
High-Density Genetic Map Construction and Quantitative Trait Locus Analysis of Fruit- and Oil-Related Traits in Camellia oleifera Based on Double Digest Restriction Site-Associated DNA Sequencing


Abstract
:1. Introduction
2. Results
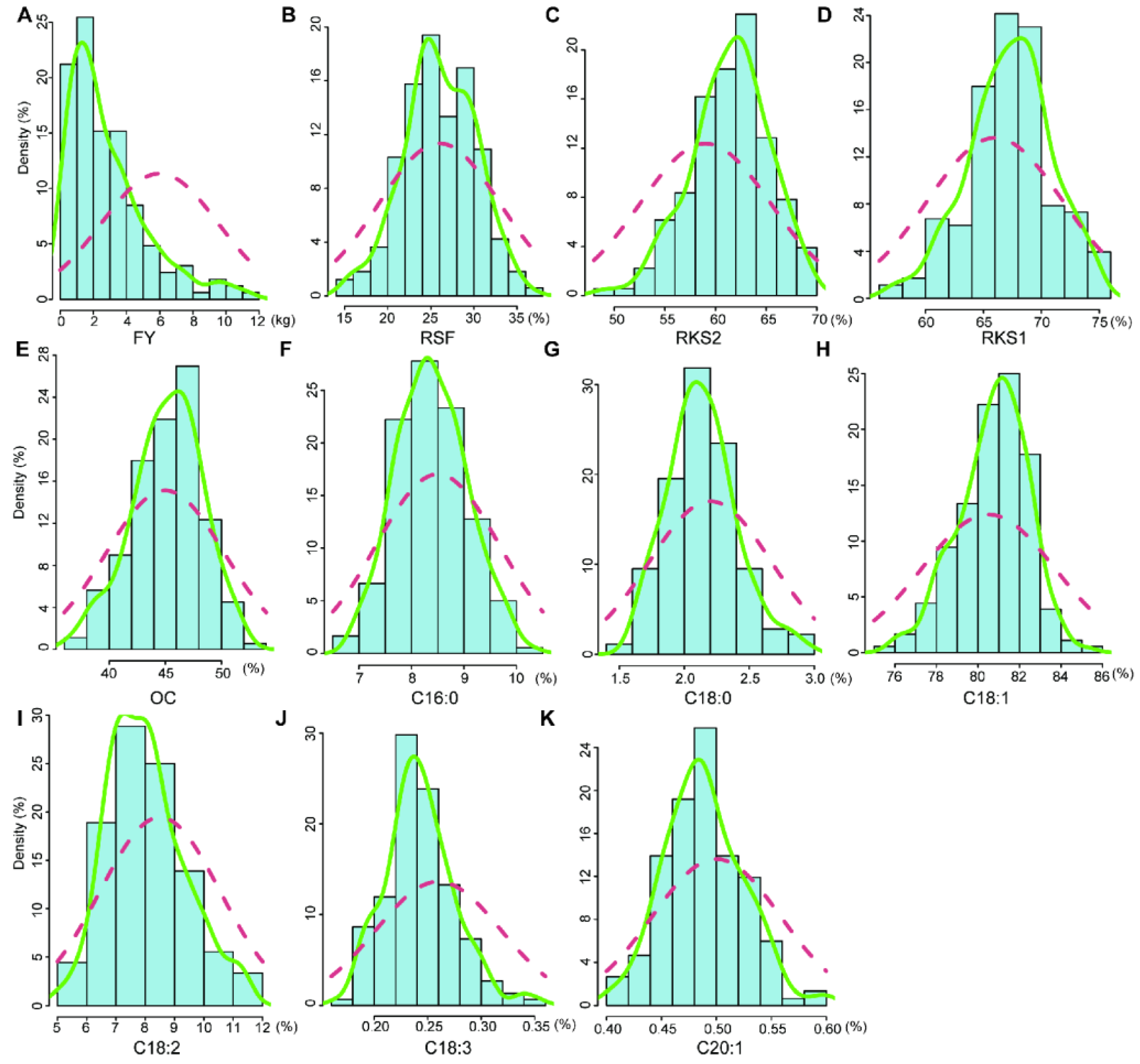
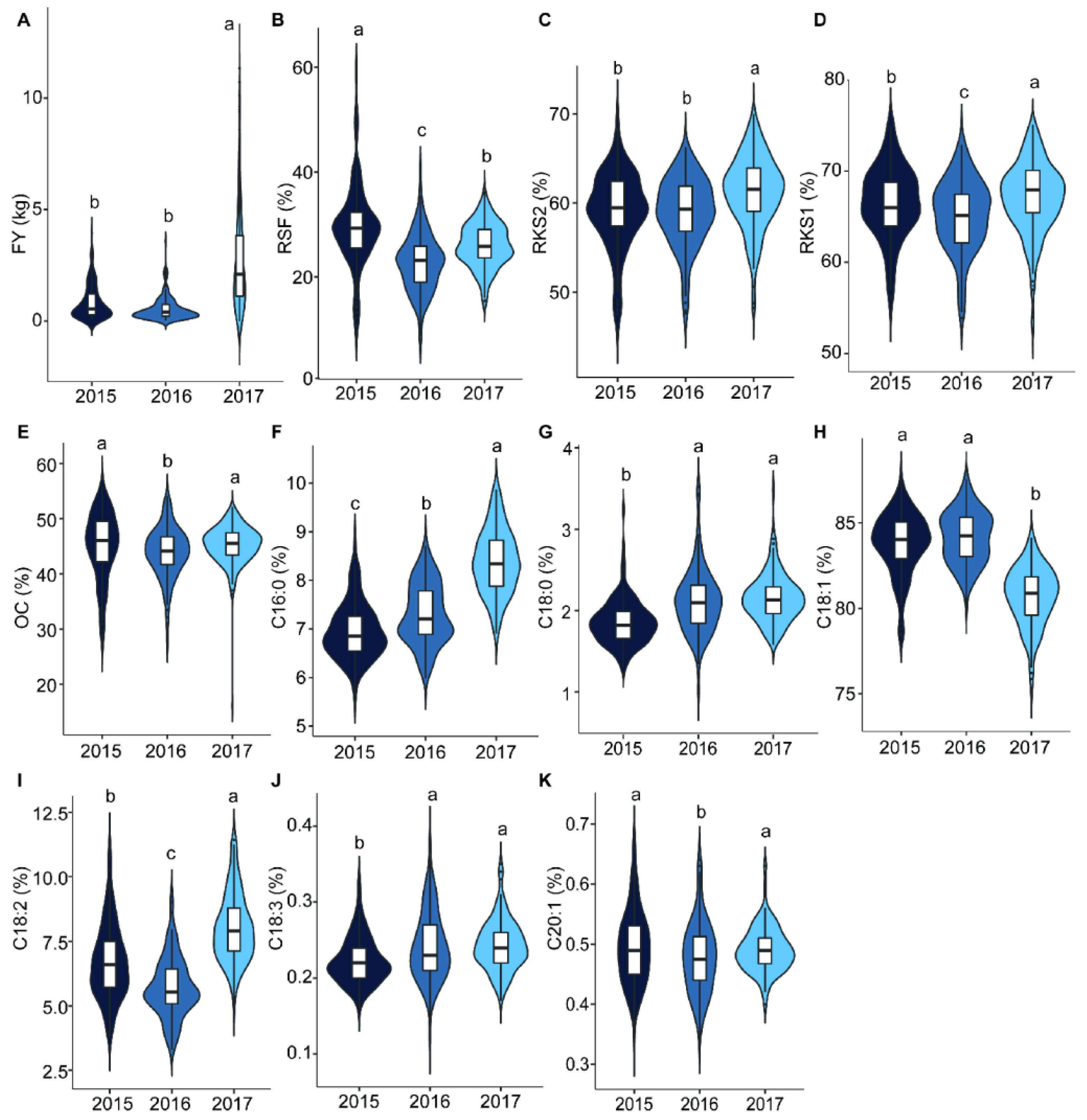
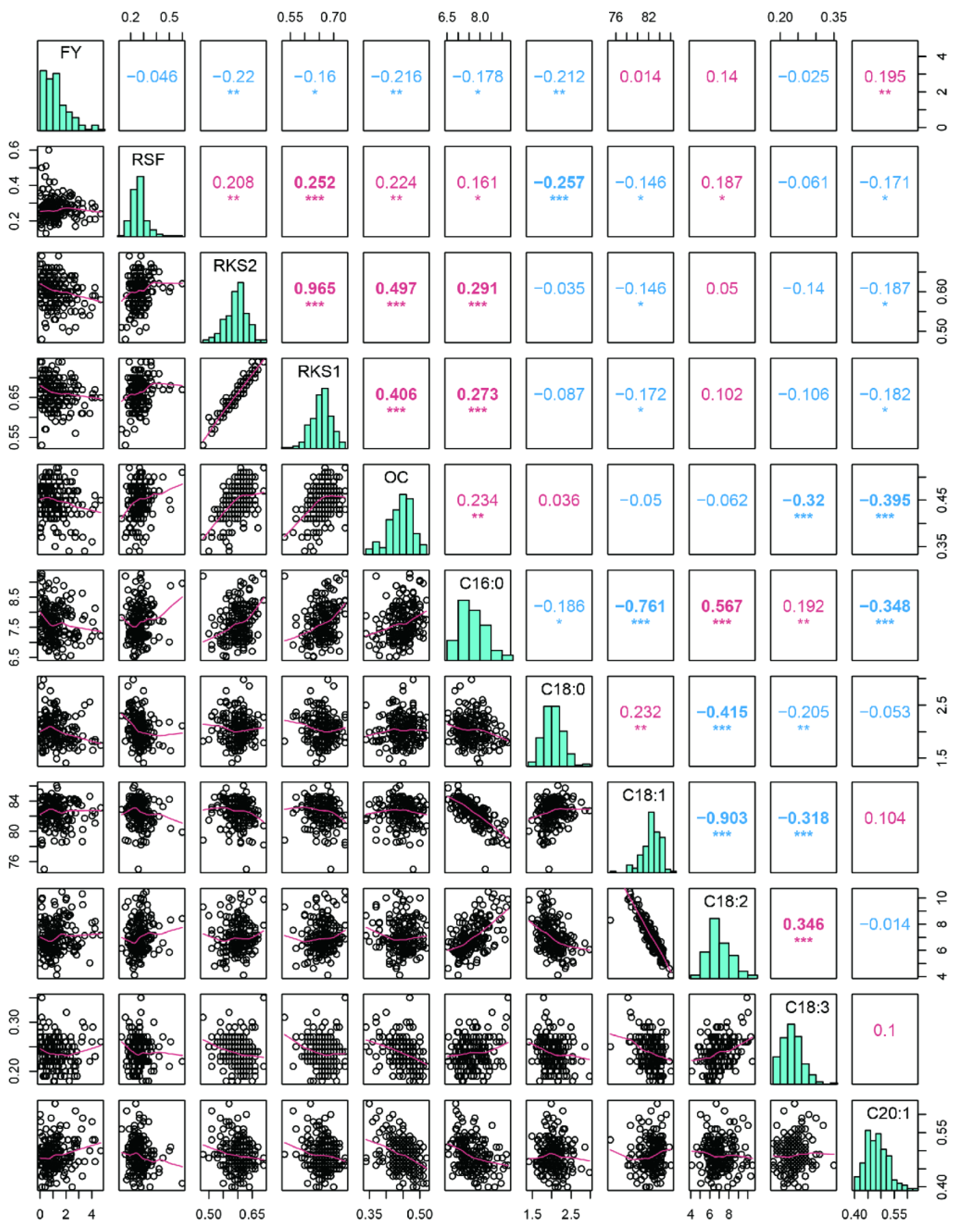
2.1. Variation and Correlation Analysis of Fruit- and Oil-Related Traits
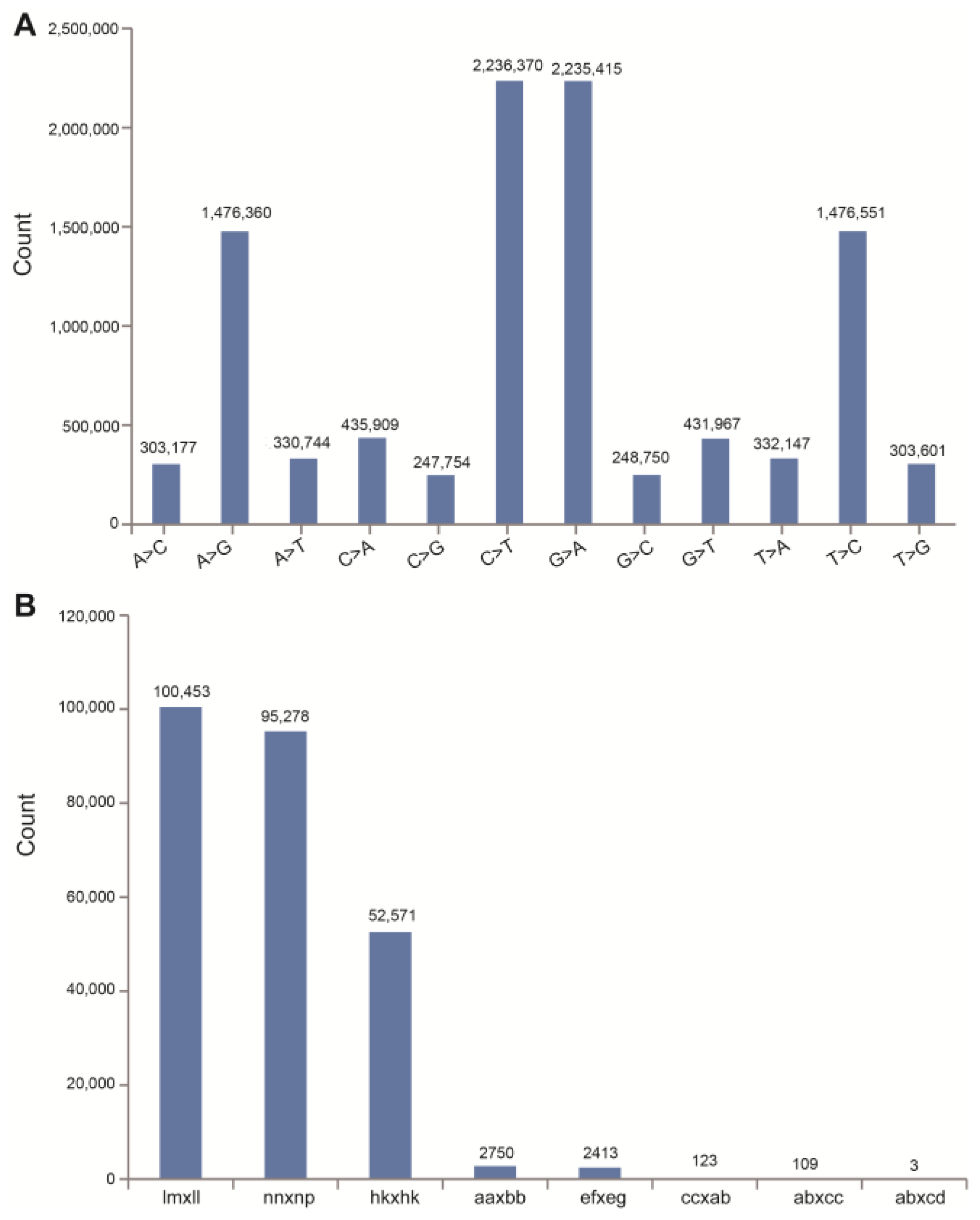
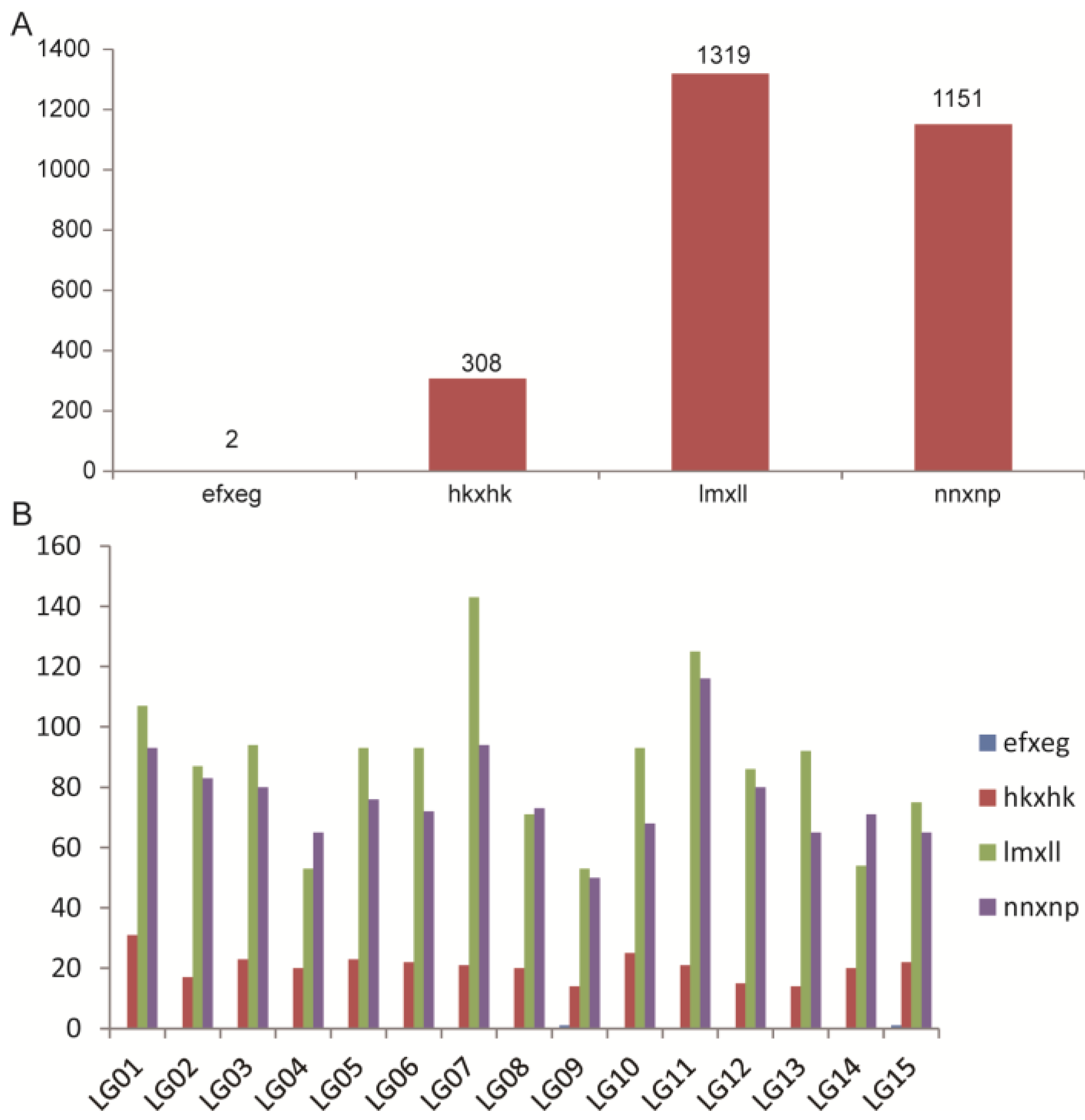
2.2. Analysis of ddRAD Data and SNP Markers
2.3. Genetic Map Construction and Its Basic Characteristics
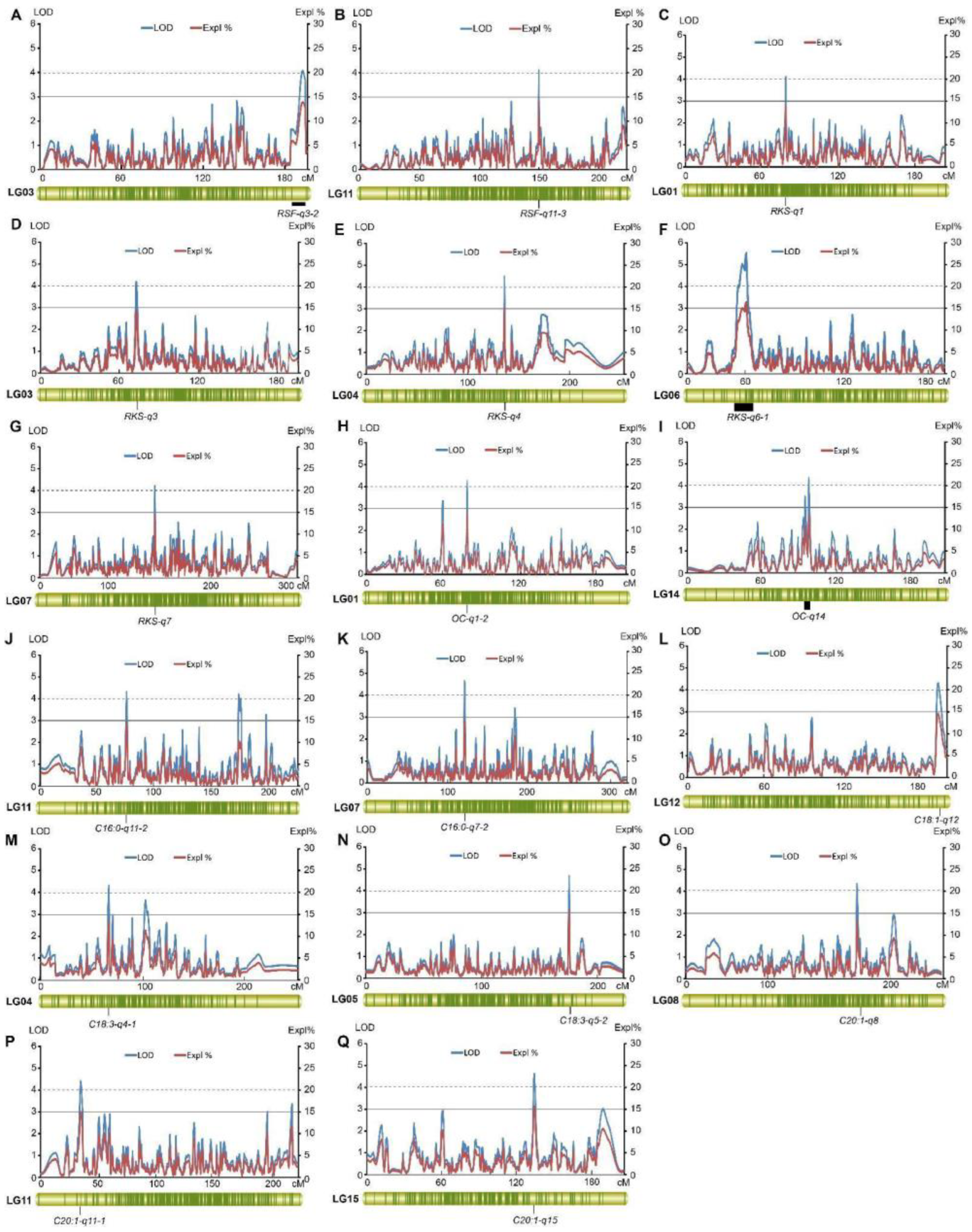
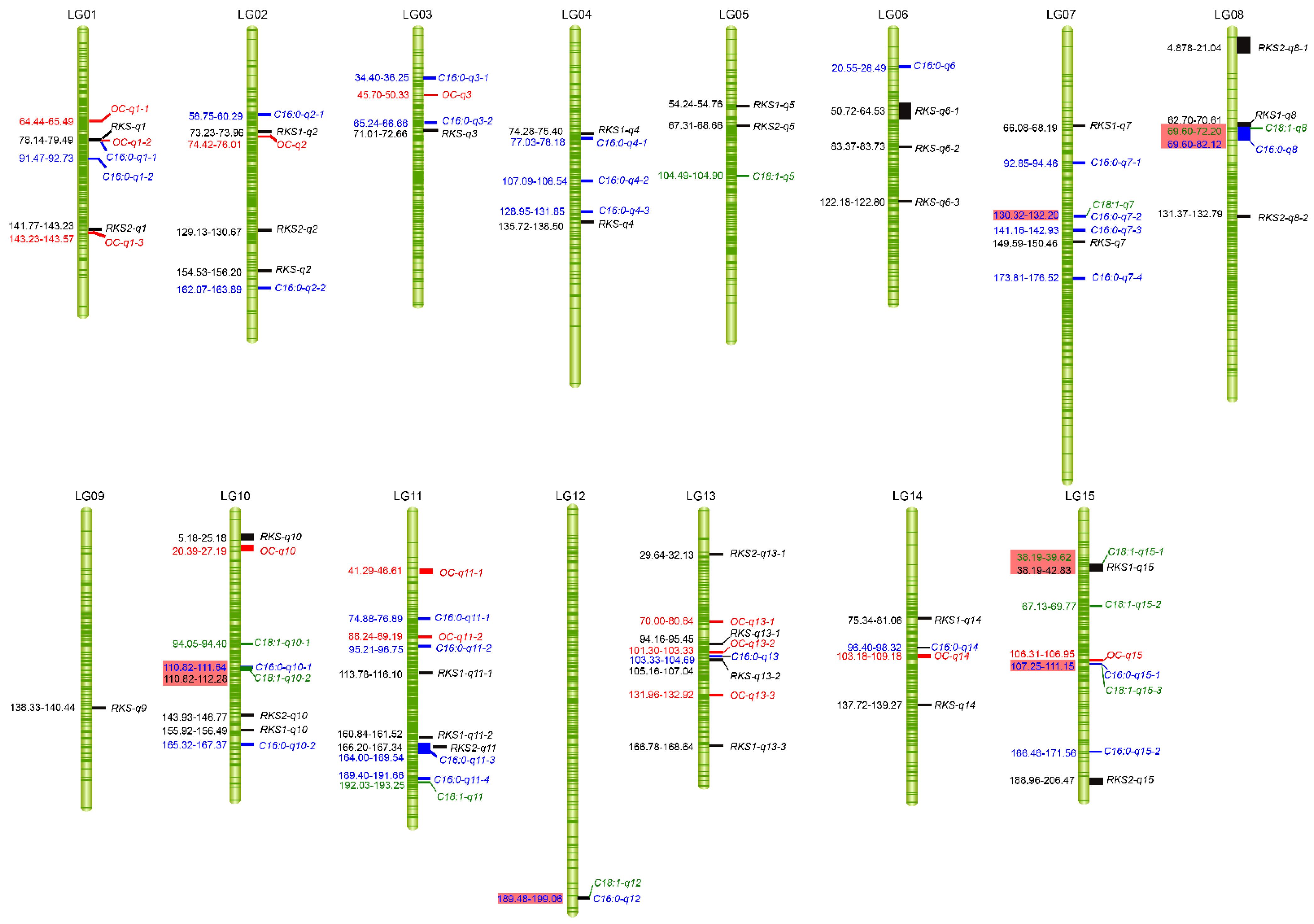
2.4. QTL Analysis of Fruit- and Oil-Related Traits Based on the High-Density Genetic Map
2.4.1. Fruit-Related Traits
2.4.2. Oil-Related Traits
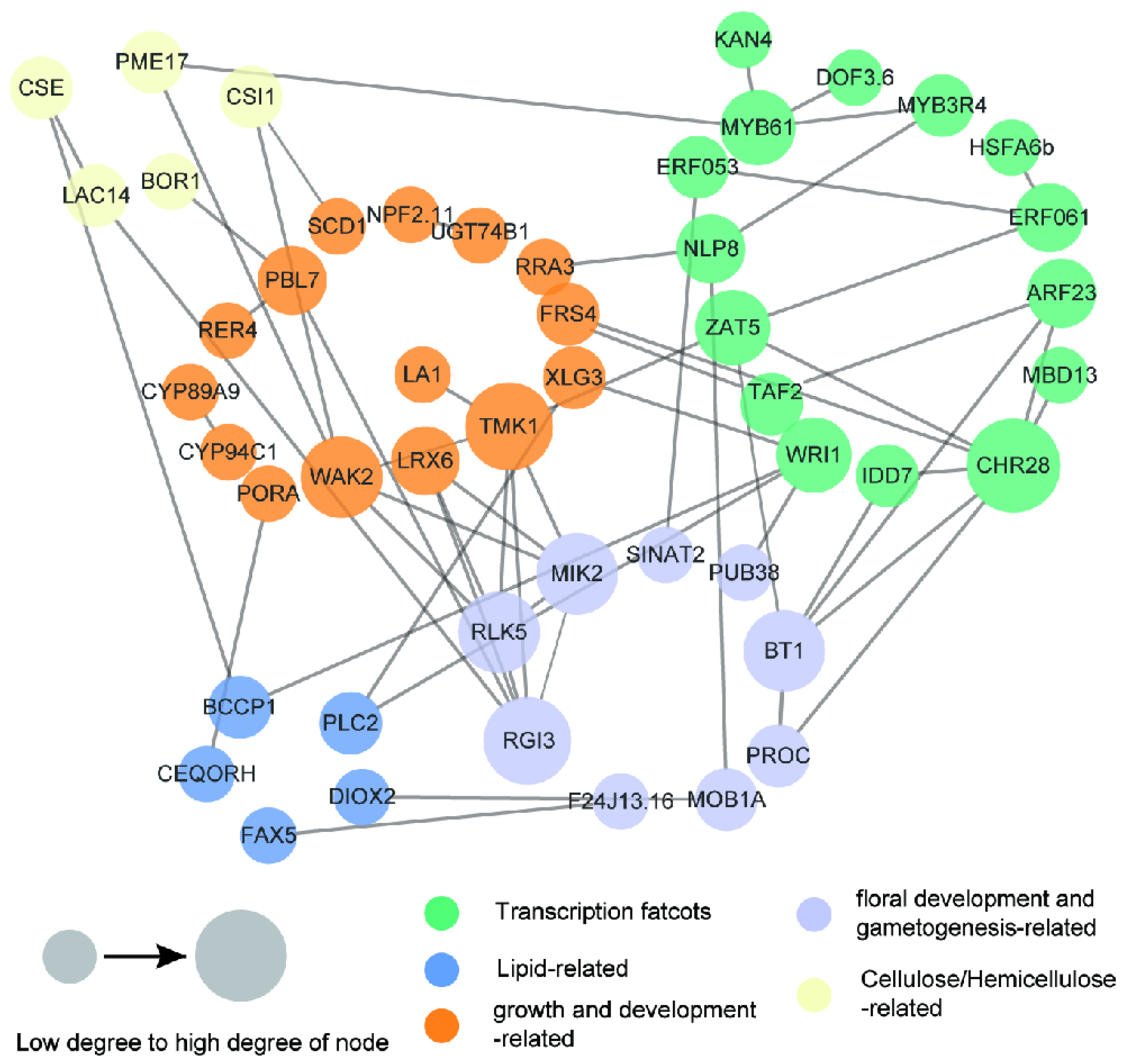
2.5. Identification of Fruit- and Oil-Related Candidate Genes and Potential Regulatory Network
3. Discussion
3.1. Mapping Population and Phenotypic Variation
3.2. Density and Accuracy of the C. oleifera Genetic Map
3.3. QTL Analysis of Economic Traits in C. oleifera
3.4. Identification and Analysis of the Candidate Genes
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Data Collection
4.3. DNA Extraction
4.4. ddRAD Libraries Sequencing and SNPs Identification
4.5. Genetic Map Construction
4.6. QTL Mapping and Genetic Mode Analysis of SNPs
4.7. Identification of Fruit- and Oil-Related Candidate Genes and Potential Regulatory Networks
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhuang, R. Oil-Tea Camellia in China, 2nd ed.; China Forestry Publishing House: Beijing, China, 2012. [Google Scholar]
- Gao, D.F.; Xu, M.; Zhao, P.; Zhang, X.Y.; Wang, Y.F.; Yang, C.R.; Zhang, Y.J. Kaempferol acetylated glycosides from the seed cake of Camellia oleifera. Food Chem. 2011, 124, 432–436. [Google Scholar] [CrossRef]
- Gong, W.; Song, Q.; Ji, K.; Gong, S.F.; Wang, L.; Chen, L.; Zhang, J.; Yuan, D. Full-length transcriptome from Camellia oleifera seed provides insight into the transcript variants involved in oil biosynthesis. J. Agric. Food Chem. 2020, 68, 14670–14683. [Google Scholar] [CrossRef]
- Yu, Y.S.; Ren, S.X.; Tan, K.Y. Study on climatic regionalization and layer and belt distribution of oil-tea Camellia quality in China. J. Nat. Resour. 1999, 14, 123–127. [Google Scholar]
- Ohlrogge, J.B. Design of new plant products: Enginnering of fatty acid metabolism. Plant Physiol. 1994, 104, 821–826. [Google Scholar] [CrossRef]
- Lin, P.; Yin, H.; Yan, C.; Yao, X.; Wang, K. Association genetics identifies single nucleotide polymorphisms related to kernel oil content and quality in Camellia oleifera. J. Agric. Food Chem. 2019, 67, 2547–2562. [Google Scholar] [CrossRef]
- He, L.; Zhou, G.Y.; Zhang, H.Y.; Liu, J.A. Research progress on the health function of tea oil. J. Med. Plants Res. 2011, 5, 485–489. [Google Scholar]
- Wu, X.H.; Huang, Y.F.; Xie, Z.F. Health functions and prospective of Camellia oil. Food Sci. Technol. 2005, 8, 94–96. [Google Scholar]
- Zhang, R.; Yang, H.; Zhou, Z.; Shen, B.; Xiao, J.; Wang, B. A high-density genetic map of Schima superba based on its chromosomal characteristics. BMC Plant Biol. 2019, 19, 41. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, J.; Luo, G.; Yuan, X.; Gong, D.; Hu, L.; Wang, S.; Chen, H.; Chen, X.; Cheng, X. Construction of a high-density adzuki bean genetic map and evaluation of its utility based on a QTL analysis of seed size. J. Integr. Agric. 2021, 7, 1753–1761. [Google Scholar] [CrossRef]
- Cai, Z.; Cheng, Y.; Ma, Z.; Liu, X.; Ma, Q.; Xia, Q.; Zhang, G.; Mu, Y.; Nian, H. Fine-mapping of QTLs for individual and total isoflavone content in soybean (Glycine max L.) using a high-density genetic map. Theor. Appl. Genet. 2018, 131, 555–568. [Google Scholar] [CrossRef]
- Zhang, W.K.; Wang, Y.J.; Luo, G.Z.; Zhang, J.S.; He, C.Y.; Wu, X.L.; Gai, J.Y.; Chen, S.Y. QTL mapping of ten agronomic traits on the soybean (Glycine max L. Merr.) genetic map and their association with EST markers. Theor. Appl. Genet. 2004, 108, 1131–1139. [Google Scholar] [CrossRef]
- Zulkifli, Y.; Kamaruddin, K.; Singh, R.; Mustafa, S.; Marjuni, M.; Ting, N.C.; Amiruddin, M.D.; Leslie, L.E.T.; Cheng-Li, O.L.; Sritharan, K.; et al. An integrated linkage map of interspecific backcross 2 (BC2) populations reveals QTLs associated with fatty acid composition and vegetative parameters influencing compactness in oil palm. BMC Plant Biol. 2020, 20, 356. [Google Scholar]
- Miao, L.; Chao, H.; Chen, L.; Wang, H.; Zhao, W.; Li, B.; Zhang, L.; Li, H.; Wang, B.; Li, M. Stable and novel QTL identification and new insights into the genetic networks affecting seed fiber traits in Brassica napus. Theor. Appl. Genet. 2019, 132, 1761–1775. [Google Scholar] [CrossRef]
- Stein, A.; Wittkop, B.; Liu, L.; Obermeier, C.; Friedt, W.; Snowdon, R.J. Dissection of a major QTL for seed colour and fibre content in Brassica napus reveals colocalization with candidate genes for phenylpropanoid biosynthesis and flavonoid deposition. Plant Breed. 2013, 132, 382–389. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Long, Y.; Li, D.; Yin, Y. Identification of QTLs associated with oil content in a high-oil Brassica napus cultivar and construction of a high-density consensus map for QTLs comparison in B. napus. PLoS ONE 2013, 8, e80569. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Long, Y.; Liu, L.; Zhao, Y.; Tian, J.; Zhao, W.; Li, B.; Chen, L.; Chao, H.; et al. Dynamic and comparative QTL analysis for plant height in different developmental stages of Brassica napus L. Theor. Appl. Genet. 2015, 128, 1175–1192. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Wang, K.; Wang, Y.; Hu, Z.; Yan, C.; Huang, H.; Ma, X.; Cao, Y.; Long, W.; Liu, W.; et al. The genome of oil-Camellia and population genomics analysis provide insights into seed oil domestication. Genome Biol. 2022, 23, 14. [Google Scholar] [CrossRef]
- Gong, W.; Xiao, S.; Wang, L.; Liao, Z.; Chang, Y.; Mo, W.; Hu, G.; Li, W.; Zhao, G.; Zhu, H.; et al. Chromosome-level genome of Camellia lanceoleosa provides a valuable resource for understanding genome evolution and self-incompatibility. Plant J. 2022, 110, 881–898. [Google Scholar] [CrossRef]
- Shen, T.; Huang, B.; Xu, M.; Zhou, P.; Ni, Z.; Gong, C.; Wen, Q.; Cao, F.; Xu, L. The reference genome of Camellia chekiangoleosa provides insights into Camellia evolution and tea oil biosynthesis. Hortic. Res. 2022, 9, uhab083. [Google Scholar] [CrossRef]
- Zhang, M.; Fan, J.; Ohlrogge, T.J.B. DGAT1 and PDAT1 acyltransferases have overlapping functions in Arabidopsis triacylglycerol biosynthesis and are essential for normal pollen and seed development. Plant Cell 2009, 21, 3885–3901. [Google Scholar] [CrossRef]
- Zhao, Y.; Cao, P.; Cui, Y.; Liu, D.; Li, J.; Zhao, Y.; Yang, S.; Zhang, B.; Zhou, R.; Sun, M. Enhanced production of seed oil with improved fatty acid composition by overexpressing NAD+-dependent glycerol-3-phosphate dehydrogenase in soybean. J. Integr. Plant Biol. 2021, 63, 18. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Mano, S.; Kondo, M.; Hayashi, M.; Nishimura, M. Extension of oil biosynthesis during the mid-phase of seed development enhances oil content in Arabidopsis seeds. Plant Biotechnol. J. 2016, 14, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.M.; Morgan, C.; Hattori, C.; Brennan, M.; Rosas, U.; Tschoep, H.; Deng, P.X.; Baker, D.; Wells, R.; Bancroft, I. Towards the genetic architecture of seed lipid biosynthesis and accumulation in Arabidopsis thaliana. Heredity 2012, 108, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Kai, W.; Nomura, K.; Fujiwara, A.; Nakamura, Y.; Yasuike, M.; Ojima, N.; Masaoka, T.; Ozaki, A.; Kazeto, Y.; Gen, K. A ddRAD-based genetic map and its integration with the genome assembly of Japanese eel (Anguilla japonica) provides insights into genome evolution after the teleost-specific genome duplication. BMC Genom. 2014, 15, 233. [Google Scholar] [CrossRef] [PubMed]
- Orlando, L.; Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for De Novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar]
- Liang, Y.; Baring, M.; Wang, S.; Septiningsih, E.M. Mapping QTLs for leafspot resistance in peanut using snp-based next-generation sequencing markers. Plant Breed. Biotechnol. 2017, 5, 115–122. [Google Scholar] [CrossRef]
- Zhou, X.; Xia, Y.; Ren, X.; Chen, Y.; Huang, L.; Huang, S.; Liao, B.; Lei, Y.; Yan, L.; Jiang, H. Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq). BMC Genom. 2014, 15, 351. [Google Scholar] [CrossRef] [PubMed]
- Scheben, A.; Severn-Ellis, A.; Patel, D.; Pradhan, A.; Edwards, D. Linkage mapping and QTL analysis of flowering time using ddRAD sequencing with genotype error correction in Brassica napus. BMC Plant Biol. 2020, 20, 546. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Wang, L.; Zhang, Y.; Lee, M.; Yue, G. Developing genome-wide SNPs and constructing an ultra high-density linkage map in oil palm. Sci. Rep. 2018, 8, 691. [Google Scholar]
- Wu, K.; Liu, H.; Yang, M.; Tao, Y.; Ma, H.; Wu, W.; Zuo, Y.; Zhao, Y. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014, 14, 274. [Google Scholar] [CrossRef]
- Shi, M.; Yu, L.; Shi, J.; Liu, J. A Conserved MYB transcription factor is involved in regulating lipid metabolic pathways for oil biosynthesis in green algae. New Phytol. 2022, 235, 576–594. [Google Scholar] [CrossRef]
- Zhang, P.; Xin, Y.; He, Y.; Tang, X.; Shen, C.; Wang, Q.; Lv, N.; Li, Y.; Hu, Q.; Xu, J. Exploring a blue-light-sensing transcription factor to double the peak productivity of oil in Nannochloropsis oceanica. Nat. Commun. 2022, 13, 1664. [Google Scholar] [CrossRef]
- Yang, J.; Chen, B.; Manan, S.; Li, P.; Liu, C.; She, G.; Zhao, S.; Zhao, J. Critical metabolic pathways and Sad/Fads, WRI1s, and DGATs cooperate for high-oleic acid oil production in developing oil tea (Camellia oleifera) seeds. Hortic. Res. 2022, 9, uhac087. [Google Scholar] [CrossRef]
- Huang, R.; Liu, M.; Gong, G.; Wu, P.; Bai, M.; Qin, H.; Wang, G.; Liao, H.; Wang, X.; Li, Y.; et al. BLISTER promotes seed maturation and fatty acid biosynthesis by interacting with WRINKLED1 to regulate chromatin dynamics in Arabidopsis. Plant Cell 2022, 34, 2242–2265. [Google Scholar] [CrossRef]
- Fenn, M.A.; Giovannoni, J.J. Phytohormones in fruit development and maturation. Plant J. 2021, 105, 446–458. [Google Scholar] [CrossRef]
- Cernac, A.; Andre, C.; Hoffmann-Benning, S.; Benning, C. WRI1 is required for seed germination and seedling establishment. Plant Physiol. 2006, 141, 745–757. [Google Scholar] [CrossRef]
- Wang, H.; Jones, B.; Li, Z.; Frasse, P.; Delalande, C.; Regad, F.; Chaabouni, S.; Latché, A.; Pech, J.C.; Bouzayen, M. The tomato Aux/IAA transcription factor IAA9 is involved in fruit development and leaf morphogenesis. Plant Cell 2005, 17, 2676–2692. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, B.; Hao, Y.; Huang, J.; Tian, A. The Soybean Dof-type transcription factor genes, GmDof4 and GmDof11, enhance lipid content in the seeds of transgenic Arabidopsis plants. Plant J. 2007, 52, 716–729. [Google Scholar] [CrossRef]
- Li, H.; Zhao, T.; Wang, Y.; Yu, D.; Chen, S.; Zhou, R.; Gai, J. Genetic structure composed of additive QTL, epistatic QTL pairs and collective unmapped minor QTL conferring oil content and fatty acid components of soybeans. Euphytica 2011, 182, 117–132. [Google Scholar] [CrossRef]
- Branham, S.E.; Levi, A.; Farnham, M.W.; Patrick Wechter, W. A GBS-SNP-based linkage map and quantitative trait loci (QTL) associated with resistance to Fusarium oxysporum f. sp. niveum race 2 identified in Citrullus lanatus var. citroides. Theor. Appl. Genet. 2017, 130, 319–330. [Google Scholar] [CrossRef]
- Emebiri, L.C.; Tan, M.K.; El-Bouhssini, M.; Wildman, O.; Ogbonnaya, F.C. QTL mapping identifies a major locus for resistance in wheat to Sunn pest (Eurygaster integriceps) feeding at the vegetative growth stage. Theor. Appl. Genet. 2016, 130, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Pek, A.; Ylmaz, K.; Skc, P.; Tangu, N.A.; Gülen, H. SNP discovery by GBS in olive and the construction of a high-density genetic linkage map. Biochem. Genet. 2016, 54, 313–325. [Google Scholar]
- Ma, J.; Huang, L.; Ma, C.; Jin, J.; Li, C.; Wang, R.; Zheng, H.; Yao, M.; Chen, L. Large-scale SNP discovery and genotyping for constructing a high-density genetic map of tea plant using specific-locus amplified fragment sequencing (SLAF-seq). PLoS ONE 2015, 10, e0128798. [Google Scholar] [CrossRef]
- Guo, Y.; Shi, G.; Liu, Z.; Zhao, Y.; Yang, X.; Zhu, J.; Li, K.; Guo, X. Using specific length amplified fragment sequencing to construct the high-density genetic map for Vitis (Vitis vinifera L. × Vitis amurensis Rupr.). Front. Plant Sci. 2015, 6, 393. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Yu, H.; Tang, Z.; Jiang, X.; Wang, L.; Wang, X.; Xu, Q.; Deng, X. Construction of a SNP-based high-density genetic map for pummelo using RAD sequencing. Tree Genet. Genomes 2015, 11, 2. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, T.; Zhong, Y.; Li, X.; Huang, J. Construction of a high-density genetic map of Ziziphus jujuba Mill. using genotyping by sequencing technology. Tree Genet. Genomes 2016, 12, 76. [Google Scholar] [CrossRef]
- Chai, J.; Wang, K.; Xie, Y.; Wang, A.; Zhong, H.; Yao, X.; Lin, P. Combining ability from complete diallel design of Camellia oleifera: Implications for the utility of GCA and SCA in oil-related traits breeding. Ind. Crops Prod. 2023, 205, 117434. [Google Scholar] [CrossRef]
- Ye, Z.; Wu, Y.; Ul Haq Muhammad, Z.; Yan, W.; Yu, J.; Zhang, J.; Yao, G.; Hu, X. Complementary transcriptome and proteome profiling in the mature seeds of Camellia oleifera from Hainan Island. PLoS ONE 2020, 15, e0226888. [Google Scholar] [CrossRef]
- Song, X.; Xu, Y.; Gao, K.; Fan, G.; Zhang, F.; Deng, C.; Dai, S.; Huang, H.; Xin, H.; Li, Y. High-density genetic map construction and identification of loci controlling flower-type traits in Chrysanthemum (Chrysanthemum × morifolium Ramat.). Hortic. Res. 2020, 7, 108. [Google Scholar] [CrossRef]
- Gutierrez-Gonzalez, J.J.; Vuong, T.D.; Zhong, R.; Yu, O.; Lee, J.D.; Shannon, G.; Ellersieck, M.; Nguyen, H.T.; Sleper, D.A. Major locus and other novel additive and epistatic loci involved in modulation of isoflavone concentration in soybean seeds. Theor. Appl. Genet. 2011, 123, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Zou, G. Identification of QTLs for eight agronomically important traits using an ultra-high-density map based on SNPs generated from high-throughput sequencing in sorghum under contrasting photoperiods. J. Exp. Bot. 2012, 63, 5451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, H.; Wang, J.; Zhang, H.; Hu, Z.; Chu, S.; Lv, H.; Yu, D. High-density genetic mapping identifies new major loci for tolerance to low-phosphorus stress in soybean. Front. Plant Sci. 2016, 7, 372. [Google Scholar] [CrossRef]
- Fu, X.; Dou, J.; Mao, J.; Su, H.; Jiao, W.; Zhang, L.; Hu, X.; Huang, X.; Wang, S.; Bao, Z. RAD typing: An integrated package for accurate de novo codominant and dominant RAD genotyping in mapping populations. PLoS ONE 2013, 8, e79960. [Google Scholar] [CrossRef]
- Amit, A.D.; Larissa, R.; Andrew, G.S.; Marwan, D.; Anoop, S.; Kirstin, B.; Thomas, D.W.; Bunyamin, T.A. Genome wide SNP identification in chickpea for use in development of a high density genetic map and improvement of chickpea reference genome assembly. BMC Genom. 2014, 15, 708. [Google Scholar]
- Yin, T.; Wang, Y.; Ning, Z.; Hu, Y.; Chen, J.; Zhao, R.; Chen, H.; Ai, N.; Guo, W.; Zhang, T. Molecular mapping of restriction-site associated DNA markers in allotetraploid upland cotton. PLoS ONE 2015, 10, e0124781. [Google Scholar]
- Liu, Z.; Zhu, H.; Liu, Y.; Kuang, J.; Zhou, K.; Liang, F.; Liu, Z.; Wang, D.; Ke, W. Construction of a high-density, high-quality genetic map of cultivated lotus (Nelumbo nucifera) using next-generation sequencing. BMC Genom. 2016, 17, 466. [Google Scholar] [CrossRef]
- Subudhi, P.K.; Fan, W.; Zong, J.; Luo, Z.; Chen, M.; Zhao, X.; Zhang, D.; Qi, Y.; Yuan, Z. Development of a RAD-Seq based DNA polymorphism identification software, agromarker finder, and its application in rice marker-assisted breeding. PLoS ONE 2016, 11, e0147187. [Google Scholar]
- Zhang, M.; Lu, N.; Zhu, T.; Yang, G.; Qu, G.; Shi, C.; Fei, Y.; Liu, B.; Ma, W.; Wang, J. A bivariate mapping model identifies major covariation QTLs for biomass allocation between leaf and stem growth of Catalpa bungei. Front. Genet. 2021, 12, 758209. [Google Scholar] [CrossRef]
- Zhang, M.; Lu, N.; Jiang, L.; Liu, B.; Fei, Y.; Ma, W.; Shi, C.; Wang, J.; Plomion, C. Multiple dynamic models reveal the genetic architecture for growth in height of Catalpa bungei in the field. Tree Physiol. 2022, 42, 1239–1255. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, B.; Fei, Y.; Yang, X.; Zhao, L.; Shi, C.; Zhang, Y.; Lu, N.; Wu, C.; Ma, W.; et al. Genetic architecture of leaf morphology revealed by integrated trait module in Catalpa bungei. Hortic. Res. 2023, 10, uhad032. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Xing, Y.; Li, S.; Ding, J.; Yue, B.; Deng, K.; Li, Y.; Zhu, Y. Molecular dissection of developmental behavior of tiller number and plant height and their relationship in rice (Oryza sativa L.). Hereditas 2006, 143, 236–245. [Google Scholar] [CrossRef]
- Pootakham, W.; Jomchai, N.; Ruang-areerate, P.; Shearman, J.R.; Sonthirod, C.; Sangsrakru, D.; Tragoonrung, S.; Tangphatsornruang, S. Genome-wide SNP discovery and identification of QTL associated with agronomic traits in oil palm using genotyping-by-sequencing (GBS). Genomics 2015, 105, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, X.; Ma, Z.; Chen, S. Quantitative trait locus mapping of fruit aroma compounds in cucumber (Cucumber sativus L.) based on a recombinant inbred line population. Hortic. Res. 2022, 9, uhac151. [Google Scholar] [CrossRef]
- Song, Q.; Ji, K.; Mo, W.; Wang, L.; Chen, L.; Gao, L.; Gong, W.; Yuan, D. Dynamics of sugars, endogenous hormones, and oil content during the development of Camellia oleifera fruit. Botany 2021, 99, 515–529. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Z.; Zhou, J.; Gu, Y.; Tan, X. Comparative study on fruit development and oil synthesis in two cultivars of Camellia oleifera. BMC Plant Biol. 2021, 21, 348. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Yu, J.; Yan, W.; Zhang, J.; Yang, D.; Yao, G.; Liu, Z.; Wu, Y.; Hou, X. Integrative iTRAQ-based proteomic and transcriptomic analysis reveals the accumulation patterns of key metabolites associated with oil quality during seed ripening of Camellia oleifera. Hortic. Res. 2021, 8, 157. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Dai, X.; Chen, Y.; Chen, J.; Shi, J.; Yin, T. Discovery and experimental analysis of microsatellites in an oil woody plant Camellia chekiangoleosa. Plant Syst. Evol. 2013, 299, 1387–1393. [Google Scholar] [CrossRef]
- Brkić, K.; Radulović, M.; Sladonja, B.; Lukić, I.; Šetić, E. Application of Soxtec apparatus for oil content determination in olive fruit. Riv. Ital. Sostanze Grasse 2006, 83, 115–119. [Google Scholar]
- Lall, R.K.; Proctor, A.; Jain, V.P. A rapid, micro FAME preparation method for vegetable oil fatty acid analysis by Gas Chromatography. J. Am. Oil Chem. Soc. 2009, 86, 309–314. [Google Scholar] [CrossRef]
- Lin, P.; Wang, K.; Zhou, C.; Xie, Y.; Yao, X.; Yin, H. Seed transcriptomics analysis in Camellia oleifera uncovers genes associated with oil content and fatty acid composition. Int. J. Mol. Sci. 2018, 19, 118. [Google Scholar] [CrossRef]
- Tang, Q.; Zhang, C. Data Processing System (DPS) software with experimental design, statistical analysis and data mining developed for use in entomological research. Insect Sci. 2013, 20, 254–260. [Google Scholar] [CrossRef]
- Grattapaglia, D.; Sederoff, R. Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: Mapping strategy and RAPD markers. Genetics 1994, 137, 1121–1137. [Google Scholar] [CrossRef]
- Liu, D.; Ma, C.; Hong, W.; Huang, L.; Liu, M.; Liu, H.; Zeng, H.; Deng, D.; Xin, H.; Song, J.; et al. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef]
- Kosambi, D.D. The estimation of map distance from recombination values. Ann. Eugen. 1943, 12, 172–175. [Google Scholar] [CrossRef]
- Wang, J.K. Inclusive composite interval mapping of quantitative trait genes. Acta Agron. Sin. 2009, 35, 239–245. [Google Scholar] [CrossRef]
- Wegrzyn, J.L.; Eckert, A.J.; Choi, M.; Lee, J.M.; Stanton, B.J.; Sykes, R.; Davis, M.F.; Tsai, C.J.; Neale, D.B. Association genetics of traits controlling lignin and cellulose biosynthesis in black cottonwood (Populus trichocarpa, Salicaceae) secondary xylem. New Phytol. 2010, 188, 515–532. [Google Scholar] [CrossRef]
- Eckert, A.J.; Bower, A.D.; Wegrzyn, J.L.; Pande, B.; Jermstad, K.D.; Krutovsky, K.V.; St. Clair, J.B.; Neale, D.B. Association genetics of coastal Douglas fir (Pseudotsuga menziesii var. menziesii, Pinaceae). I. Cold-Hardiness related traits. Genetics 2009, 182, 1289–1302. [Google Scholar] [CrossRef]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Bader, G.D.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sources of Variation | Year | Genotypes | Error | |
|---|---|---|---|---|
| df | 2 | 179 | 358 | |
| FY | MS | 269.4273 | 2.8638 | 1.8835 |
| F value | 143.049 ** | 1.521 ** | ||
| RSF | MS | 3488.9907 | 72.3878 | 37.9708 |
| F value | 91.886 ** | 1.906 ** | ||
| RKS2 | MS | 291.5984 | 27.5792 | 6.5039 |
| F value | 44.834 ** | 4.24 ** | ||
| RKS1 | MS | 268.7622 | 26.0167 | 5.7166 |
| F value | 47.015 ** | 4.551 ** | ||
| OC | MS | 75.0375 | 27.4846 | 15.3854 |
| F value | 4.877 ** | 1.786 ** | ||
| C16:0 | MS | 102.5323 | 0.5838 | 0.1932 |
| F value | 530.75 ** | 3.022 ** | ||
| C18:0 | MS | 4.6355 | 0.1403 | 0.0551 |
| F value | 84.199 ** | 2.549 ** | ||
| C18:1 | MS | 657.5575 | 4.0707 | 1.4652 |
| F value | 448.777 ** | 2.778 ** | ||
| C18:2 | MS | 245.2011 | 2.6365 | 0.8135 |
| F value | 301.403 ** | 3.241 ** | ||
| C18:3 | MS | 0.0264 | 0.0018 | 0.0008 |
| F value | 33.577 ** | 2.266 ** | ||
| C20:1 | MS | 0.0143 | 0.004 | 0.0015 |
| F value | 9.765 ** | 2.754 ** | ||
| LG ID | Total Markers | Total Distance (cM) | Average Distance (cM) | Gaps > 5 cM(%) | Max Gap (cM) |
|---|---|---|---|---|---|
| LG01 | 231 | 203.79 | 0.89 | 1.74 | 20.62 |
| LG02 | 187 | 220.89 | 1.19 | 5.91 | 14.28 |
| LG03 | 197 | 196.62 | 1.00 | 2.04 | 10.63 |
| LG04 | 138 | 253.02 | 1.85 | 5.84 | 40.67 |
| LG05 | 192 | 222.04 | 1.16 | 4.71 | 26.96 |
| LG06 | 187 | 196.19 | 1.05 | 2.69 | 12.75 |
| LG07 | 258 | 319.60 | 1.24 | 4.28 | 19.77 |
| LG08 | 164 | 262.68 | 1.61 | 7.98 | 16.16 |
| LG09 | 118 | 211.33 | 1.81 | 5.98 | 19.35 |
| LG10 | 186 | 205.40 | 1.11 | 2.70 | 15.22 |
| LG11 | 262 | 224.30 | 0.86 | 2.30 | 22.44 |
| LG12 | 181 | 201.07 | 1.12 | 2.22 | 10.37 |
| LG13 | 171 | 196.04 | 1.15 | 2.35 | 19.84 |
| LG14 | 145 | 207.60 | 1.44 | 5.56 | 15.29 |
| LG15 | 163 | 206.47 | 1.27 | 5.56 | 17.51 |
| Total | 2780 | 3327.02 | 1.20 | 3.88 | 40.67 |
| Average | 185 | 415.88 | 1.20 | 3.88 | --- |
| Traits | Year | Number of QTLs | Linkage Group | PVE a (%) | LOD Value |
|---|---|---|---|---|---|
| FY | 2015 | 10 | 1, 2, 5, 6, 10, 13 | 7.4–9.4 | 3.0–3.87 |
| 2016 | 5 | 3, 5, 7, 9 | 7.4–9.0 | 3.02–3.7 | |
| 2017 | 8 | 3, 4, 7, 11, 13, 15 | 7.6–9.9 | 3.07–4.07 | |
| RSF | 2015 | 14 | 2, 4, 6, 7, 9, 10, 11, 14 | 9.3–11.4 | 3.0–3.72 |
| 2016 | 8 | 2, 3, 6, 7, 8, 11 | 10.5–14.0 | 3.01–4.1 | |
| 2017 | 10 | 3, 9, 10, 12, 13, 14 | 8.2–11.2 | 3.06–4.25 | |
| RKS2 | 2015 | 5 | 1, 6, 8, 10, 11 | 9.8–16.6 | 3.17–5.57 |
| 2016 | 14 | 1, 2, 3, 4, 6, 7, 8, 9, 13, 14, 15 | 10.4–15.1 | 3.0–4.48 | |
| 2017 | 5 | 4, 5, 6, 10, 13 | 7.5–9.0 | 3.02–3.66 | |
| RKS1 | 2015 | 5 | 2, 6, 10, 11 | 9.6–14.3 | 3.09–4.71 |
| 2016 | 14 | 1, 2, 3, 4, 6, 7, 9, 13, 14, 15 | 10.6–14.5 | 3.08–4.27 | |
| 2017 | 8 | 4, 5, 6, 7, 8, 10, 14 | 7.5–10.0 | 3.0–4.08 | |
| OC | 2015 | 6 | 3, 13, 14, 16 | 9.4–13.3 | 3.03–4.37 |
| 2016 | 4 | 1, 10, 11 | 11.5–14.5 | 3.36–4.28 | |
| 2017 | 3 | 1, 2, 11 | 7.5–8.3 | 3.03–3.35 | |
| C16:0 | 2015 | 9 | 1, 2, 7, 8, 11, 13, 15 | 9.5–13.9 | 3.07–4.58 |
| 2016 | 11 | 1, 3, 4, 7, 10, 11, 12, 14, 15 | 10.4–14.6 | 3.02–4.31 | |
| 2017 | 7 | 3, 6, 7, 10, 11 | 7.9–11.9 | 3.17–4.89 | |
| C18:0 | 2015 | 5 | 2, 5, 7, 13 | 9.7–12.5 | 3.13–4.09 |
| 2016 | 5 | 3, 4, 12, 15 | 10.4–12.6 | 3.01–3.67 | |
| 2017 | 4 | 2, 9, 13, 14 | 7.7–8.8 | 3.09–3.56 | |
| C18:1 | 2015 | 4 | 5, 7, 8, 15 | 10.1–12.2 | 3.26–3.99 |
| 2016 | 6 | 10, 11, 12, 15 | 10.4–14.5 | 3.0–4.3 | |
| 2017 | 0 | ||||
| C18:2 | 2015 | 5 | 3, 5, 7, 11, 15 | 9.6–10.8 | 3.07–3.5 |
| 2016 | 7 | 2, 6, 10, 11, 12, 13 | 10.5–13.4 | 3.05–3.95 | |
| 2017 | 4 | 5, 10, 11 | 7.5–9.8 | 3.0–3.99 | |
| C18:3 | 2015 | 14 | 1, 3, 4, 8, 9, 10, 11, 14, 15 | 9.4–13.0 | 3.02–4.26 |
| 2016 | 9 | 3, 4, 5, 7, 11, 12, 13 | 10.5–15.3 | 3.05–4.54 | |
| 2017 | 6 | 2, 3, 5, 11, 13, 15 | 8.0–9.7 | 3.2–3.93 | |
| C20:1 | 2015 | 5 | 1, 7, 8, 10, 13 | 9.5–10.2 | 3.05–3.31 |
| 2016 | 9 | 4, 7, 9, 10, 11, 13, 15 | 10.5–15.4 | 3.02–4.57 | |
| 2017 | 2 | 1, 11 | 7.7–8.1 | 3.09–3.27 |
| Trait | QTL | Start | End | Length (cM) | Year | LOD | PVE (%) | Marker | Cross Type | CL53 (♀) | CL81 (♂) | Genotype | Individual Numbers | Phenotype Means(%) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2015 | 2016 | 2017 | ||||||||||||||
| RKS2 | RKS2-q4 | 135.721 | 138.501 | 2.78 | 2016 | 4.48 | 15.1 | Chr04_77743434 | lm × ll | GT | GG | GG | 101 | 59.70 | 62.05 | |
| 2017 | 3.01 | 7.5 | GT | 76 | 58.24 | 60.33 | ||||||||||
| - | 3 | 57.78 | 61.94 | |||||||||||||
| RKS2-q6-1 | 50.723 | 64.534 | 13.811 | 2015 | 5.57 | 16.6 | Chr06_142599472 | lm × ll | GA | GG | GG | 97 | 59.25 | 61.07 | ||
| 2017 | 3.66 | 9.0 | GA | 81 | 59.12 | 61.56 | ||||||||||
| - | 1 | 62.77 | 62.97 | |||||||||||||
| Chr06_112292496 | lm × ll | GA | GG | GG | 97 | 59.31 | 61.22 | |||||||||
| GA | 80 | 59.33 | 61.58 | |||||||||||||
| - | 3 | 54.52 | 56.69 | |||||||||||||
| Chr06_88890750 | lm × ll | CT | CC | CC | 100 | 59.57 | 61.35 | |||||||||
| CT | 77 | 58.85 | 61.29 | |||||||||||||
| - | 3 | 56.83 | 60.14 | |||||||||||||
| Chr06_25329856 | hk × hk | AG | AG | AA | 35 | 57.95 | 61.19 | |||||||||
| AG | 96 | 59.58 | 61.24 | |||||||||||||
| GG | 30 | 59.01 | 61.50 | |||||||||||||
| - | 19 | 59.93 | 61.57 | |||||||||||||
| Chr06_111554923 | lm × ll | GA | GG | GG | 91 | 59.44 | 61.36 | |||||||||
| GA | 86 | 58.87 | 61.20 | |||||||||||||
| - | 3 | 61.85 | 62.65 | |||||||||||||
| RSK1 | RKS1-q4-2 | 135.721 | 138.501 | 2.78 | 2016 | 4.27 | 14.5 | Chr04_77743434 | lm × ll | GT | GG | GG | 101 | 64.78 | 67.21 | |
| 2017 | 3.28 | 8.1 | GT | 76 | 64.65 | 66.97 | ||||||||||
| - | 3 | 63.40 | 67.17 | |||||||||||||
| RKS1-q6-1 | 54.072 | 64.534 | 10.462 | 2015 | 4.71 | 14.3 | Chr06_142599472 | lm × ll | GA | GG | GG | 97 | 65.67 | 64.48 | ||
| 2016 | 3.37 | 11.6 | GA | 81 | 66.71 | 65.26 | ||||||||||
| - | 1 | 69.27 | 68.23 | |||||||||||||
| Chr06_112292496 | lm × ll | GA | GG | GG | 97 | 66.14 | 64.75 | |||||||||
| GA | 80 | 66.23 | 64.72 | |||||||||||||
| - | 3 | 62.76 | 61.66 | |||||||||||||
| Chr06_88890750 | lm × ll | CT | CC | CC | 100 | 66.31 | 65.23 | |||||||||
| CT | 77 | 65.99 | 64.04 | |||||||||||||
| - | 3 | 62.91 | 61.79 | |||||||||||||
| Chr06_25329856 | hk × hk | AG | AG | AA | 35 | 65.24 | 63.62 | |||||||||
| AG | 96 | 66.25 | 65.11 | |||||||||||||
| GG | 30 | 66.19 | 64.90 | |||||||||||||
| - | 19 | 66.77 | 64.22 | |||||||||||||
| C18:3 | C18:3-q11-2 | 84.52 | 85.695 | 1.175 | 2015 | 3.16 | 9.8 | Chr11_27083024 | nn × np | CC | CT | CC | 86 | 22.24 | 24.45 | |
| 2017 | 3.93 | 9.7 | CT | 92 | 22.23 | 24.53 | ||||||||||
| - | 1 | 31.47 | 33.96 | |||||||||||||
| C18:2 | C18:2-q11-1 | 162.869 | 166.196 | 3.327 | 2015 | 3.07 | 9.6 | Chr11_114235680 | nn × np | CC | CT | CC | 98 | 6.54 | 7.96 | |
| 2017 | 3.02 | 7.5 | CT | 80 | 6.90 | 8.05 | ||||||||||
| - | 2 | 6.23 | 9.21 | |||||||||||||
| Chr11_65073730 | lm × ll | AG | AA | AA | 78 | 6.78 | 8.08 | |||||||||
| AG | 98 | 6.59 | 7.91 | |||||||||||||
| - | 4 | 8.16 | 9.35 | |||||||||||||
| C18:2-q5-1 | 104.487 | 105.646 | 1.159 | 2015 | 3.5 | 10.8 | Chr05_107820202 | lm × ll | AG | GG | GG | 95 | 6.84 | 8.22 | ||
| 2017 | 3.0 | 7.5 | AG | 79 | 6.55 | 7.76 | ||||||||||
| - | 4 | 7.00 | 7.62 | |||||||||||||
| Chr05_93231328 | hk × hk | TC | TC | TT | 56 | 6.75 | 7.88 | |||||||||
| TC | 67 | 6.87 | 8.13 | |||||||||||||
| CC | 32 | 6.37 | 8.15 | |||||||||||||
| - | 25 | 6.58 | 7.84 | |||||||||||||
| C16:0 | C16:0-q11-3 | 164.001 | 169.537 | 5.536 | 2015 | 3.73 | 11.5 | Chr11_39972487 | lm × ll | AG | AA | AA | 84 | 6.87 | 8.37 | |
| 2017 | 4.2 | 10.3 | AG | 94 | 7.02 | 8.39 | ||||||||||
| - | 2 | 6.68 | 8.10 | |||||||||||||
| Chr11_39972501 | lm × ll | AG | AA | AA | 83 | 6.85 | 8.35 | |||||||||
| AG | 95 | 7.03 | 8.41 | |||||||||||||
| - | 2 | 6.68 | 8.10 | |||||||||||||
| Chr11_39972457 | lm × ll | CA | CC | CC | 82 | 6.85 | 8.35 | |||||||||
| CA | 96 | 7.03 | 8.40 | |||||||||||||
| - | 2 | 6.68 | 8.10 | |||||||||||||
| Chr11_128763300 | nn × np | TT | TA | TT | 98 | 6.88 | 8.33 | |||||||||
| TA | 79 | 7.04 | 8.44 | |||||||||||||
| - | 3 | 6.47 | 8.37 | |||||||||||||
| Chr11_10782110 | nn × np | GG | GA | GG | 78 | 6.95 | 8.38 | |||||||||
| GA | 100 | 6.95 | 8.38 | |||||||||||||
| - | 2 | 6.34 | 8.36 | |||||||||||||
| Chr11_64280835 | nn × np | CC | CT | CC | 99 | 6.96 | 8.38 | |||||||||
| CT | 78 | 6.93 | 8.37 | |||||||||||||
| - | 3 | 6.68 | 8.46 | |||||||||||||
| Chr11_146396217 | lm × ll | GA | GG | GG | 79 | 6.91 | 8.37 | |||||||||
| GA | 100 | 6.97 | 8.38 | |||||||||||||
| - | 1 | 6.84 | 8.44 | |||||||||||||
| C18:0 | C18:0-q2 | 78.596 | 82.114 | 3.518 | 2015 | 3.13 | 9.7 | Chr02_116850822 | lm × ll | AG | GG | GG | 99 | 1.85 | 2.13 | |
| 2017 | 3.44 | 8.5 | GA | 76 | 1.87 | 2.17 | ||||||||||
| - | 5 | 1.75 | 1.94 | |||||||||||||
| Chr02_98558620 | hk × hk | AG | AG | GG | 41 | 1.79 | 2.11 | |||||||||
| GA | 86 | 1.89 | 2.14 | |||||||||||||
| AA | 39 | 1.85 | 2.15 | |||||||||||||
| - | 14 | 1.81 | 2.21 | |||||||||||||
| Trait | SNP | 2a 1 | d 2 | d/a | Frequency 3 | a4 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2015 | 2016 | 2017 | 2015 | 2016 | 2017 | 2015 | 2016 | 2017 | 2015 | 2016 | 2017 | ||||
| RKS2 | Chr06_25329856 | 1.06 | 0.93 | 0.31 | 1.10 | 0.95 | −0.11 | 2.08 | 2.05 | −0.68 | 0.48 | (G) | −0.76 | −0.51 | 0.04 |
| RKS1 | Chr06_25329856 | 0.95 | 1.28 | 0.34 | 0.53 | 0.85 | 0.04 | 1.13 | 1.33 | 0.24 | 0.48 | (G) | −0.42 | −0.46 | −0.07 |
| C18:2 | Chr05_93231328 | 0.38 | 0.16 | 0.27 | 0.31 | 0.37 | 0.12 | 1.63 | 4.62 | 0.85 | 0.42 | (C) | −0.11 | −0.17 | −0.02 |
| C18:0 | Chr02_98558620 | 0.06 | 0.14 | 0.04 | 0.07 | −0.06 | 0.01 | 2.33 | −0.86 | 0.50 | 0.49 | (A) | −0.03 | 0.04 | −0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, P.; Chai, J.; Wang, A.; Zhong, H.; Wang, K. High-Density Genetic Map Construction and Quantitative Trait Locus Analysis of Fruit- and Oil-Related Traits in Camellia oleifera Based on Double Digest Restriction Site-Associated DNA Sequencing. Int. J. Mol. Sci. 2024, 25, 8840. https://doi.org/10.3390/ijms25168840
Lin P, Chai J, Wang A, Zhong H, Wang K. High-Density Genetic Map Construction and Quantitative Trait Locus Analysis of Fruit- and Oil-Related Traits in Camellia oleifera Based on Double Digest Restriction Site-Associated DNA Sequencing. International Journal of Molecular Sciences. 2024; 25(16):8840. https://doi.org/10.3390/ijms25168840
Chicago/Turabian StyleLin, Ping, Jingyu Chai, Anni Wang, Huiqi Zhong, and Kailiang Wang. 2024. "High-Density Genetic Map Construction and Quantitative Trait Locus Analysis of Fruit- and Oil-Related Traits in Camellia oleifera Based on Double Digest Restriction Site-Associated DNA Sequencing" International Journal of Molecular Sciences 25, no. 16: 8840. https://doi.org/10.3390/ijms25168840