
The Discovery of a Specific CKIP-1 Ligand for the Potential Treatment of Disuse Osteoporosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. CKIP-1 Protein Level Is Upregulated under Microgravity Stimulation
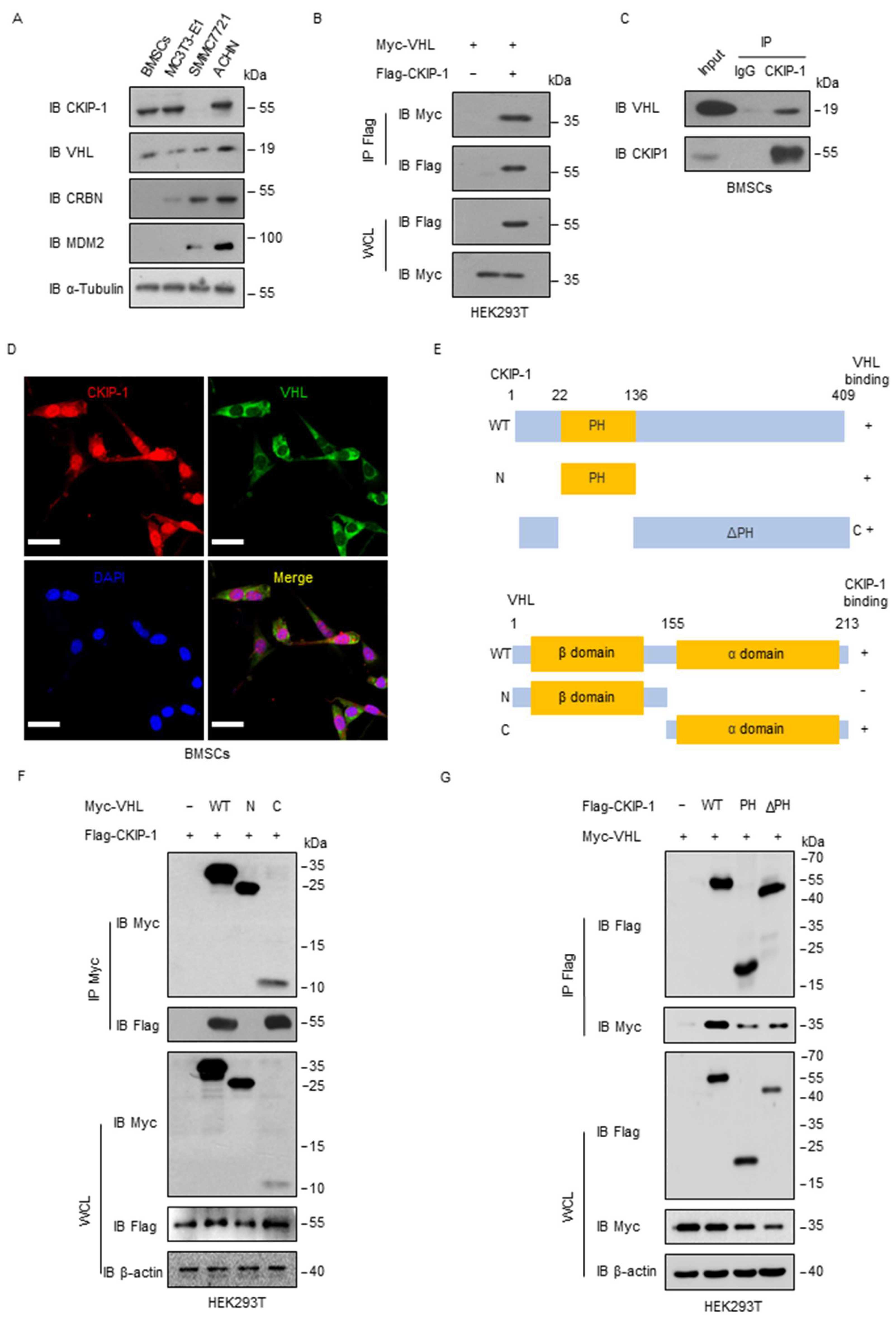
2.2. VHL Interacts with CKIP-1
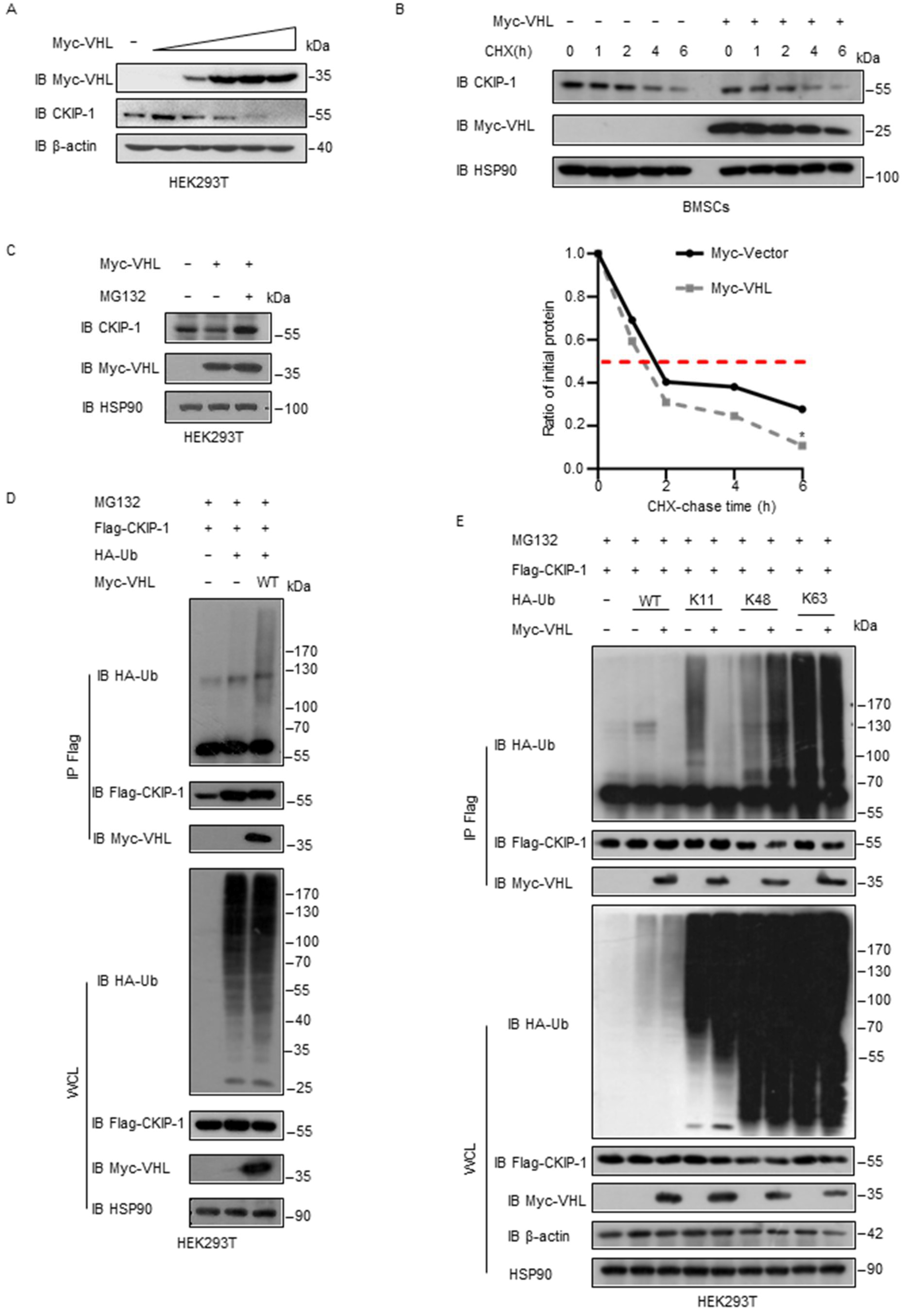
2.3. VHL Promotes Ubiquitylation of CKIP-1
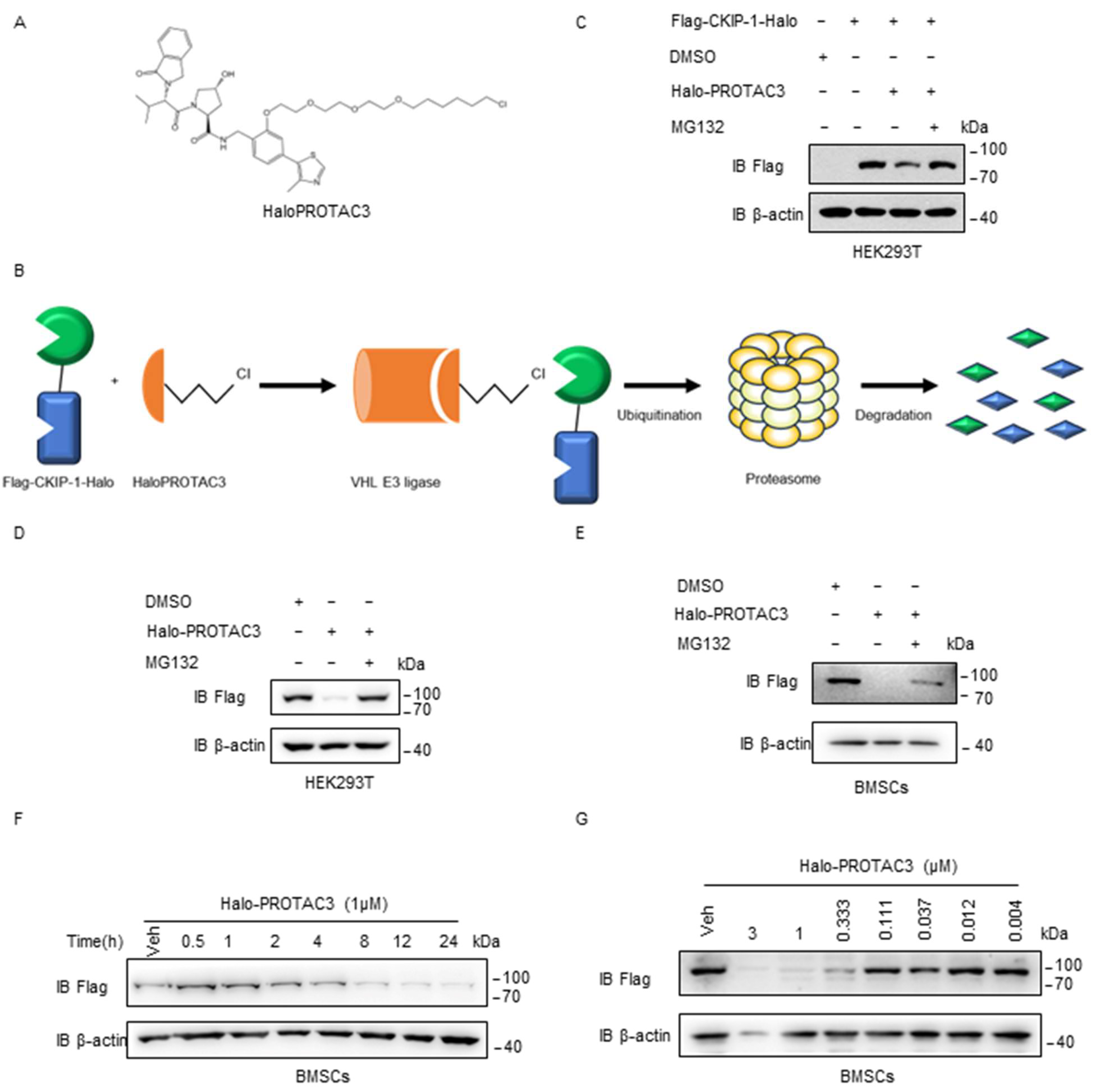
2.4. HaloPROTAC3 Degrades CKIP-1
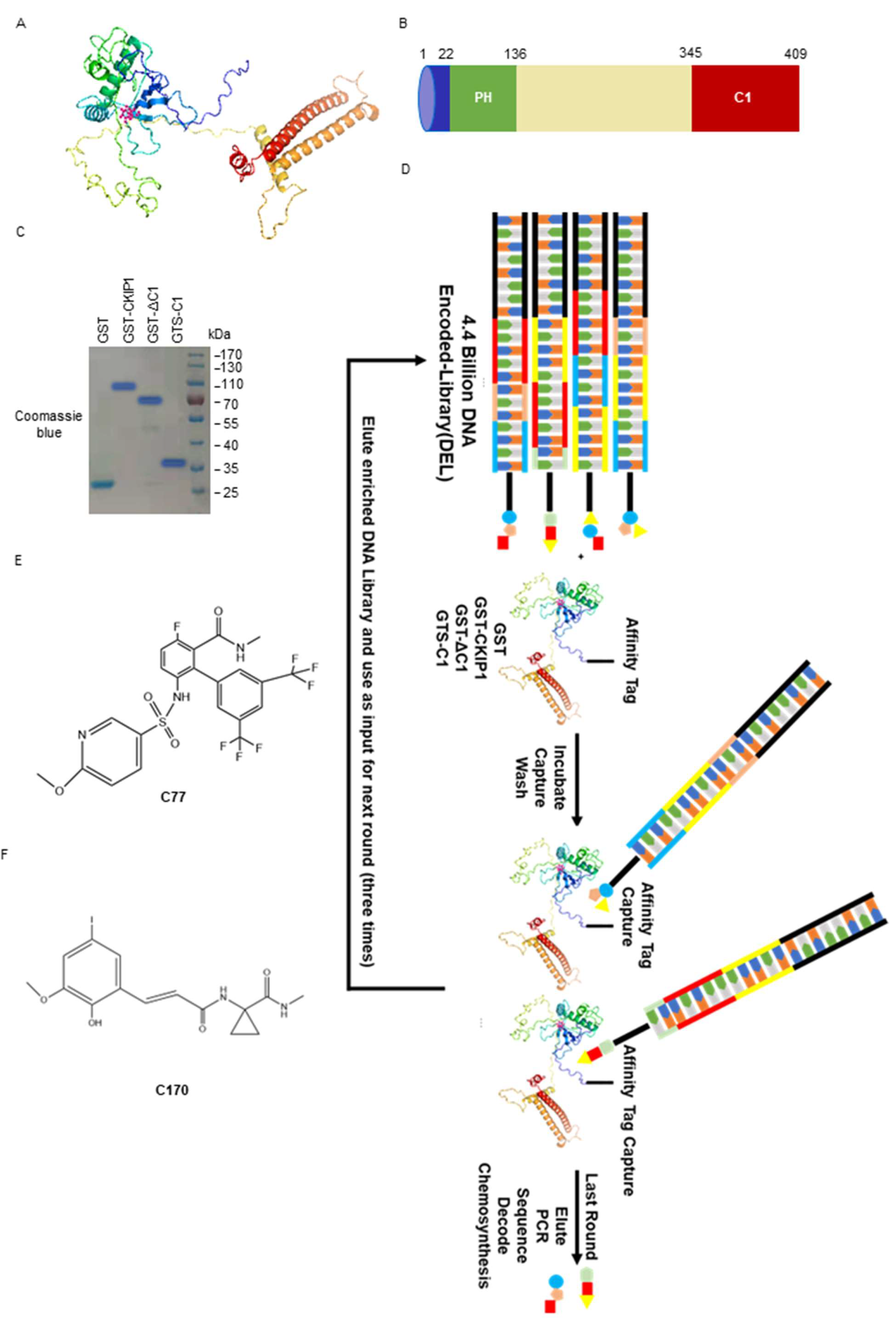
2.5. Hit Generation from DELs Screening Data
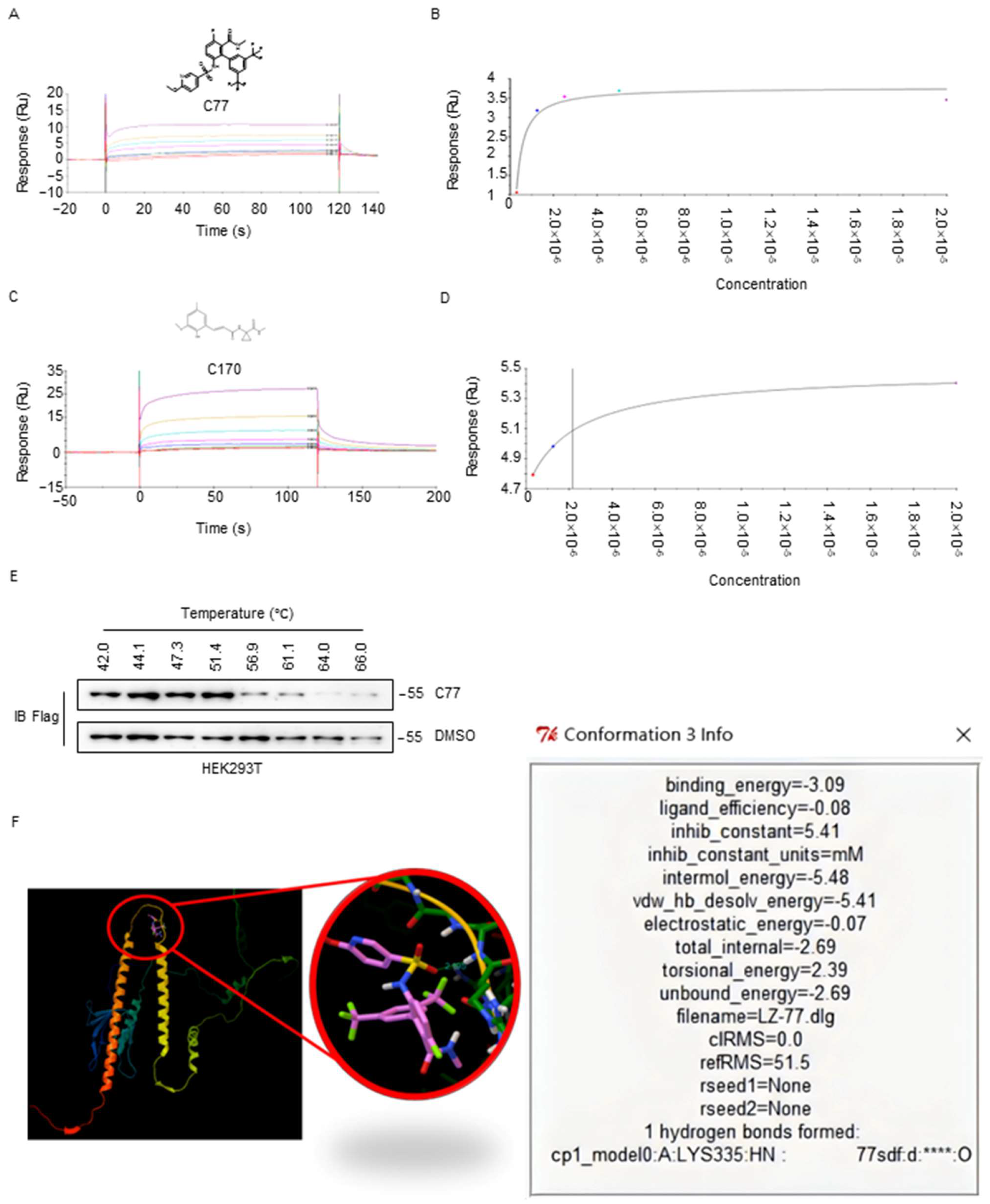
2.6. Biophysical Characterization of Compounds with CKIP-1
3. Discussion
4. Materials and Methods
4.1. Animals Model
4.2. Rotary Cell Culture System (RCCS)
4.3. Quantitative Real-Time PCR Analysis
4.4. Immunofluorescence
4.5. Immunoblotting
4.6. Plasmid Constructs
4.7. Mass Spectrometry
4.8. Cell Transfection and Immunoprecipitation
4.9. In Vivo Ubiquitylation Assays
4.10. Surface Plasmon Resonance (SPR)
4.11. Cellular Thermal Shift Assay (CETSA)
4.12. Micro-Computed Tomography (Micro-CT) Analysis
4.13. Mechanical Properties
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salhotra, A.; Shah, H.N.; Levi, B.; Longaker, M.T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 2020, 21, 696–711. [Google Scholar] [CrossRef]
- Vico, L.; Hargens, A. Skeletal changes during and after spaceflight. Nat. Rev. Rheumatol. 2018, 14, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Lehar, A.; Meir, J.U.; Koch, C.; Morgan, A.; Warren, L.E.; Rydzik, R.; Youngstrom, D.W.; Chandok, H.; George, J.; et al. Targeting myostatin/activin A protects against skeletal muscle and bone loss during spaceflight. Proc. Natl. Acad. Sci. 2020, 117, 23942–23951. [Google Scholar] [CrossRef] [PubMed]
- Mochi, F.; Scatena, E.; Rodriguez, D.; Ginebra, M.-P.; Del Gaudio, C. Scaffold-based bone tissue engineering in microgravity: Potential, concerns and implications. NPJ Microgravity 2022, 8, 45. [Google Scholar] [CrossRef]
- Sibonga, J.; Matsumoto, T.; Jones, J.; Shapiro, J.; Lang, T.; Shackelford, L.; Smith, S.M.; Young, M.; Keyak, J.; Kohri, K.; et al. Resistive exercise in astronauts on prolonged spaceflights provides partial protection against spaceflight-induced bone loss. Bone 2019, 128, 112037. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wentworth, K.; Shoback, D.M. New Frontiers in Osteoporosis Therapy. Annu. Rev. Med. 2020, 71, 277–288. [Google Scholar] [CrossRef]
- Lu, K.; Yin, X.; Weng, T.; Xi, S.; Li, L.; Xing, G.; Cheng, X.; Yang, X.; Zhang, L.; He, F. Targeting WW domains linker of HECT-type ubiquitin ligase Smurf1 for activation by CKIP-1. Nat. Cell Biol. 2008, 10, 994–1002. [Google Scholar] [CrossRef]
- Fu, L.; Zhang, L. Physiological functions of CKIP-1: From molecular mechanisms to therapy implications. Ageing Res. Rev. 2019, 53, 100908. [Google Scholar] [CrossRef]
- Liu, J.; Lu, C.; Wu, X.; Zhang, Z.; Li, J.; Guo, B.; Li, D.; Liang, C.; Dang, L.; Pan, X. Targeting osteoblastic casein kinase-2 interacting protein-1 to enhance Smad-dependent BMP signaling and reverse bone formation reduction in glucocorticoid-induced osteoporosis. Sci. Rep. 2017, 7, 141295. [Google Scholar] [CrossRef]
- Rosen, C.J. Building bones by knocking down genes. Nat. Med. 2012, 18, 202–204. [Google Scholar] [CrossRef]
- Tian, X.G.; Gong, F.F.; Li, X.; Meng, F.H.; Zhou, Z.; Zhang, H.Z. Inflammation-mediated age-dependent effects of casein kinase 2-interacting protein-1 on osteogenesis in mesenchymal stem cells. Chin. Med. J. 2020, 133, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Shen, S.; He, J.; Wang, L. CKIP-1 contributes to osteogenic differentiation of mouse bone marrow mesenchymal stem cells. Regen. Med. 2021, 16, 847–859. [Google Scholar] [CrossRef]
- Zhang, G.; Guo, B.; Wu, H.; Tang, T.; Zhang, B.-T.; Zheng, L.; He, Y.; Yang, Z.; Pan, X.; Chow, H.; et al. A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy. Nat. Med. 2012, 18, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, C.; Guo, B.; Wu, X.; Li, D.; Zhang, Z.; Zheng, K.; Dang, L.; He, X.; Lu, C. Increased PLEKHO1 within osteoblasts suppresses Smad-dependent BMP signaling to inhibit bone formation during aging. Aging Cell 2017, 16, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Guo, B.; Wu, H.; Shao, N.; Li, D.; Liu, J.; Dang, L.; Wang, C.; Li, H.; Li, S.; et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference–based bone anabolic strategy. Nat. Med. 2015, 21, 288–294. [Google Scholar] [CrossRef]
- Guo, B.; Zhang, B.; Zheng, L.; Tang, T.; Liu, J.; Wu, H.; Yang, Z.; Peng, S.; He, X.; Zhang, H. Therapeutic RNA interference targeting CKIP-1 with a cross-species sequence to stimulate bone formation. Bone 2014, 59, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Wu, X.; Zhang, J.; Zhang, G.; Li, G.; Pan, X. The role of CKIP-1 in osteoporosis development and treatment. Bone Jt. Res. 2018, 7, 173–178. [Google Scholar] [CrossRef]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Q.; Wan, Z.; Li, J.; Liu, L.; Zhang, X. CKIP-1 knockout offsets osteoporosis induced by simulated microgravity. Prog. Biophys. Mol. Biol. 2016, 122, 140–148. [Google Scholar] [CrossRef]
- Mullard, A. Targeted protein degraders crowd into the clinic. Nat. Reviews. Drug Discov. 2021, 20, 247–250. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sosič, I.; Bricelj, A.; Steinebach, C. E3 ligase ligand chemistries: From building blocks to protein degraders. Chem. Soc. Rev. 2022, 51, 3487–3534. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Liu, L.; He, F.; Fu, X.; Han, W.; Zhang, L. CKIP-1: A scaffold protein and potential therapeutic target integrating multiple signaling pathways and physiological functions. Ageing Res. Rev. 2013, 12, 276–281. [Google Scholar] [CrossRef]
- Wang, L.; You, X.; Lotinun, S.; Zhang, L.; Wu, N.; Zou, W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat. Commun. 2020, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Cardote, T.A.F.; Gadd, M.S.; Ciulli, A. Crystal Structure of the Cul2-Rbx1-EloBC-VHL Ubiquitin Ligase Complex. Structure 2017, 25, 901–911.e3. [Google Scholar] [CrossRef] [PubMed]
- Caine, E.A.; Mahan, S.D.; Johnson, R.L.; Nieman, A.N.; Lam, N.; Warren, C.R.; Riching, K.M.; Urh, M.; Daniels, D.L. Targeted Protein Degradation Phenotypic Studies Using HaloTag CRISPR/Cas9 Endogenous Tagging Coupled with HaloPROTAC3. Curr. Protoc. Pharmacol. 2020, 91, e81. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xu, Y.; Li, X.; Bartlam, M. Crystallization and preliminary X-ray crystallographic analysis of the human CKIP-1 pleckstrin homology domain. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef]
- Sözen, T.; Özışık, L.; Başaran, N.Ç. An overview and management of osteoporosis. Eur. J. Rheumatol. 2017, 4, 46–56. [Google Scholar] [CrossRef]
- LeBlanc, A.D.; Spector, E.R.; Evans, H.J.; Sibonga, J.D. Skeletal responses to space flight and the bed rest analog: A review. J. Musculoskelet. Neuronal Interact. 2007, 7, 33–47. [Google Scholar]
- Fu, J.; Goldsmith, M.; Crooks, S.D.; Condon, S.F.; Morris, M.; Komarova, S.V. Bone health in spacefaring rodents and primates: Systematic review and meta-analysis. NPJ Microgravity 2021, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nie, J.; Wang, Y.; Zhang, L.; Lu, K.; Xing, G.; Xie, P.; He, F.; Zhang, L. CKIP-1 couples Smurf1 ubiquitin ligase with Rpt6 subunit of proteasome to promote substrate degradation. EMBO Rep. 2012, 13, 1004–1011. [Google Scholar] [CrossRef]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Hamann, L.G. Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef]
- Guenette, R.G.; Yang, S.W.; Min, J.; Pei, B.; Potts, P.R. Target and tissue selectivity of PROTAC degraders. Chem. Soc. Rev. 2022, 51, 5740–5756. [Google Scholar] [CrossRef]
- Zorba, A.; Nguyen, C.; Xu, Y.; Starr, J.; Borzilleri, K.; Smith, J.; Zhu, H.; Farley, K.A.; Ding, W.; Schiemer, J.; et al. Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. USA 2018, 115, E7285–E7292. [Google Scholar] [CrossRef]
- Yang, Y.; Xiao, H.; Lin, Z.; Chen, R.; Li, S.; Li, C.; Sun, X.; Hei, Z.; Gong, W.; Huang, H. The ubiquitination of CKIP-1 mediated by Src aggravates diabetic renal fibrosis (original article). Biochem. Pharmacol. 2022, 206, 115339. [Google Scholar] [CrossRef]
- Neri, D.; Lerner, R.A. DNA-Encoded Chemical Libraries: A Selection System Based on Endowing Organic Compounds with Amplifiable Information. Annu. Rev. Biochem. 2018, 87, 479–502. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Sun, Q.; Li, Y.; Zhang, L.; Zhang, P.; Wang, X.; Tian, C.; Li, Q.; Song, J.; Liu, H. CKIP-1 inhibits cardiac hypertrophy by regulating class II histone deacetylase phosphorylation through recruiting PP2A. Circulation 2012, 126, 3028–3040. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Li, Y.; Zhong, G.; Zheng, Y.; Xu, Q.; Zhao, D.; Sun, W.; Jin, X.; Li, H.; Li, J. Myocardial CKIP-1 overexpression protects from simulated microgravity-induced cardiac remodeling. Front. Physiol. 2018, 9, 40. [Google Scholar] [CrossRef]
- Schneider, M.; Radoux, C.J.; Hercules, A.; Ochoa, D.; Dunham, I.; Zalmas, L.P.; Hessler, G.; Ruf, S.; Shanmugasundaram, V.; Hann, M.M.; et al. The PROTACtable genome. Nat. Rev. Drug Discov. 2021, 20, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; He, M.; Wang, L.; He, Y.; Rao, Y. Chemistries of bifunctional PROTAC degraders. Chem. Soc. Rev. 2022, 51, 7066–7114. [Google Scholar] [CrossRef] [PubMed]
- Morabito, C.; Steimberg, N.; Mazzoleni, G.; Guarnieri, S.; Fanò-Illic, G.; Mariggiò, M.A. RCCS bioreactor-based modelled microgravity induces significant changes on in vitro 3D neuroglial cell cultures. Biomed. Res. Int. 2015, 2015, 754283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xing, G.; Tie, Y.; Tang, Y.; Tian, C.; Li, L.; Sun, L.; Wei, H.; Zhu, Y.; He, F. Role for the pleckstrin homology domain-containing protein CKIP-1 in AP-1 regulation and apoptosis. EMBO J. 2005, 24, 766–778. [Google Scholar] [CrossRef]
- Zhang, L.; Tie, Y.; Tian, C.; Xing, G.; Song, Y.; Zhu, Y.; Sun, Z.; He, F. CKIP-1 recruits nuclear ATM partially to the plasma membrane through interaction with ATM. Cell. Signal. 2006, 18, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Peng, Z.; Li, C.; Wang, Z.; Wang, C.; Deng, Z.; Wu, B.; Cui, Y.; Wang, Z. Deubiquitylase OTUD6B governs pVHL stability in an enzyme-independent manner and suppresses hepatocellular carcinoma metastasis. Adv. Sci. 2020, 7, 1902040. [Google Scholar] [CrossRef]
- Zhu, Q.; Fu, Y.; Cui, C.P.; Ding, Y.; Deng, Z.; Ning, C.; Hu, F.; Qiu, C.; Yu, B.; Zhou, X.; et al. OTUB1 promotes osteoblastic bone formation through stabilizing FGFR2. Signal Transduct. Target. Ther. 2023, 8, 142. [Google Scholar] [CrossRef]
- Chen, G.; Long, C.; Wang, S.; Wang, Z.; Chen, X.; Tang, W.; He, X.; Bao, Z.; Tan, B.; Zhao, J.; et al. Circular RNA circStag1 promotes bone regeneration by interacting with HuR. Bone Res. 2022, 10. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Wu, B.; Liu, M.; Cui, C.-P. The Discovery of a Specific CKIP-1 Ligand for the Potential Treatment of Disuse Osteoporosis. Int. J. Mol. Sci. 2024, 25, 8870. https://doi.org/10.3390/ijms25168870
Wei Y, Wu B, Liu M, Cui C-P. The Discovery of a Specific CKIP-1 Ligand for the Potential Treatment of Disuse Osteoporosis. International Journal of Molecular Sciences. 2024; 25(16):8870. https://doi.org/10.3390/ijms25168870
Chicago/Turabian StyleWei, Yange, Bo Wu, Mingqiu Liu, and Chun-Ping Cui. 2024. "The Discovery of a Specific CKIP-1 Ligand for the Potential Treatment of Disuse Osteoporosis" International Journal of Molecular Sciences 25, no. 16: 8870. https://doi.org/10.3390/ijms25168870
APA StyleWei, Y., Wu, B., Liu, M., & Cui, C.-P. (2024). The Discovery of a Specific CKIP-1 Ligand for the Potential Treatment of Disuse Osteoporosis. International Journal of Molecular Sciences, 25(16), 8870. https://doi.org/10.3390/ijms25168870