
Accurate Long-Read RNA Sequencing Analysis Reveals the Key Pathways and Candidate Genes under Drought Stress in the Seed Germination Stage in Faba Bean

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
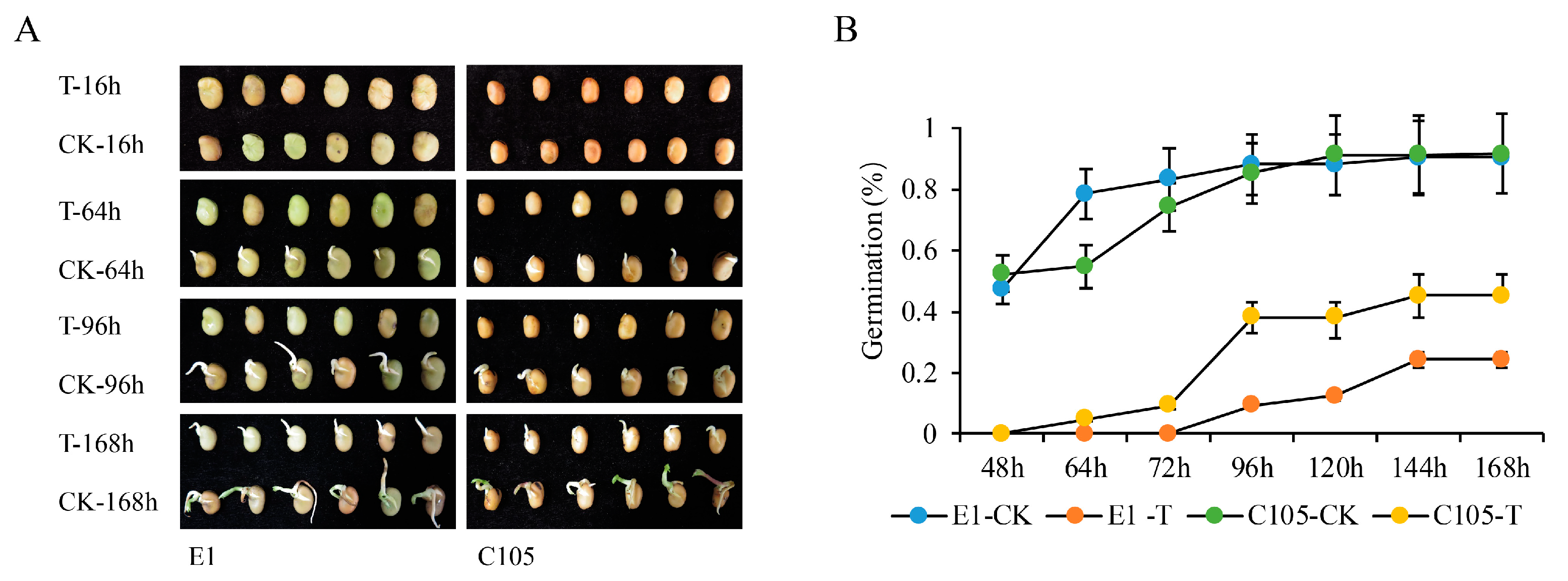
2.1. The Phenotypes of E1 and C105 under Drought Stress at the Germination Stage
2.2. Identification of Full-Length Transcripts and Annotation under Drought Stress
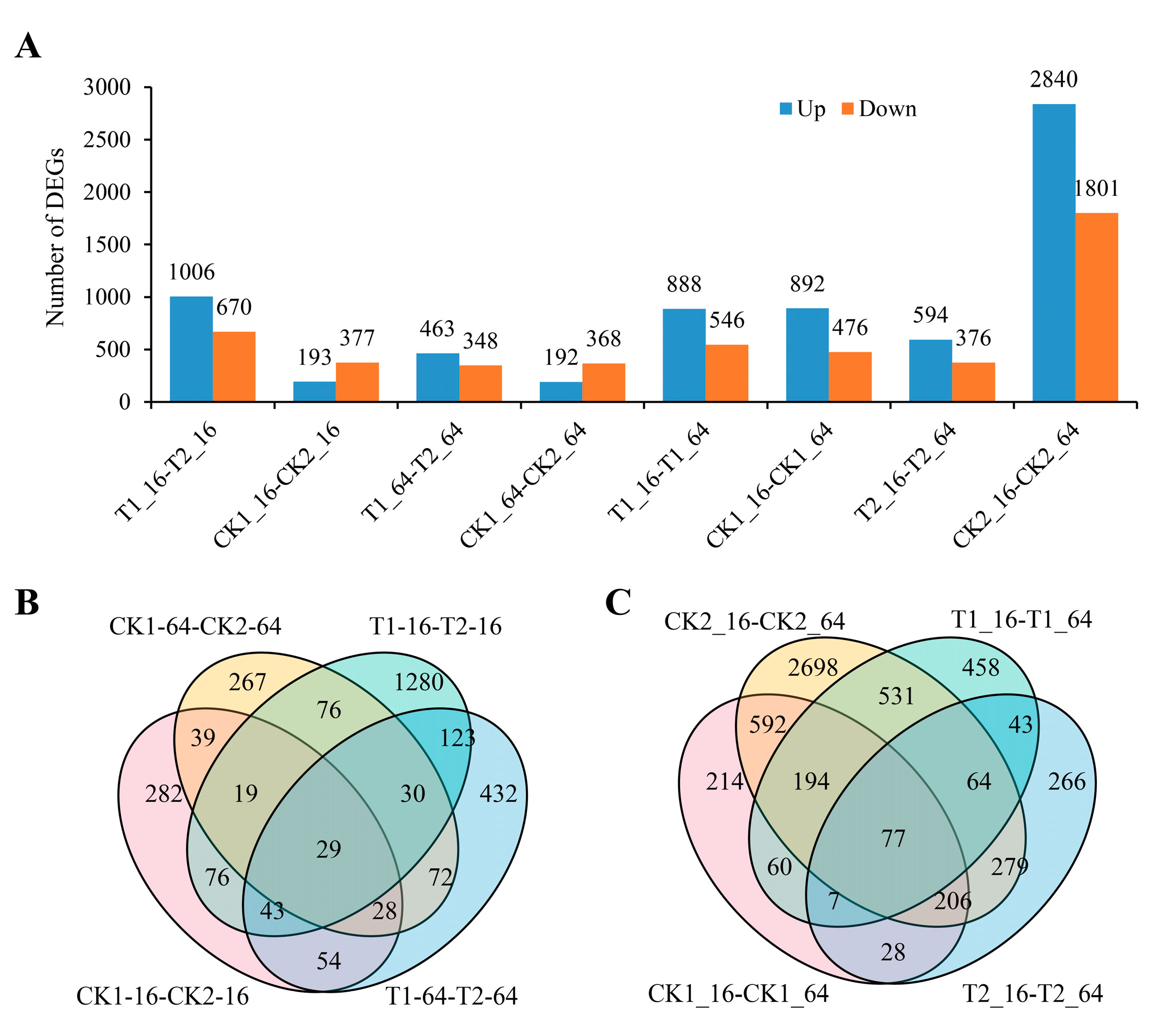
2.3. Identification of Differentially Expressed Genes (DEGs) of C105 and E1 under Drought Stress
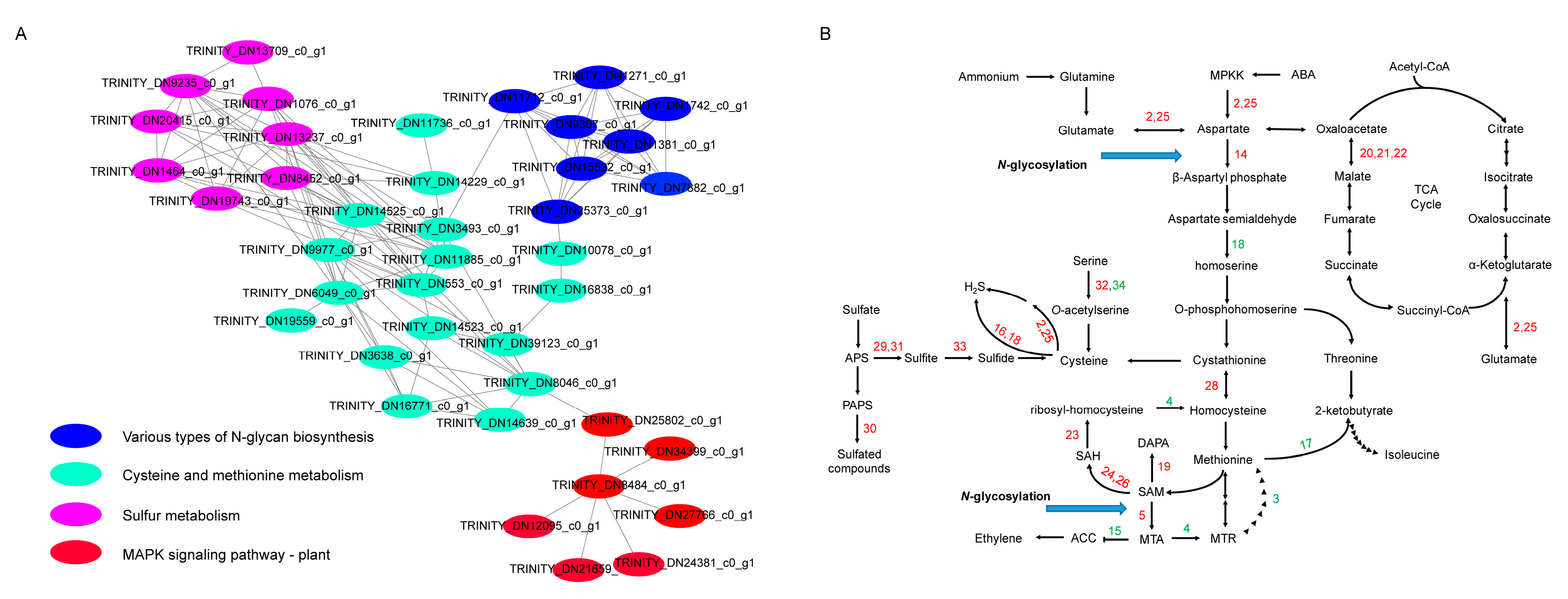
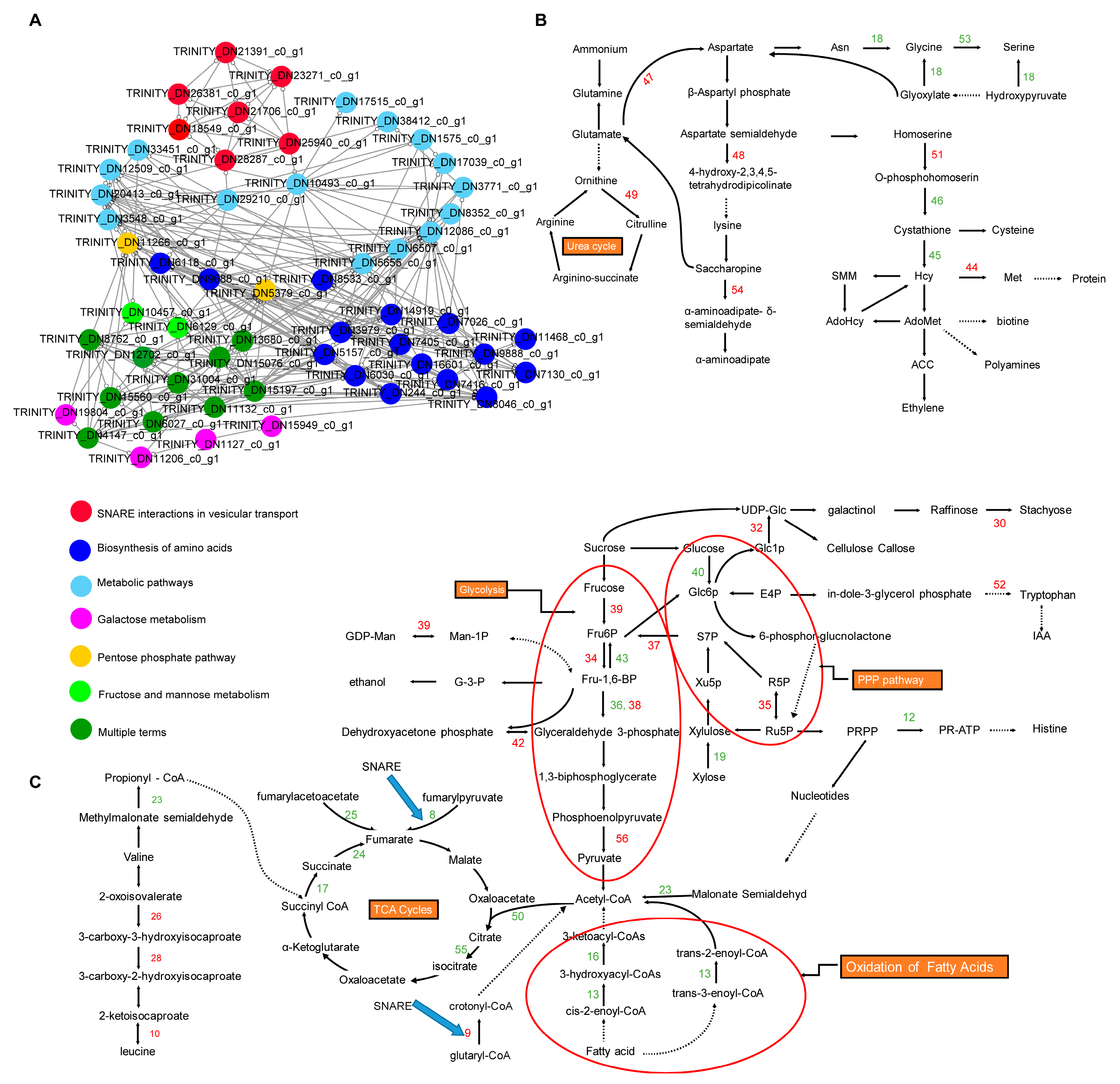
2.4. Pathway Enrichment Analysis of DEGs at 16 h and 64 h of Drought Stress
2.5. PPI Network Analysis of DEGs at 16 h and 64 h of Drought Stress
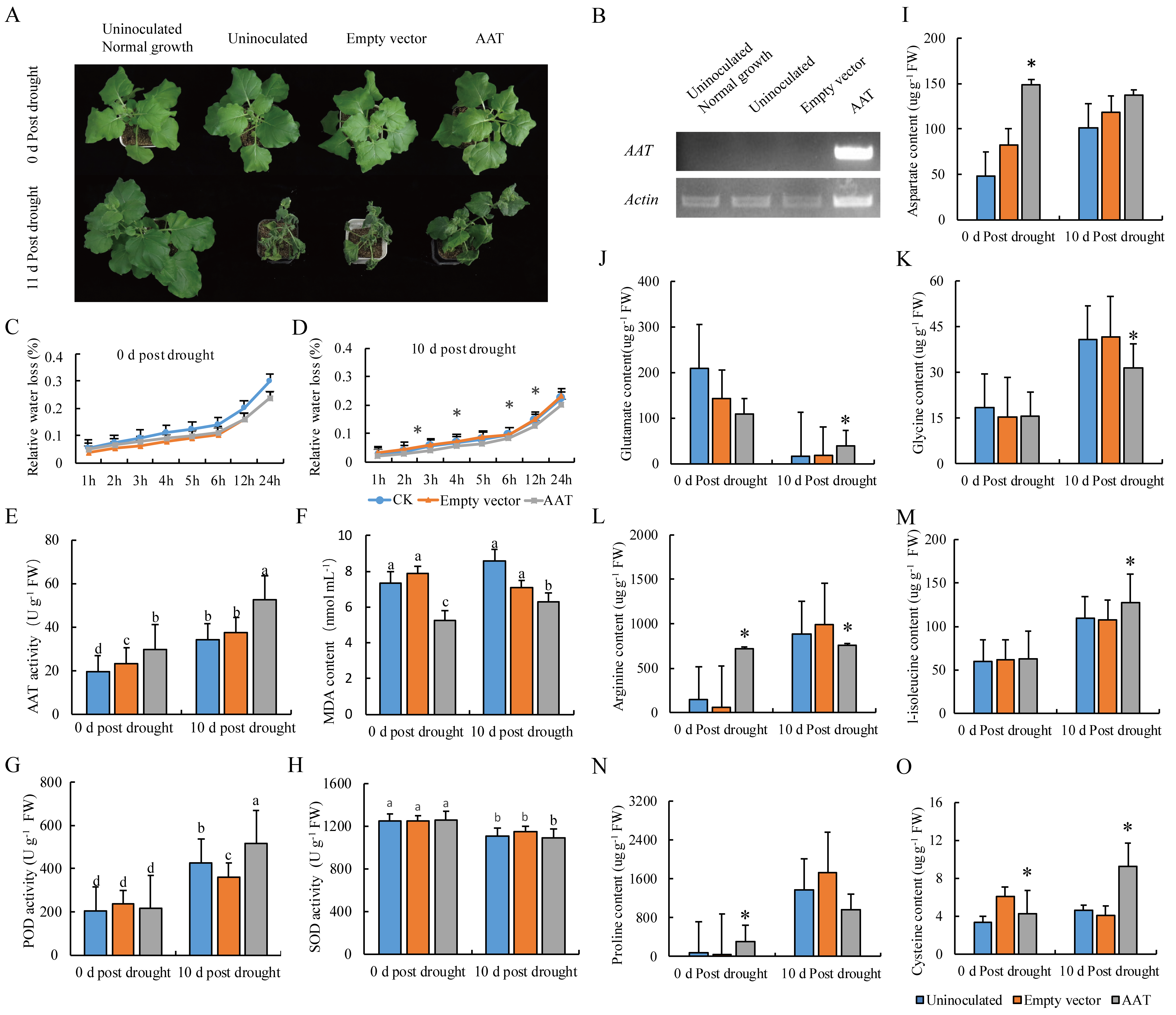
2.6. Ectopic Overexpression of AAT Enhanced Drought Tolerance in Tobacco
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Drought Stress Treatment
4.2. Library Construction for NGS
4.3. Library Construction for Pacbio SMRT Sequencing
4.4. Illumina and Pacbio SMRT Sequencing
4.5. Work Pipeline for Full-Length Transcript
4.6. GO Enrichment Analysis
4.7. Pathway Enrichment Analysis
4.8. PPI Network Analysis
4.9. qRT-PCR Analysis
4.10. Ectopic Overexpression of AAT and RT-PCR Detection
4.11. Physiological Parameter Measurements
4.12. Data Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Witcombe, J.R. Genetic Resources of Faba Beans. In Faba Bean Improvement: Proceedings of the Faba Bean Conference Held in Cairo, Egypt, 7–11 March 1981; Hawtin, G., Webb, C., Eds.; Springer: Dordrecht, The Netherlands, 1982; pp. 1–13. [Google Scholar]
- FAOSTAT Database. Available online: http://www.fao.org/faostat/en/#home (accessed on 29 November 2019).
- Badjona, A.; Bradshaw, R.; Millman, C.; Howarth, M.; Dubey, B. Faba Bean Flavor Effects from Processing to Consumer Acceptability. Foods 2023, 12, 2237. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.; Zanghelini, G.; Zotzel, J.; Bonerz, D.; Aschoff, J.; Saint-Eve, A.; Maillard, M.-N. Fava bean (Vicia faba L.) for food applications: From seed to ingredient processing and its effect on functional properties, antinutritional factors, flavor, and color. Compr. Rev. Food Sci. Food Saf. 2021, 20, 401–428. [Google Scholar] [CrossRef] [PubMed]
- Gangola, M.P.; Ramadoss, B.R.; Jaiswal, S.; Fabek, H.; Tulbek, M.; Anderson, G.H.; Chibbar, R.N. Nutritional Composition and In Vitro Starch Digestibility of Crackers Supplemented with Faba Bean Whole Flour, Starch Concentrate, Protein Concentrate and Protein Isolate. Foods 2022, 11, 645. [Google Scholar] [CrossRef]
- Martineau-Côté, D.; Achouri, A.; Karboune, S.; L’Hocine, L. Faba Bean: An Untapped Source of Quality Plant Proteins and Bioactives. Nutrients 2022, 14, 1541. [Google Scholar] [CrossRef]
- Foyer, C.H.; Lam, H.-M.; Nguyen, H.T.; Siddique, K.H.M.; Varshney, R.K.; Colmer, T.D.; Cowling, W.; Bramley, H.; Mori, T.A.; Hodgson, J.M.; et al. Neglecting legumes has compromised human health and sustainable food production. Nat. Plants 2016, 2, 16112. [Google Scholar] [CrossRef]
- Cooper, J.W.; Wilson, M.H.; Derks, M.F.L.; Smit, S.; Kunert, K.J.; Cullis, C.; Foyer, C.H. Enhancing faba bean (Vicia faba L.) genome resources. J. Exp. Bot. 2017, 68, 1941–1953. [Google Scholar] [CrossRef]
- Jayakodi, M.; Golicz, A.A.; Kreplak, J.; Fechete, L.I.; Angra, D.; Bednář, P.; Bornhofen, E.; Zhang, H.; Boussageon, R.; Kaur, S.; et al. The giant diploid faba genome unlocks variation in a global protein crop. Nature 2023, 615, 652–659. [Google Scholar] [CrossRef]
- Yang, F.; Chen, H.; Liu, C.; Li, L.; Liu, L.; Han, X.; Wan, Z.; Sha, A. Transcriptome profile analysis of two Vicia faba cultivars with contrasting salinity tolerance during seed germination. Sci. Rep. 2020, 10, 7250. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, R.; Liu, Y.; Hou, W.; Wang, X.; Miao, Y.; He, Y.; Ma, Y.; Li, G.; Wang, D.; et al. Development and application of the Faba_bean_130K targeted next-generation sequencing SNP genotyping platform based on transcriptome sequencing. Theor. Appl. Genet. 2021, 134, 3195–3207. [Google Scholar] [CrossRef]
- Yuan, X.; Wang, Q.; Yan, B.; Zhang, J.; Xue, C.; Chen, J.; Lin, Y.; Zhang, X.; Shen, W.; Chen, X. Single-Molecule Real-Time and Illumina-Based RNA Sequencing Data Identified Vernalization-Responsive Candidate Genes in Faba Bean (Vicia faba L.). Front. Genet. 2021, 12, 656137. [Google Scholar] [CrossRef]
- Björnsdotter, E.; Nadzieja, M.; Chang, W.; Escobar-Herrera, L.; Mancinotti, D.; Angra, D.; Xia, X.; Tacke, R.; Khazaei, H.; Crocoll, C.; et al. VC1 catalyses a key step in the biosynthesis of vicine in faba bean. Nat. Plants 2021, 7, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Asmann, Y.W.; Klee, E.W.; Thompson, E.A.; Perez, E.A.; Middha, S.; Oberg, A.L.; Therneau, T.M.; Smith, D.I.; Poland, G.A.; Wieben, E.D.; et al. 3′ tag digital gene expression profiling of human brain and universal reference RNA using Illumina Genome Analyzer. BMC Genom. 2009, 10, 531. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz, V.; Sela, N.; Taha-Salaime, L.; Liu, J.; Rafael, G.; Kessler, C.; Aly, R.; Levy, M.; Wisniewski, M.; Droby, S. De-novo assembly and characterization of the transcriptome of Metschnikowia fructicola reveals differences in gene expression following interaction with Penicillium digitatumand grapefruit peel. BMC Genom. 2013, 14, 168. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Furtado, A.; Henry, R.J. Long-read sequencing of the coffee bean transcriptome reveals the diversity of full-length transcripts. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L.; et al. A global survey of alternative splicing in allopolyploid cotton: Landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef]
- Marthandan, V.; Geetha, R.; Kumutha, K.; Renganathan, V.G.; Karthikeyan, A.; Ramalingam, J. Seed Priming: A Feasible Strategy to Enhance Drought Tolerance in Crop Plants. Int. J. Mol. Sci. 2020, 21, 8258. [Google Scholar] [CrossRef] [PubMed]
- Thabet, S.G.; Moursi, Y.S.; Karam, M.A.; Graner, A.; Alqudah, A.M. Genetic basis of drought tolerance during seed germination in barley. PLoS ONE 2018, 13, e0206682. [Google Scholar] [CrossRef]
- Gad, M.; Chao, H.; Li, H.; Zhao, W.; Lu, G.; Li, M. QTL Mapping for Seed Germination Response to Drought Stress in Brassica napus. Front. Plant Sci. 2020, 11, 629970. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, S.; Guo, X.; Jia, J.; Yang, W.; Liu, Z.; Chen, S.; Li, X.; Qi, D.; Liu, G.; et al. A MYB-related transcription factor from sheepgrass, LcMYB2, promotes seed germination and root growth under drought stress. BMC Plant Biol. 2019, 19, 564. [Google Scholar] [CrossRef]
- Yu, A.; Zhao, J.; Wang, Z.; Cheng, K.; Zhang, P.; Tian, G.; Liu, X.; Guo, E.; Du, Y.; Wang, Y. Transcriptome and metabolite analysis reveal the drought tolerance of foxtail millet significantly correlated with phenylpropanoids-related pathways during germination process under PEG stress. BMC Plant Biol. 2020, 20, 274. [Google Scholar] [CrossRef]
- Zhu, H.; Yang, X.; Wang, X.; Li, Q.; Guo, J.; Ma, T.; Zhao, C.; Tang, Y.; Qiao, L.; Wang, J.; et al. The sweetpotato β-amylase gene IbBAM1.1 enhances drought and salt stress resistance by regulating ROS homeostasis and osmotic balance. Plant Physiol. Biochem. 2021, 168, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ruan, M.; Wan, Q.; He, W.; Yang, L.; Liu, X.; He, L.; Yan, L.; Bi, Y. Nitric oxide and hydrogen peroxide increase glucose-6-phosphate dehydrogenase activities and expression upon drought stress in soybean roots. Plant Cell Rep. 2020, 39, 63–73. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.; Liu, Y.; Li, X.; Hao, G.; Han, Q.; Dirk, L.M.A.; Downie, A.B.; Ruan, Y.L.; Wang, J.; et al. Raffinose synthase enhances drought tolerance through raffinose synthesis or galactinol hydrolysis in maize and Arabidopsis plants. J. Biol. Chem. 2020, 295, 8064–8077. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, F.; Zhao, Y.; Chen, H.; Wan, Z.; Li, L.; Sun, L.; Liu, L.; Jiao, C.; Liu, C.; et al. iTRAQ based protein profile analysis revealed key proteins involved in regulation of drought-tolerance during seed germination in Adzuki bean. Sci. Rep. 2021, 11, 23725. [Google Scholar] [CrossRef]
- Han, M.; Zhang, C.; Suglo, P.; Sun, S.; Wang, M.; Su, T. l-Aspartate: An Essential Metabolite for Plant Growth and Stress Acclimation. Molecules 2021, 26, 1887. [Google Scholar] [CrossRef]
- Jin, Z.; Shen, J.; Qiao, Z.; Yang, G.; Wang, R.; Pei, Y. Hydrogen sulfide improves drought resistance in Arabidopsis thaliana. Biochem. Biophys. Res. Commun. 2011, 414, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Bekturova, A.; Oshanova, D.; Tiwari, P.; Nurbekova, Z.; Kurmanbayeva, A.; Soltabayeva, A.; Yarmolinsky, D.; Srivastava, S.; Turecková, V.; Strnad, M.; et al. Adenosine 5′ phosphosulfate reductase and sulfite oxidase regulate sulfite-induced water loss in Arabidopsis. J. Exp. Bot. 2021, 72, 6447–6466. [Google Scholar] [CrossRef]
- Chen, J.; Wei, B.; Li, G.; Fan, R.; Zhong, Y.; Wang, X.; Zhang, X. TraeALDH7B1-5A, encoding aldehyde dehydrogenase 7 in wheat, confers improved drought tolerance in Arabidopsis. Planta 2015, 242, 137–151. [Google Scholar] [CrossRef]
- Li, G.Z.; Li, H.X.; Xu, M.J.; Wang, P.F.; Xiao, X.H.; Kang, G.Z. Functional characterization and regulatory mechanism of wheat CPK34 kinase in response to drought stress. BMC Genom. 2020, 21, 577. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Song, R.F.; Zheng, S.Q.; Li, T.T.; Zhang, B.L.; Gao, X.; Lu, Y.T. Coordination of plant growth and abiotic stress responses by tryptophan synthase β subunit 1 through modulation of tryptophan and ABA homeostasis in Arabidopsis. Mol. Plant 2022, 15, 973–990. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Vikal, Y.; Kaur, L.; Kalia, A.; Mittal, A.; Kaur, D.; Yadav, I. Elucidating the morpho-physiological adaptations and molecular responses under long-term waterlogging stress in maize through gene expression analysis. Plant Sci. 2021, 304, 110823. [Google Scholar] [CrossRef] [PubMed]
- Nan, N.; Wang, J.; Shi, Y.; Qian, Y.; Jiang, L.; Huang, S.; Liu, Y.; Wu, Y.; Liu, B.; Xu, Z.-Y. Rice plastidial NAD-dependent malate dehydrogenase 1 negatively regulates salt stress response by reducing the vitamin B6 content. Plant Biotechnol. J. 2020, 18, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Mishra, S.K.; Misra, S.; Pandey, V.; Agrawal, L.; Nautiyal, C.S.; Chauhan, P.S. Revealing the complexity of protein abundance in chickpea root under drought-stress using a comparative proteomics approach. Plant Physiol. Biochem. 2020, 151, 88–102. [Google Scholar] [CrossRef] [PubMed]
- AbdElgawad, H.; Avramova, V.; Baggerman, G.; Van Raemdonck, G.; Valkenborg, D.; Van Ostade, X.; Guisez, Y.; Prinsen, E.; Asard, H.; Van den Ende, W.; et al. Starch biosynthesis contributes to the maintenance of photosynthesis and leaf growth under drought stress in maize. Plant Cell Environ. 2020, 43, 2254–2271. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, J.; Li, C.; Zhang, Z.; Ma, F.; Li, M. Response of sugar metabolism in apple leaves subjected to short-term drought stress. Plant Physiol. Biochem. 2019, 141, 164–171. [Google Scholar] [CrossRef]
- Hewage, K.A.H.; Yang, J.F.; Wang, D.; Hao, G.F.; Yang, G.F.; Zhu, J.K. Chemical Manipulation of Abscisic Acid Signaling: A New Approach to Abiotic and Biotic Stress Management in Agriculture. Adv. Sci. 2020, 7, 2001265. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Park, Y.; Hwang, I. Abscisic acid: Biosynthesis, inactivation, homoeostasis and signalling. Essays Biochem. 2015, 58, 29–48. [Google Scholar]
- Song, J.; Shang, L.; Wang, X.; Xing, Y.; Xu, W.; Zhang, Y.; Wang, T.; Li, H.; Zhang, J.; Ye, Z. MAPK11 regulates seed germination and ABA signaling in tomato by phosphorylating SnRKs. J. Exp. Bot. 2021, 72, 1677–1690. [Google Scholar] [CrossRef]
- Gangl, R.; Tenhaken, R. Raffinose Family Oligosaccharides Act As Galactose Stores in Seeds and Are Required for Rapid Germination of Arabidopsis in the Dark. Front. Plant Sci. 2016, 7, 1115. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, B.; Plaxton, W.C. Multifaceted functions of post-translational enzyme modifications in the control of plant glycolysis. Curr. Opin. Plant Biol. 2020, 55, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Beard, K.F.M.; Swart, C.; Bergmann, S.; Krahnert, I.; Nikoloski, Z.; Graf, A.; Ratcliffe, R.G.; Sweetlove, L.J.; Fernie, A.R.; et al. Protein-protein interactions and metabolite channelling in the plant tricarboxylic acid cycle. Nat. Commun. 2017, 8, 15212. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, T.D. Pentose Phosphate Pathway Reactions in Photosynthesizing Cells. Cells 2021, 10, 1547. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J. The multiple evolutionary origins of the eukaryotic N-glycosylation pathway. Biol. Direct 2016, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.X.; Li, S.T.; Wang, N.; Kitajima, T.; Yoko, O.T.; Fujita, M.; Nakanishi, H.; Gao, X.D. Structural and functional analysis of Alg1 beta-1,4 mannosyltransferase reveals the physiological importance of its membrane topology. Glycobiology 2018, 28, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Brennan, A.; Wei, W.; Guo, G.; Lindsey, K. Vesicle Transport in Plants: A Revised Phylogeny of SNARE Proteins. Evol. Bioinform. 2020, 16, 1176934320956575. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.I.; Ramekar, R.; Kim, J.M.; Hung, N.N.; Seo, J.S.; Kim, J.-B.; Choi, I.-Y.; Park, K.-C.; Kwon, S.-J. Unraveling the complexity of faba bean (Vicia faba L.) transcriptome to reveal cold-stress-responsive genes using long-read isoform sequencing technology. Sci. Rep. 2021, 11, 21094. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, C.; Chen, H.; L., L.; Sun, L.; Han, X.; Jiao, C.; Sha, A. Analysis of ASPAT gene family based on drought-stressed transcriptome sequencing in Vicia faba L. Acta Agron. Sin. 2023, 49, 1871–1881. [Google Scholar]
- Chen, H.-C.; Cheng, W.-H.; Hong, C.-Y.; Chang, Y.-S.; Chang, M.-C. The transcription factor OsbHLH035 mediates seed germination and enables seedling recovery from salt stress through ABA-dependent and ABA-independent pathways, respectively. Rice 2018, 11, 50. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M.; et al. Widespread Polycistronic Transcripts in Fungi Revealed by Single-Molecule mRNA Sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sahu, D.K.; Chowdhry, R.; Mishra, A.; Goel, M.M.; Faheem, M.; Srivastava, C.; Ojha, B.K.; Gupta, D.K.; Kant, R. IsoSeq analysis and functional annotation of the infratentorial ependymoma tumor tissue on PacBio RSII platform. Meta Gene 2016, 7, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Jo, Y.; Lian, S.; Cho, J.K.; Choi, H.; Chu, H.; Cho, W.K. De novo transcriptome assembly of two different Prunus mume cultivars. Genom Data 2015, 6, 273–274. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, X.; Liu, C.; Yang, F.; Wei, Z.; Li, L.; Chen, H.; Han, X.; Jiao, C.; Sha, A. Accurate Long-Read RNA Sequencing Analysis Reveals the Key Pathways and Candidate Genes under Drought Stress in the Seed Germination Stage in Faba Bean. Int. J. Mol. Sci. 2024, 25, 8875. https://doi.org/10.3390/ijms25168875
Wen X, Liu C, Yang F, Wei Z, Li L, Chen H, Han X, Jiao C, Sha A. Accurate Long-Read RNA Sequencing Analysis Reveals the Key Pathways and Candidate Genes under Drought Stress in the Seed Germination Stage in Faba Bean. International Journal of Molecular Sciences. 2024; 25(16):8875. https://doi.org/10.3390/ijms25168875
Chicago/Turabian StyleWen, Xin, Changyan Liu, Fangwen Yang, Zhengxin Wei, Li Li, Hongwei Chen, Xuesong Han, Chunhai Jiao, and Aihua Sha. 2024. "Accurate Long-Read RNA Sequencing Analysis Reveals the Key Pathways and Candidate Genes under Drought Stress in the Seed Germination Stage in Faba Bean" International Journal of Molecular Sciences 25, no. 16: 8875. https://doi.org/10.3390/ijms25168875