol. 1989, 13, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Asgari, M.; Latifi, N.; Heris, H.K.; Vali, H.; Mongeau, L. In vitro fibrillogenesis of tropocollagen type III in collagen type I affects its relative fibrillar topology and mechanics. Sci. Rep. 2017, 7, 1392. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F. Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol. 2018, 71–72, 380–395. [Google Scholar] [CrossRef]
- Germain, D.P. Ehlers-Danlos syndrome type IV. Orphanet. J. Rare Dis. 2007, 2, 32. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers–Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Bartlett, M.; Baker, D.; Berlin, C.; Bowen, J.; Cummings, C.; Fallows, C.; Green, C.; Griffin, J.; Julier, K.; et al. An exemplary model of genetic counselling for highly specialised services. J. Community Genet. 2023, 14, 115–119. [Google Scholar] [CrossRef]
- Murray, M.L.; Pepin, M.; Peterson, S.; Byers, P.H. Pregnancy-related deaths and complications in women with vascular Ehlers-Danlos syndrome. Genet. Med. 2014, 16, 874–880. [Google Scholar] [CrossRef] [PubMed]
- Matthew, J.E. Arterial complications of vascular Ehlers-Danlos syndrome. J. Vasc. Surg. 2016, 64, 1869–1880. [Google Scholar]
- Schwarze, U.; Schievink, W.I.; Petty, E.; Jaff, M.R.; Babovic-Vuksanovic, D.; Cherry, K.J.; Pepin, M.; Byers, P.H. Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers-Danlos syndrome, Ehlers-Danlos syndrome type IV. Am. J. Hum. Genet. 2001, 69, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Leistritz, D.F.; Pepin, M.G.; Schwarze, U.; Byers, P.H. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet. Med. 2011, 13, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.; Albuisson, J.; Ranque, B.; Golmard, L.; Mazzella, J.-M.; Bal-Theoleyre, L.; Fauret, A.-L.; Mirault, T.; Denarié, N.; Mousseaux, E.; et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers–Danlos syndrome. Eur. J. Hum. Genet. 2015, 23, 1657–1664. [Google Scholar] [CrossRef]
- Shalhub, S.; Black, J.H., 3rd; Cecchi, A.C.; Xu, Z.; Griswold, B.F.; Safi, H.J.; Milewicz, D.M.; McDonnell, N.B. Molecular diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of arterial involvement and outcomes. J. Vasc. Surg. 2014, 60, 160–169. [Google Scholar] [CrossRef]
- Pichavant, C.; Aartsma-Rus, A.; Clemens, P.R.; Davies, K.E.; Dickson, G.; Takeda, S.; Wilton, S.D.; Wolff, J.A.; Wooddell, C.I.; Xiao, X.; et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol. Ther. 2011, 19, 830–840. [Google Scholar] [CrossRef]
- Li, D.; Adams, A.M.; Johnsen, R.D.; Fletcher, S.; Wilton, S.D. Morpholino Oligomer-Induced Dystrophin Isoforms to Map the Functional Domains in the Dystrophin Protein. Mol. Ther. Nucleic Acids 2020, 22, 263–272. [Google Scholar] [CrossRef]
- Pepin, M.G.; Schwarze, U.; Rice, K.M.; Liu, M.; Leistritz, D.; Byers, P.H. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet. Med. 2014, 16, 881–888. [Google Scholar] [CrossRef]
- Cale, J.M.; Greer, K.; Fletcher, S.; Wilton, S.D. Proof-of-Concept: Antisense Oligonucleotide Mediated Skipping of Fibrillin-1 Exon 52. Int. J. Mol. Sci. 2021, 22, 3479. [Google Scholar] [CrossRef]
- Bremer, J.; Bornert, O.; Nyström, A.; Gostynski, A.; Jonkman, M.F.; Aartsma-Rus, A.; van den Akker, P.C.; Pasmooij, A.M. Antisense Oligonucleotide-mediated Exon Skipping as a Systemic Therapeutic Approach for Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. Nucleic Acids 2016, 5, e379. [Google Scholar] [CrossRef] [PubMed]
- Turczynski, S.; Titeux, M.; Tonasso, L.; Décha, A.; Ishida-Yamamoto, A.; Hovnanian, A. Targeted Exon Skipping Restores Type VII Collagen Expression and Anchoring Fibril Formation in an In Vivo RDEB Model. J. Investig. Dermatol. 2016, 136, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, T.; Horinouchi, T.; Adachi, T.; Terakawa, M.; Takaoka, Y.; Omachi, K.; Takasato, M.; Takaishi, K.; Shoji, T.; Onishi, Y.; et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat. Commun. 2020, 11, 2777. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.K.; Taga, Y.; Sakai, T.; Ito, S.; Hattori, S.; Nagata, K.; Koide, T. Lowering the culture temperature corrects collagen abnormalities caused by HSP47 gene knockout. Sci. Rep. 2019, 9, 17433. [Google Scholar] [CrossRef] [PubMed]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Mays, P.K.; Bishop, J.E.; Laurent, G.J. Age-related changes in the proportion of types I and III collagen. Mech. Ageing Dev. 1988, 45, 203–212. [Google Scholar] [CrossRef]
- Cheng, W.; Rong, Y.-H.; Ning, F.-G.; Zhang, G.-A. The content and ratio of type I and III collagen in skin differ with age and injury. Afr. J. Biotechnol. 2011, 10, 2524–2529. [Google Scholar]
- Khoshnoodi, J.; Pedchenko, V.; Hudson, B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008, 71, 357–370. [Google Scholar] [CrossRef]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef]
- Berg, R.A.; Prockop, D.J. The thermal transition of a non-hydroxylated form of collagen. Evidence for a role for hydroxyproline in stabilizing the triple-helix of collagen. Biochem. Biophys. Res. Commun. 1973, 52, 115–120. [Google Scholar] [CrossRef]
- Sakakibara, S.; Inouye, K.; Shudo, K.; Kishida, Y.; Kobayashi, Y.; Prockop, D.J. Synthesis of (Pro-Hyp-Gly) n of defined molecular weights. Evidence for the stabilization of collagen triple helix by hydroxypyroline. Biochim. Biophys. Acta 1973, 303, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.D.; Page, A.P. Prolyl 4-hydroxylase is an essential procollagen-modifying enzyme required for exoskeleton formation and the maintenance of body shape in the nematode Caenorhabditis elegans. Mol. Cell. Biol. 2000, 20, 4084–4093. [Google Scholar] [CrossRef] [PubMed]
- Jürgensen, H.J.; Madsen, D.H.; Ingvarsen, S.; Melander, M.C.; Gårdsvoll, H.; Patthy, L.; Engelholm, L.H.; Behrendt, N. A novel functional role of collagen glycosylation: Interaction with the endocytic collagen receptor uparap/ENDO180. J. Biol. Chem. 2011, 286, 32736–32748. [Google Scholar] [CrossRef] [PubMed]
- Hua, C.; Zhu, Y.; Xu, W.; Ye, S.; Zhang, R.; Lu, L.; Jiang, S. Characterization by high-resolution crystal structure analysis of a triple-helix region of human collagen type III with potent cell adhesion activity. Biochem. Biophys. Res. Commun. 2019, 508, 1018–1023. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.N.; Brandts, J.F. Determination of cis-trans proline isomerization by trypsin proteolysis. Application to a model pentapeptide and to oxidized ribonuclease A. Biochemistry 1983, 22, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, K.; Boudko, S.; Engel, J.; Bächinger, H.P. Vascular Ehlers-Danlos syndrome mutations in type III collagen differently stall the triple helical folding. J. Biol. Chem. 2013, 288, 19166–19176. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Mizuno, K.; Bächinger, H.P. Ziploc-ing the structure 2.0: Endoplasmic reticulum-resident peptidyl prolyl isomerases show different activities toward hydroxyproline. J. Biol. Chem. 2017, 292, 9273–9282. [Google Scholar] [CrossRef] [PubMed]
- Aung-Htut, M.T.; McIntosh, C.S.; West, K.A.; Fletcher, S.; Wilton, S.D. In Vitro Validation of Phosphorodiamidate Morpholino Oligomers. Molecules 2019, 24, 2922. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Lim, L.; Dye, D.; Laing, N. Bandstab: A PCR-based alternative to cloning PCR products. Biotechniques 1997, 22, 642–645. [Google Scholar] [CrossRef]
- Kumar, P.; Satyam, A.; Fan, X.; Collin, E.; Rochev, Y.; Rodriguez, B.J.; Gorelov, A.; Dillon, S.; Joshi, L.; Raghunath, M.; et al. Macromolecularly crowded in vitro microenvironments accelerate the production of extracellular matrix-rich supramolecular assemblies. Sci. Rep. 2015, 5, 8729. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | ASO Sequence 5′ --> 3′ | Length (bp) | Target COL3A1 Exon |
|---|---|---|---|
| COL3A1_H10A(−13+12) | ACCUUUGAUACCCUAAAAAUAAUGA | 25 | 10 |
| COL3A1_H10A(+15+39) | ACCAGGGAAUCCAGGUAUCCCAGCU | 25 | 10 |
| COL3A1_H10D(+12−13) | AAACUCUACUUACUCUGUGUCCUUU | 25 | 10 |
| COL3A1_H15A(−43−19) | AUUUCAAGAGCUCUUUUUCAACUAA | 25 | 15 |
| COL3A1_H15A(−15+10) | CAGGAGGGCCCCGAAAAAAUUAAAU | 25 | 15 |
| COL3A1_H15A(+20+44) | GGGGAUCCAGGGAAUCCGGCAGUUC | 25 | 15 |
| COL3A1_H15D(+6−19) | UAUAGAAACACAUGUUUACCUUAGC | 25 | 15 |
| COL3A1_H15A(+25+49) | CACCAGGGGAUCCAGGGAAUCCGGC | 25 | 15 |
| COL3A1_H15A(+15+39) | UCCAGGGAAUCCGGCAGUUCCAGGA | 25 | 15 |
| COL3A1_H15A(+20+39) | UCCAGGGAAUCCGGCAGUUC | 20 | 15 |
| COL3A1_H15A(+25+44) | GGGGAUCCAGGGAAUCCGGC | 20 | 15 |
| Control * | CCTCTTACCTCAGTTACAATTTATA | 25 | None |
| Name | Sequence 5′ --> 3′ | Amplicon Size (bp) | Optimized Condition |
|---|---|---|---|
| COL3A1_Ex1F | GCAGGGAACAACTTGATGGTGC | 1079 | 58 °C 30 min |
| 94 °C 2 min | |||
| 94 °C 10 s | |||
| COL3A1_Ex14R | GGTTGACCATCACTGCCTCGA | 58 °C 10 s | |
| 68 °C 1 min | |||
| for 20 cycles | |||
| COL3A1_Ex9F | GACCTGGAGAGCGAGGATTG | 687 | 58 °C 30 min |
| 94 °C 2 min | |||
| 94 °C 10 s | |||
| COL3A1_Ex20R | CCTTGCCATCTTCGCCTTTA | 58 °C 10 s | |
| 68 °C 1 min | |||
| for 20 cycles | |||
| hGAPDH Ex1F | ATGCTGGCGCTGAGTACGT | 874 | 94 °C 2 min |
| 94 °C 30 s | |||
| hGAPDH Ex3R | AGGGGTCTACATGGCAACTG | 58 °C 30 s | |
| 68 °C 1 min | |||
| for 20 cycles |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Utama, S.; Cale, J.M.; Mitrpant, C.; Fletcher, S.; Wilton, S.D.; Aung-Htut, M.T. Is Exon Skipping a Viable Therapeutic Approach for Vascular Ehlers–Danlos Syndrome with Mutations in COL3A1 Exon 10 or 15? Int. J. Mol. Sci. 2024, 25, 8816. https://doi.org/10.3390/ijms25168816
Utama S, Cale JM, Mitrpant C, Fletcher S, Wilton SD, Aung-Htut MT. Is Exon Skipping a Viable Therapeutic Approach for Vascular Ehlers–Danlos Syndrome with Mutations in COL3A1 Exon 10 or 15? International Journal of Molecular Sciences. 2024; 25(16):8816. https://doi.org/10.3390/ijms25168816
Chicago/Turabian StyleUtama, Sasiwimon, Jessica M. Cale, Chalermchai Mitrpant, Sue Fletcher, Steve D. Wilton, and May T. Aung-Htut. 2024. "Is Exon Skipping a Viable Therapeutic Approach for Vascular Ehlers–Danlos Syndrome with Mutations in COL3A1 Exon 10 or 15?" International Journal of Molecular Sciences 25, no. 16: 8816. https://doi.org/10.3390/ijms25168816
APA StyleUtama, S., Cale, J. M., Mitrpant, C., Fletcher, S., Wilton, S. D., & Aung-Htut, M. T. (2024). Is Exon Skipping a Viable Therapeutic Approach for Vascular Ehlers–Danlos Syndrome with Mutations in COL3A1 Exon 10 or 15? International Journal of Molecular Sciences, 25(16), 8816. https://doi.org/10.3390/ijms25168816