
Metabolic Reprogramming of Astrocytes in Pathological Conditions: Implications for Neurodegenerative Diseases

 ,
, 
 , and
, and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Metabolism of Astrocytes in Physiological Conditions
3. Metabolic Reprogramming of Astrocytes in Pathological Conditions
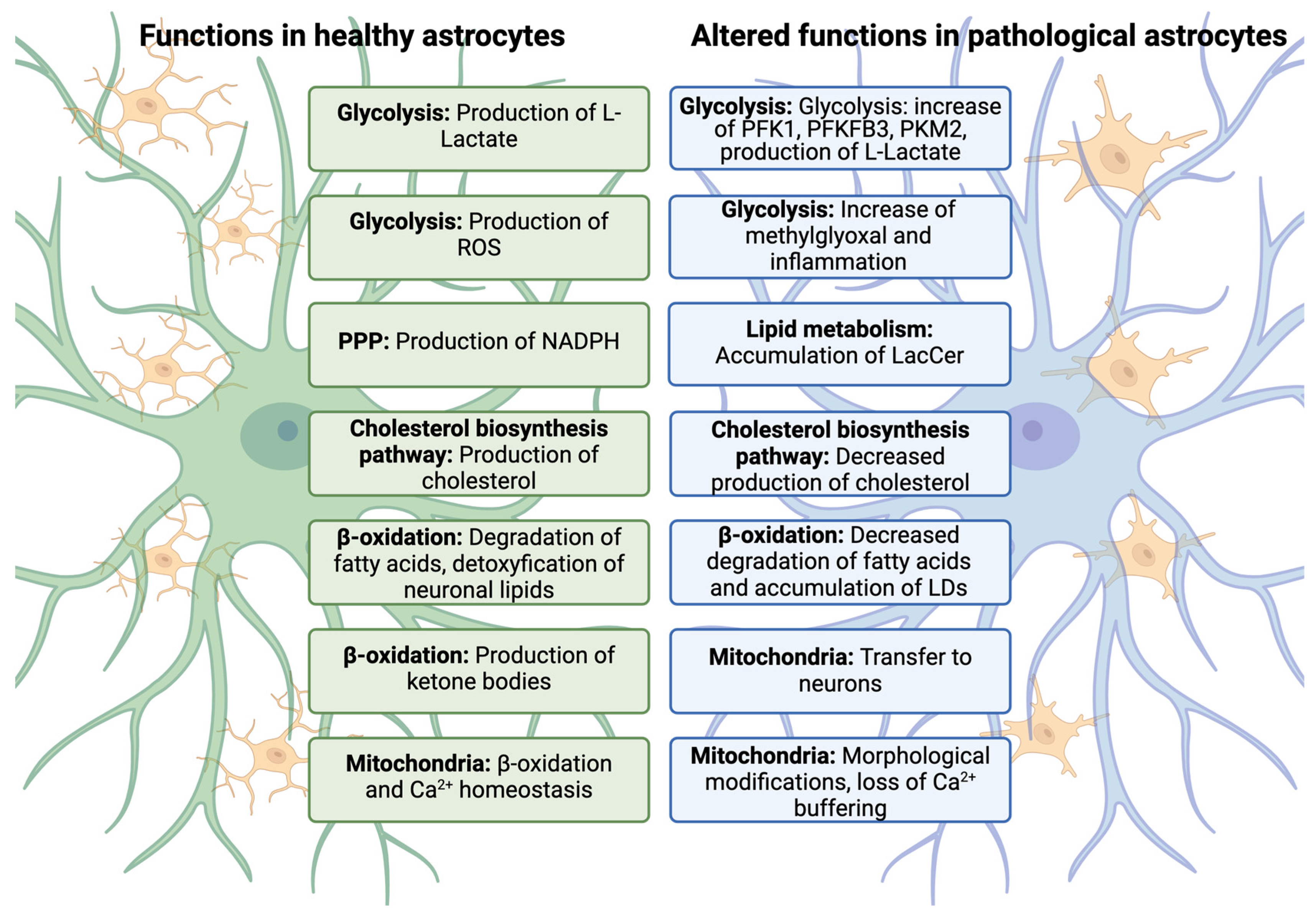
3.1. Alteration of Glycolytic Pathways in Reactive Astrocytes
3.2. Lipid Metabolism Alteration in Reactive Astrocytes
3.3. Mitochondria Metabolism Alteration in Reactive Astrocytes
4. Conclusions
5. Future Directions and Key Questions in Astrocyte Metabolism Research
Author Contributions
Funding
Conflicts of Interest
References
- Clarke, D.D.; Sokoloff, L. Circulation and Energy Metabolism of the Brain. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Lippincott-Raven: New York, NY, USA, 1999. [Google Scholar]
- Siegel, G.J.; Agranoff, B.W.; Albers, R.W.; Molinoff, P.B. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects; Raven Press: New York, NY, USA, 1994; ISBN 978-0-7817-0104-4. [Google Scholar]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of Central Nervous System to Body Metabolism in Vertebrates: Its Constancy and Functional Basis. Am. J. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef] [PubMed]
- Ward, J. Oxygen Delivery and Demand. Surgery 2006, 24, 354–360. [Google Scholar] [CrossRef]
- Barrett, K.E.; Barman, S.M.; Boitano, S.; Brooks, H.L. Circulation through Special Regions. In Ganong’s Review of Medical Physiology; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Barrett, K.E.; Barman, S.M.; Boitano, S.; Brooks, H. Ganong’s Review of Medical Physiology, 24th ed.; McGraw Hill Professional: New York, NY, USA, 2012; ISBN 978-0-07-178003-2. [Google Scholar]
- Bonvento, G.; Bolaños, J.P. Astrocyte-Neuron Metabolic Cooperation Shapes Brain Activity. Cell Metab. 2021, 33, 1546–1564. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain Metabolism in Health, Aging, and Neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a Risk Factor for Neurodegenerative Disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Scheiblich, H.; Trombly, M.; Ramirez, A.; Heneka, M.T. Neuroimmune Connections in Aging and Neurodegenerative Diseases. Trends Immunol. 2020, 41, 300–312. [Google Scholar] [CrossRef]
- Kennedy, C.; Sokoloff, L. An Adaptation of the Nitrous Oxide Method to the Study of the Cerebral Circulation in Children; Normal Values for Cerebral Blood Flow and Cerebral Metabolic Rate in Childhood. J. Clin. Investig. 1957, 36, 1130–1137. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Quantitative Growth and Development of Human Brain. Arch. Dis. Child. 1973, 48, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic Glycolysis in the Human Brain Is Associated with Development and Neotenous Gene Expression. Cell Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef]
- Hyder, F.; Herman, P.; Bailey, C.J.; Møller, A.; Globinsky, R.; Fulbright, R.K.; Rothman, D.L.; Gjedde, A. Uniform Distributions of Glucose Oxidation and Oxygen Extraction in Gray Matter of Normal Human Brain: No Evidence of Regional Differences of Aerobic Glycolysis. J. Cereb. Blood Flow. Metab. 2016, 36, 903–916. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Cantando, I.; Centofanti, C.; D’Alessandro, G.; Limatola, C.; Bezzi, P. Metabolic Dynamics in Astrocytes and Microglia during Post-Natal Development and Their Implications for Autism Spectrum Disorders. Front. Cell Neurosci. 2024, 18, 1354259. [Google Scholar] [CrossRef] [PubMed]
- Sokoloff, L. Localization of Functional Activity in the Central Nervous System by Measurement of Glucose Utilization with Radioactive Deoxyglucose. J. Cereb. Blood Flow. Metab. 1981, 1, 7–36. [Google Scholar] [CrossRef]
- Chugani, H.T.; Phelps, M.E.; Mazziotta, J.C. Positron Emission Tomography Study of Human Brain Functional Development. Ann. Neurol. 1987, 22, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Barros, L.F. Metabolic Signaling by Lactate in the Brain. Trends Neurosci. 2013, 36, 396–404. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite Synapses: Glia, the Unacknowledged Partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite Synapses: Astrocytes Process and Control Synaptic Information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Calì, C. Regulated Exocytosis from Astrocytes: A Matter of Vesicles? Front. Neurosci. 2024, 18, 1393165. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef]
- Bezzi, P.; Volterra, A. A Neuron–Glia Signalling Network in the Active Brain. Curr. Opin. Neurobiol. 2001, 11, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Calì, C.; Bezzi, P. Gliotransmission and the Tripartite Synapse. In Synaptic Plasticity: Dynamics, Development and Disease; Kreutz, M.R., Sala, C., Eds.; Advances in Experimental Medicine and Biology; Springer: Vienna, Austria, 2012; pp. 307–331. ISBN 978-3-7091-0932-8. [Google Scholar]
- Bezzi, P.; Carmignoto, G.; Pasti, L.; Vesce, S.; Rossi, D.; Rizzini, B.L.; Pozzan, T.; Volterra, A. Prostaglandins Stimulate Calcium-Dependent Glutamate Release in Astrocytes. Nature 1998, 391, 281–285. [Google Scholar] [CrossRef]
- Bezzi, P.; Domercq, M.; Brambilla, L.; Galli, R.; Schols, D.; De Clercq, E.; Vescovi, A.; Bagetta, G.; Kollias, G.; Meldolesi, J.; et al. CXCR4-Activated Astrocyte Glutamate Release via TNFα: Amplification by Microglia Triggers Neurotoxicity. Nat. Neurosci. 2001, 4, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Bezzi, P.; Gundersen, V.; Galbete, J.L.; Seifert, G.; Steinhäuser, C.; Pilati, E.; Volterra, A. Astrocytes Contain a Vesicular Compartment That Is Competent for Regulated Exocytosis of Glutamate. Nat. Neurosci. 2004, 7, 613–620. [Google Scholar] [CrossRef]
- Haydon, P.G.; Carmignoto, G. Astrocyte Control of Synaptic Transmission and Neurovascular Coupling. Physiol. Rev. 2006, 86, 1009–1031. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Gonzalo, M.; Zehnder, T.; Requie, L.M.; Bezzi, P.; Carmignoto, G. Insights into the Release Mechanism of Astrocytic Glutamate Evoking in Neurons NMDA Receptor-Mediated Slow Depolarizing Inward Currents. Glia 2018, 66, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Cali, C.; Lopatar, J.; Petrelli, F.; Pucci, L.; Bezzi, P. G-Protein Coupled Receptor-Evoked Glutamate Exocytosis from Astrocytes: Role of Prostaglandins. Neural Plast. 2014, 2014, e254574. [Google Scholar] [CrossRef]
- Sultan, S.; Li, L.; Moss, J.; Petrelli, F.; Cassé, F.; Gebara, E.; Lopatar, J.; Pfrieger, F.W.; Bezzi, P.; Bischofberger, J.; et al. Synaptic Integration of Adult-Born Hippocampal Neurons Is Locally Controlled by Astrocytes. Neuron 2015, 88, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Dallérac, G.; Pucci, L.; Calì, C.; Zehnder, T.; Sultan, S.; Lecca, S.; Chicca, A.; Ivanov, A.; Asensio, C.S.; et al. Dysfunction of Homeostatic Control of Dopamine by Astrocytes in the Developing Prefrontal Cortex Leads to Cognitive Impairments. Mol. Psychiatry 2020, 25, 732–749. [Google Scholar] [CrossRef]
- Petrelli, F.; Bezzi, P. Novel Insights into Gliotransmitters. Curr. Opin. Pharmacol. 2016, 26, 138–145. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- van Deijk, A.-L.F.; Camargo, N.; Timmerman, J.; Heistek, T.; Brouwers, J.F.; Mogavero, F.; Mansvelder, H.D.; Smit, A.B.; Verheijen, M.H.G. Astrocyte Lipid Metabolism Is Critical for Synapse Development and Function in Vivo. Glia 2017, 65, 670–682. [Google Scholar] [CrossRef]
- Edmond, J.; Higa, T.A.; Korsak, R.A.; Bergner, E.A.; Lee, W.N. Fatty Acid Transport and Utilization for the Developing Brain. J. Neurochem. 1998, 70, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.-H.; Chang, C.-L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535. [Google Scholar] [CrossRef]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy Contribution of Octanoate to Intact Rat Brain Metabolism Measured by 13C Nuclear Magnetic Resonance Spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef]
- Morant-Ferrando, B.; Jimenez-Blasco, D.; Alonso-Batan, P.; Agulla, J.; Lapresa, R.; Garcia-Rodriguez, D.; Yunta-Sanchez, S.; Lopez-Fabuel, I.; Fernandez, E.; Carmeliet, P.; et al. Fatty Acid Oxidation Organizes Mitochondrial Supercomplexes to Sustain Astrocytic ROS and Cognition. Nat. Metab. 2023, 5, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Le Foll, C.; Levin, B.E. Fatty Acid-Induced Astrocyte Ketone Production and the Control of Food Intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R1186–R1192. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty Acids in Energy Metabolism of the Central Nervous System. BioMed Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Qi, Y.-B.; Gao, Y.-N.; Chen, W.-G.; Zhou, T.; Zang, Y.; Li, J. Astrocyte Metabolism and Signaling Pathways in the CNS. Front. Neurosci. 2023, 17, 1217451. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J.; Auestad, N.; Robbins, R.A.; Bergstrom, J.D. Ketone Body Metabolism in the Neonate: Development and the Effect of Diet. Fed. Proc. 1985, 44, 2359–2364. [Google Scholar] [PubMed]
- Martin, P.M.; Gopal, E.; Ananth, S.; Zhuang, L.; Itagaki, S.; Prasad, B.M.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Identity of SMCT1 (SLC5A8) as a Neuron-Specific Na+-Coupled Transporter for Active Uptake of L-Lactate and Ketone Bodies in the Brain. J. Neurochem. 2006, 98, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, G.M.I.; Jiang, L.; Rothman, D.L.; Behar, K.L. The Contribution of Ketone Bodies to Basal and Activity-Dependent Neuronal Oxidation in Vivo. J. Cereb. Blood Flow. Metab. 2014, 34, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Mantha, O.L.; Schor, J.; Pascual, A.; Plaçais, P.-Y.; Pavlowsky, A.; Preat, T. Glia Fuel Neurons with Locally Synthesized Ketone Bodies to Sustain Memory under Starvation. Nat. Metab. 2022, 4, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, T.; Petrelli, F.; Romanos, J.; De Oliveira Figueiredo, E.C.; Lewis, T.L.; Déglon, N.; Polleux, F.; Santello, M.; Bezzi, P. Mitochondrial Biogenesis in Developing Astrocytes Regulates Astrocyte Maturation and Synapse Formation. Cell Rep. 2021, 35, 108952. [Google Scholar] [CrossRef] [PubMed]
- Motori, E.; Puyal, J.; Toni, N.; Ghanem, A.; Angeloni, C.; Malaguti, M.; Cantelli-Forti, G.; Berninger, B.; Conzelmann, K.-K.; Götz, M.; et al. Inflammation-Induced Alteration of Astrocyte Mitochondrial Dynamics Requires Autophagy for Mitochondrial Network Maintenance. Cell Metab. 2013, 18, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Göbel, J.; Engelhardt, E.; Pelzer, P.; Sakthivelu, V.; Jahn, H.M.; Jevtic, M.; Folz-Donahue, K.; Kukat, C.; Schauss, A.; Frese, C.K.; et al. Mitochondria-Endoplasmic Reticulum Contacts in Reactive Astrocytes Promote Vascular Remodeling. Cell Metab. 2020, 31, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Hamberger, A.; Hyden, H. Inverse Enzymatic Changes in Neurons and Glia during Increased Function and Hypoxia. J. Cell Biol. 1963, 16, 521–525. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate Uptake into Astrocytes Stimulates Aerobic Glycolysis: A Mechanism Coupling Neuronal Activity to Glucose Utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Blüml, S.; Moreno-Torres, A.; Shic, F.; Nguy, C.-H.; Ross, B.D. Tricarboxylic Acid Cycle of Glia in the in Vivo Human Brain. NMR Biomed. 2002, 15, 1–5. [Google Scholar] [CrossRef]
- Dienel, G.A.; Cruz, N.F. Aerobic Glycolysis during Brain Activation: Adrenergic Regulation and Influence of Norepinephrine on Astrocytic Metabolism. J. Neurochem. 2016, 138, 14–52. [Google Scholar] [CrossRef]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 Improves the Supportive Ability of Astrocytes to Neurons by Promoting Aerobic Glycolysis in Alzheimer’s Disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Gao, V.; Suzuki, A.; Magistretti, P.J.; Lengacher, S.; Pollonini, G.; Steinman, M.Q.; Alberini, C.M. Astrocytic Β2-Adrenergic Receptors Mediate Hippocampal Long-Term Memory Consolidation. Proc. Natl. Acad. Sci. USA 2016, 113, 8526–8531. [Google Scholar] [CrossRef]
- Coggan, J.S.; Keller, D.; Calì, C.; Lehväslaiho, H.; Markram, H.; Schürmann, F.; Magistretti, P.J. Norepinephrine Stimulates Glycogenolysis in Astrocytes to Fuel Neurons with Lactate. PLoS Comput. Biol. 2018, 14, e1006392. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-Neuron Lactate Transport Is Required for Long-Term Memory Formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Vezzoli, E.; Calì, C.; De Roo, M.; Ponzoni, L.; Sogne, E.; Gagnon, N.; Francolini, M.; Braida, D.; Sala, M.; Muller, D.; et al. Ultrastructural Evidence for a Role of Astrocytes and Glycogen-Derived Lactate in Learning-Dependent Synaptic Stabilization. Cereb. Cortex 2020, 30, 2114–2127. [Google Scholar] [CrossRef]
- Benarroch, E.E. Brain Glucose Transporters: Implications for Neurologic Disease. Neurology 2014, 82, 1374–1379. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The Bioenergetic and Antioxidant Status of Neurons Is Controlled by Continuous Degradation of a Key Glycolytic Enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Halim, N.D.; Mcfate, T.; Mohyeldin, A.; Okagaki, P.; Korotchkina, L.G.; Patel, M.S.; Jeoung, N.H.; Harris, R.A.; Schell, M.J.; Verma, A. Phosphorylation Status of Pyruvate Dehydrogenase Distinguishes Metabolic Phenotypes of Cultured Rat Brain Astrocytes and Neurons. Glia 2010, 58, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Veloz Castillo, M.F.; Magistretti, P.J.; Calì, C. L-Lactate: Food for Thoughts, Memory and Behavior. Metabolites 2021, 11, 548. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I Assembly into Supercomplexes Determines Differential Mitochondrial ROS Production in Neurons and Astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic Mitochondrial ROS Modulate Brain Metabolism and Mouse Behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Lovatt, D.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A.; He, W.; Lin, J.H.-C.; Han, X.; Takano, T.; Wang, S.; Sim, F.J.; et al. The Transcriptome and Metabolic Gene Signature of Protoplasmic Astrocytes in the Adult Murine Cortex. J. Neurosci. 2007, 27, 12255–12266. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A Transcriptome Database for Astrocytes, Neurons, and Oligodendrocytes: A New Resource for Understanding Brain Development and Function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy Metabolism in Astrocytes: High Rate of Oxidative Metabolism and Spatiotemporal Dependence on Glycolysis/Glycogenolysis. J. Cereb. Blood Flow. Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Semyanov, A.; Henneberger, C.; Agarwal, A. Making Sense of Astrocytic Calcium Signals—From Acquisition to Interpretation. Nat. Rev. Neurosci. 2020, 21, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Stephen, T.-L.; Higgs, N.F.; Sheehan, D.F.; Al Awabdh, S.; López-Doménech, G.; Arancibia-Carcamo, I.L.; Kittler, J.T. Miro1 Regulates Activity-Driven Positioning of Mitochondria within Astrocytic Processes Apposed to Synapses to Regulate Intracellular Calcium Signaling. J. Neurosci. 2015, 35, 15996–16011. [Google Scholar] [CrossRef]
- Bindocci, E.; Savtchouk, I.; Liaudet, N.; Becker, D.; Carriero, G.; Volterra, A. Three-Dimensional Ca2+ Imaging Advances Understanding of Astrocyte Biology. Science 2017, 356, eaai8185. [Google Scholar] [CrossRef]
- Jackson, J.G.; Robinson, M.B. Regulation of Mitochondrial Dynamics in Astrocytes: Mechanisms, Consequences, and Unknowns. Glia 2018, 66, 1213–1234. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Wu, P.-H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605. [Google Scholar] [CrossRef] [PubMed]
- Gunter, T.E.; Yule, D.I.; Gunter, K.K.; Eliseev, R.A.; Salter, J.D. Calcium and Mitochondria. FEBS Lett. 2004, 567, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Parnis, J.; Montana, V.; Delgado-Martinez, I.; Matyash, V.; Parpura, V.; Kettenmann, H.; Sekler, I.; Nolte, C. Mitochondrial Exchanger NCLX Plays a Major Role in the Intracellular Ca2+ Signaling, Gliotransmission, and Proliferation of Astrocytes. J. Neurosci. 2013, 33, 7206–7219. [Google Scholar] [CrossRef]
- Panatier, A.; Vallée, J.; Haber, M.; Murai, K.K.; Lacaille, J.-C.; Robitaille, R. Astrocytes Are Endogenous Regulators of Basal Transmission at Central Synapses. Cell 2011, 146, 785–798. [Google Scholar] [CrossRef]
- Civenni, G.; Bezzi, P.; Trotti, D.; Volterra, A.; Racagni, G. Inhibitory Effect of the Neuroprotective Agent Idebenone on Arachidonic Acid Metabolism in Astrocytes. Eur. J. Pharmacol. 1999, 370, 161–167. [Google Scholar] [CrossRef]
- Lee, J.A.; Hall, B.; Allsop, J.; Alqarni, R.; Allen, S.P. Lipid Metabolism in Astrocytic Structure and Function. Semin. Cell Dev. Biol. 2021, 112, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Valenza, M.; Birolini, G.; Cattaneo, E. The Translational Potential of Cholesterol-Based Therapies for Neurological Disease. Nat. Rev. Neurol. 2023, 19, 583–598. [Google Scholar] [CrossRef]
- Nieweg, K.; Schaller, H.; Pfrieger, F.W. Marked Differences in Cholesterol Synthesis between Neurons and Glial Cells from Postnatal Rats. J. Neurochem. 2009, 109, 125–134. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-Regulated Lipid Metabolism: Convergent Physiology—Divergent Pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Ferris, H.A.; Perry, R.J.; Moreira, G.V.; Shulman, G.I.; Horton, J.D.; Kahn, C.R. Loss of Astrocyte Cholesterol Synthesis Disrupts Neuronal Function and Alters Whole-Body Metabolism. Proc. Natl. Acad. Sci. USA 2017, 114, 1189–1194. [Google Scholar] [CrossRef]
- Camargo, N.; Brouwers, J.F.; Loos, M.; Gutmann, D.H.; Smit, A.B.; Verheijen, M.H.G. High-Fat Diet Ameliorates Neurological Deficits Caused by Defective Astrocyte Lipid Metabolism. FASEB J. 2012, 26, 4302–4315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Q. Cholesterol Metabolism and Homeostasis in the Brain. Protein Cell 2015, 6, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef]
- Montesinos, J.; Guardia-Laguarta, C.; Area-Gomez, E. The Fat Brain. Curr. Opin. Clin. Nutr. Metab. Care 2020, 23, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Cabodevilla, A.G.; Sánchez-Caballero, L.; Nintou, E.; Boiadjieva, V.G.; Picatoste, F.; Gubern, A.; Claro, E. Cell Survival during Complete Nutrient Deprivation Depends on Lipid Droplet-Fueled β-Oxidation of Fatty Acids. J. Biol. Chem. 2013, 288, 27777–27788. [Google Scholar] [CrossRef] [PubMed]
- Auestad, N.; Korsak, R.A.; Morrow, J.W.; Edmond, J. Fatty Acid Oxidation and Ketogenesis by Astrocytes in Primary Culture. J. Neurochem. 1991, 56, 1376–1386. [Google Scholar] [CrossRef]
- Blázquez, C.; Sánchez, C.; Velasco, G.; Guzmán, M. Role of Carnitine Palmitoyltransferase I in the Control of Ketogenesis in Primary Cultures of Rat Astrocytes. J. Neurochem. 1998, 71, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Bertoni, A.G.; Crago, O.L.; Hoffman, K.L.; Wood, A.C.; Arzumanyan, Z.; Lam, L.-S.K.; Roll, K.; Sandow, K.; Wu, M.; et al. Rationale, Design and Baseline Characteristics of the Microbiome and Insulin Longitudinal Evaluation Study (MILES). Diabetes Obes. Metab. 2020, 22, 1976–1984. [Google Scholar] [CrossRef]
- Pontzer, H.; Brown, M.H.; Raichlen, D.A.; Dunsworth, H.; Hare, B.; Walker, K.; Luke, A.; Dugas, L.R.; Durazo-Arvizu, R.; Schoeller, D.; et al. Metabolic Acceleration and the Evolution of Human Brain Size and Life History. Nature 2016, 533, 390–392. [Google Scholar] [CrossRef]
- Lee, I.; Lee, S.-J.; Kang, W.K.; Park, C. Inhibition of Monocarboxylate Transporter 2 Induces Senescence-Associated Mitochondrial Dysfunction and Suppresses Progression of Colorectal Malignancies in Vivo. Mol. Cancer Ther. 2012, 11, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, K.K.; Gupta, S. Biochemistry, Ketogenesis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Cunnane, S.C.; Crawford, M.A. Energetic and Nutritional Constraints on Infant Brain Development: Implications for Brain Expansion during Human Evolution. J. Hum. Evol. 2014, 77, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Wang, T.; Li, M.; Guan, T.; Guo, Y.; Zhang, X.; Zhang, G.; Kong, J. Ketogenic Diet Protects Myelin and Axons in Diffuse Axonal Injury. Nutr. Neurosci. 2022, 25, 1534–1547. [Google Scholar] [CrossRef] [PubMed]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A Modern Review of a Century-Old Therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Magistretti, P.J.; Pellerin, L. MCT2 Is a Major Neuronal Monocarboxylate Transporter in the Adult Mouse Brain. J. Cereb. Blood Flow. Metab. 2002, 22, 586–595. [Google Scholar] [CrossRef]
- Guzmán, M.; Blázquez, C. Is There an Astrocyte-Neuron Ketone Body Shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173. [Google Scholar] [CrossRef]
- Guzmán, M.; Blázquez, C. Ketone Body Synthesis in the Brain: Possible Neuroprotective Effects. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 287–292. [Google Scholar] [CrossRef]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic Diet Metabolites Reduce Firing in Central Neurons by Opening K(ATP) Channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Balland, E.; Prevot, V.; Luquet, S.; Woods, S.C.; Koch, M.; Horvath, T.L.; Yi, C.-X.; Chowen, J.A.; Verkhratsky, A.; et al. Role of Astrocytes, Microglia, and Tanycytes in Brain Control of Systemic Metabolism. Nat. Neurosci. 2019, 22, 7–14. [Google Scholar] [CrossRef]
- Clasadonte, J.; Prevot, V. The Special Relationship: Glia-Neuron Interactions in the Neuroendocrine Hypothalamus. Nat. Rev. Endocrinol. 2018, 14, 25–44. [Google Scholar] [CrossRef]
- Chowen, J.A.; Argente-Arizón, P.; Freire-Regatillo, A.; Frago, L.M.; Horvath, T.L.; Argente, J. The Role of Astrocytes in the Hypothalamic Response and Adaptation to Metabolic Signals. Prog. Neurobiol. 2016, 144, 68–87. [Google Scholar] [CrossRef]
- Kim, J.G.; Suyama, S.; Koch, M.; Jin, S.; Argente-Arizon, P.; Argente, J.; Liu, Z.-W.; Zimmer, M.R.; Jeong, J.K.; Szigeti-Buck, K.; et al. Leptin Signaling in Astrocytes Regulates Hypothalamic Neuronal Circuits and Feeding. Nat. Neurosci. 2014, 17, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Varela, L.; Stutz, B.; Song, J.E.; Kim, J.G.; Liu, Z.-W.; Gao, X.-B.; Horvath, T.L. Hunger-Promoting AgRP Neurons Trigger an Astrocyte-Mediated Feed-Forward Autoactivation Loop in Mice. J. Clin. Investig. 2021, 131, e144239. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Layritz, C.; Legutko, B.; Eichmann, T.O.; Laperrousaz, E.; Moullé, V.S.; Cruciani-Guglielmacci, C.; Magnan, C.; Luquet, S.; Woods, S.C.; et al. Disruption of Lipid Uptake in Astroglia Exacerbates Diet-Induced Obesity. Diabetes 2017, 66, 2555–2563. [Google Scholar] [CrossRef]
- Varela, L.; Kim, J.G.; Fernández-Tussy, P.; Aryal, B.; Liu, Z.W.; Fernández-Hernando, C.; Horvath, T.L. Astrocytic Lipid Metabolism Determines Susceptibility to Diet-Induced Obesity. Sci. Adv. 2021, 7, eabj2814. [Google Scholar] [CrossRef]
- Kreft, M.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A. Aspects of Astrocyte Energy Metabolism, Amino Acid Neurotransmitter Homoeostasis and Metabolic Compartmentation. ASN Neuro 2012, 4, e00086. [Google Scholar] [CrossRef]
- Nampoothiri, S.; Nogueiras, R.; Schwaninger, M.; Prevot, V. Glial Cells as Integrators of Peripheral and Central Signals in the Regulation of Energy Homeostasis. Nat. Metab. 2022, 4, 813–825. [Google Scholar] [CrossRef]
- Friedman, J.M. Leptin and the Endocrine Control of Energy Balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Glaum, S.R.; Hara, M.; Bindokas, V.P.; Lee, C.C.; Polonsky, K.S.; Bell, G.I.; Miller, R.J. Leptin, the Obese Gene Product, Rapidly Modulates Synaptic Transmission in the Hypothalamus. Mol. Pharmacol. 1996, 50, 230–235. [Google Scholar]
- Banks, W.A. The Blood-Brain Barrier as an Endocrine Tissue. Nat. Rev. Endocrinol. 2019, 15, 444–455. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Fuente-Martín, E.; Burgos-Ramos, E.; Granado, M.; Frago, L.M.; Barrios, V.; Horvath, T.; Argente, J.; Chowen, J.A. Differential Acute and Chronic Effects of Leptin on Hypothalamic Astrocyte Morphology and Synaptic Protein Levels. Endocrinology 2011, 152, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Fuente-Martín, E.; García-Cáceres, C.; Granado, M.; de Ceballos, M.L.; Sánchez-Garrido, M.Á.; Sarman, B.; Liu, Z.-W.; Dietrich, M.O.; Tena-Sempere, M.; Argente-Arizón, P.; et al. Leptin Regulates Glutamate and Glucose Transporters in Hypothalamic Astrocytes. J. Clin. Investig. 2012, 122, 3900–3913. [Google Scholar] [CrossRef]
- Boumezbeur, F.; Petersen, K.F.; Cline, G.W.; Mason, G.F.; Behar, K.L.; Shulman, G.I.; Rothman, D.L. The Contribution of Blood Lactate to Brain Energy Metabolism in Humans Measured by Dynamic 13C Nuclear Magnetic Resonance Spectroscopy. J. Neurosci. 2010, 30, 13983–13991. [Google Scholar] [CrossRef]
- Bouzier-Sore, A.-K.; Voisin, P.; Bouchaud, V.; Bezancon, E.; Franconi, J.-M.; Pellerin, L. Competition between Glucose and Lactate as Oxidative Energy Substrates in Both Neurons and Astrocytes: A Comparative NMR Study. Eur. J. Neurosci. 2006, 24, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Nedergaard, M. Glial Regulation of the Cerebral Microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef]
- Bak, L.K.; Schousboe, A.; Waagepetersen, H.S. The Glutamate/GABA-Glutamine Cycle: Aspects of Transport, Neurotransmitter Homeostasis and Ammonia Transfer. J. Neurochem. 2006, 98, 641–653. [Google Scholar] [CrossRef]
- McKenna, M.C. The Glutamate-Glutamine Cycle Is Not Stoichiometric: Fates of Glutamate in Brain. J. Neurosci. Res. 2007, 85, 3347–3358. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O’Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive Astrocyte Nomenclature, Definitions, and Future Directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- Lemoine, L.; Saint-Aubert, L.; Nennesmo, I.; Gillberg, P.-G.; Nordberg, A. Cortical Laminar Tau Deposits and Activated Astrocytes in Alzheimer’s Disease Visualised by 3H-THK5117 and 3H-Deprenyl Autoradiography. Sci. Rep. 2017, 7, 45496. [Google Scholar] [CrossRef] [PubMed]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic Reactive Astrocytes Are Induced by Activated Microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte Molecular Signatures in Huntington’s Disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State That Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Sanmarco, L.M.; Wheeler, M.A.; Gutiérrez-Vázquez, C.; Polonio, C.M.; Linnerbauer, M.; Pinho-Ribeiro, F.A.; Li, Z.; Giovannoni, F.; Batterman, K.V.; Scalisi, G.; et al. Gut-Licensed IFNγ+ NK Cells Drive LAMP1+TRAIL+ Anti-Inflammatory Astrocytes. Nature 2021, 590, 473–479. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-Driven Astrocytes Promote CNS Inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Hasel, P.; Rose, I.V.L.; Sadick, J.S.; Kim, R.D.; Liddelow, S.A. Neuroinflammatory Astrocyte Subtypes in the Mouse Brain. Nat. Neurosci. 2021, 24, 1475–1487. [Google Scholar] [CrossRef]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe Reactive Astrocytes Precipitate Pathological Hallmarks of Alzheimer’s Disease via H2O2- Production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef]
- O’Shea, T.M.; Ao, Y.; Wang, S.; Ren, Y.; Cheng, A.L.; Kawaguchi, R.; Shi, Z.; Swarup, V.; Sofroniew, M.V. Derivation and Transcriptional Reprogramming of Border-Forming Wound Repair Astrocytes after Spinal Cord Injury or Stroke in Mice. Nat. Neurosci. 2024, 27, 1505–1521. [Google Scholar] [CrossRef]
- Santello, M.; Volterra, A. TNFα in Synaptic Function: Switching Gears. Trends Neurosci. 2012, 35, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Zorec, R.; Araque, A.; Carmignoto, G.; Haydon, P.G.; Verkhratsky, A.; Parpura, V. Astroglial Excitability and Gliotransmission: An Appraisal of Ca2+ as a Signalling Route. ASN Neuro 2012, 4, e00080. [Google Scholar] [CrossRef]
- Lee, S.; Yoon, B.-E.; Berglund, K.; Oh, S.-J.; Park, H.; Shin, H.-S.; Augustine, G.J.; Lee, C.J. Channel-Mediated Tonic GABA Release from Glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.G.; Charles, A.C. Intercellular Calcium Signaling in Astrocytes via ATP Release through Connexin Hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from Reactive Astrocytes Impairs Memory in Mouse Models of Alzheimer’s Disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic Inhibition in Dentate Gyrus Impairs Long-Term Potentiation and Memory in an Alzheimer’s [Corrected] Disease Model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.R.; Fuertes, C.J.A.; Dunn, A.K.; Drew, M.R.; Jones, T.A. Reactive Astrocytes Facilitate Vascular Repair and Remodeling after Stroke. Cell Rep. 2021, 35, 109048. [Google Scholar] [CrossRef]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of Astrocyte Activation by Glycolipids Drives Chronic CNS Inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte Scar Formation Aids Central Nervous System Axon Regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ikeshima-Kataoka, H.; Kreft, M.; Vardjan, N.; Zorec, R.; Noda, M. Metabolic Plasticity of Astrocytes and Aging of the Brain. Int. J. Mol. Sci. 2019, 20, 941. [Google Scholar] [CrossRef]
- Xiong, X.-Y.; Tang, Y.; Yang, Q.-W. Metabolic Changes Favor the Activity and Heterogeneity of Reactive Astrocytes. Trends Endocrinol. Metab. 2022, 33, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Shi, D.; Westaway, D.; Jhamandas, J.H. Bioenergetic Mechanisms in Astrocytes May Contribute to Amyloid Plaque Deposition and Toxicity. J. Biol. Chem. 2015, 290, 12504–12513. [Google Scholar] [CrossRef]
- Pamies, D.; Sartori, C.; Schvartz, D.; González-Ruiz, V.; Pellerin, L.; Nunes, C.; Tavel, D.; Maillard, V.; Boccard, J.; Rudaz, S.; et al. Neuroinflammatory Response to TNFα and IL1β Cytokines Is Accompanied by an Increase in Glycolysis in Human Astrocytes In Vitro. Int. J. Mol. Sci. 2021, 22, 4065. [Google Scholar] [CrossRef] [PubMed]
- Robb, J.L.; Hammad, N.A.; Weightman Potter, P.G.; Chilton, J.K.; Beall, C.; Ellacott, K.L.J. The Metabolic Response to Inflammation in Astrocytes Is Regulated by Nuclear Factor-Kappa B Signaling. Glia 2020, 68, 2246–2263. [Google Scholar] [CrossRef]
- Gavillet, M.; Allaman, I.; Magistretti, P.J. Modulation of Astrocytic Metabolic Phenotype by Proinflammatory Cytokines. Glia 2008, 56, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Véga, C.; Pellerin, L.; Dantzer, R.; Magistretti, P.J. Long-Term Modulation of Glucose Utilization by IL-1 Alpha and TNF-Alpha in Astrocytes: Na+ Pump Activity as a Potential Target via Distinct Signaling Mechanisms. Glia 2002, 39, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Maciejewski-Lenoir, D.; Bloom, F.E.; Magistretti, P.J. Tumor Necrosis Factor-Alpha and Interleukin-1 Alpha Enhance Glucose Utilization by Astrocytes: Involvement of Phospholipase A2. Mol. Pharmacol. 1995, 48, 550–558. [Google Scholar] [PubMed]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the Dark Side of Glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef]
- Wautier, J.L.; Guillausseau, P.J. Advanced Glycation End Products, Their Receptors and Diabetic Angiopathy. Diabetes Metab. 2001, 27, 535–542. [Google Scholar] [CrossRef]
- Münch, G.; Westcott, B.; Menini, T.; Gugliucci, A. Advanced Glycation Endproducts and Their Pathogenic Roles in Neurological Disorders. Amino Acids 2012, 42, 1221–1236. [Google Scholar] [CrossRef]
- Patil, G.; Kulsange, S.; Kazi, R.; Chirmade, T.; Kale, V.; Mote, C.; Aswar, M.; Koratkar, S.; Agawane, S.; Kulkarni, M. Behavioral and Proteomic Studies Reveal Methylglyoxal Activate Pathways Associated with Alzheimer’s Disease. ACS Pharmacol. Transl. Sci. 2023, 6, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Lissner, L.J.; Wartchow, K.M.; Rodrigues, L.; Bobermin, L.D.; Borba, E.; Dias, V.G.; Hansen, F.; Quincozes-Santos, A.; Gonçalves, C.-A. Acute Methylglyoxal-Induced Damage in Blood-Brain Barrier and Hippocampal Tissue. Neurotox. Res. 2022, 40, 1337–1347. [Google Scholar] [CrossRef]
- Schuster, S.; Boley, D.; Möller, P.; Stark, H.; Kaleta, C. Mathematical Models for Explaining the Warburg Effect: A Review Focussed on ATP and Biomass Production. Biochem. Soc. Trans. 2015, 43, 1187–1194. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.W.; et al. TLR-Driven Early Glycolytic Reprogramming via the Kinases TBK1-IKKε Supports the Anabolic Demands of Dendritic Cell Activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Lauro, C.; Limatola, C. Metabolic Reprograming of Microglia in the Regulation of the Innate Inflammatory Response. Front. Immunol. 2020, 11, 493. [Google Scholar] [CrossRef]
- Bernier, L.-P.; York, E.M.; Kamyabi, A.; Choi, H.B.; Weilinger, N.L.; MacVicar, B.A. Microglial Metabolic Flexibility Supports Immune Surveillance of the Brain Parenchyma. Nat. Commun. 2020, 11, 1559. [Google Scholar] [CrossRef]
- Gimeno-Bayón, J.; López-López, A.; Rodríguez, M.J.; Mahy, N. Glucose Pathways Adaptation Supports Acquisition of Activated Microglia Phenotype. J. Neurosci. Res. 2014, 92, 723–731. [Google Scholar] [CrossRef]
- Cheng, J.; Zhang, R.; Xu, Z.; Ke, Y.; Sun, R.; Yang, H.; Zhang, X.; Zhen, X.; Zheng, L.-T. Early Glycolytic Reprogramming Controls Microglial Inflammatory Activation. J. Neuroinflamm. 2021, 18, 129. [Google Scholar] [CrossRef]
- Rider, M.H.; Bertrand, L.; Vertommen, D.; Michels, P.A.; Rousseau, G.G.; Hue, L. 6-Phosphofructo-2-Kinase/Fructose-2,6-Bisphosphatase: Head-to-Head with a Bifunctional Enzyme That Controls Glycolysis. Biochem. J. 2004, 381, 561–579. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhang, B.; Zhai, C.; Qiu, J.; Zhang, Y.; Yao, W.; Zhang, C. PFKFB3-Mediated Glycolysis Is Involved in Reactive Astrocyte Proliferation after Oxygen-Glucose Deprivation/Reperfusion and Is Regulated by Cdh1. Neurochem. Int. 2015, 91, 26–33. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Wei, Y.; Lu, M.; Mei, M.; Wang, H.; Han, Z.; Chen, M.; Yao, H.; Song, N.; Ding, X.; Ding, J.; et al. Pyridoxine Induces Glutathione Synthesis via PKM2-Mediated Nrf2 Transactivation and Confers Neuroprotection. Nat. Commun. 2020, 11, 941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, G.; Bao, G.; Xu, G.; Sun, Y.; Li, W.; Wang, L.; Chen, J.; Jin, H.; Cui, Z. Nuclear Translocation of PKM2 Modulates Astrocyte Proliferation via P27 and -Catenin Pathway after Spinal Cord Injury. Cell Cycle 2015, 14, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Israelsen, W.J.; Vander Heiden, M.G. Pyruvate Kinase: Function, Regulation and Role in Cancer. Semin. Cell Dev. Biol. 2015, 43, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 Regulates the Warburg Effect and Promotes HMGB1 Release in Sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef] [PubMed]
- Vianey-Saban, C.; Fouilhoux, A.; Vockley, J.; Acquaviva-Bourdain, C.; Guffon, N. Improving Diagnosis of Mitochondrial Fatty-Acid Oxidation Disorders. Eur. J. Hum. Genet. 2023, 31, 265–272. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, D.; Giménez-Cassina, A. Ketone Bodies in the Brain Beyond Fuel Metabolism: From Excitability to Gene Expression and Cell Signaling. Front. Mol. Neurosci. 2021, 14, 732120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Chai, H.; Diaz-Castro, B.; Shigetomi, E.; Monte, E.; Octeau, J.C.; Yu, X.; Cohn, W.; Rajendran, P.S.; Vondriska, T.M.; Whitelegge, J.P.; et al. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron 2017, 95, 531–549. [Google Scholar] [CrossRef]
- Kadish, I.; Thibault, O.; Blalock, E.M.; Chen, K.-C.; Gant, J.C.; Porter, N.M.; Landfield, P.W. Hippocampal and Cognitive Aging across the Lifespan: A Bioenergetic Shift Precedes and Increased Cholesterol Trafficking Parallels Memory Impairment. J. Neurosci. 2009, 29, 1805–1816. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Avgerinos, K.I. Brain Glucose and Ketone Utilization in Brain Aging and Neurodegenerative Diseases. Int. Rev. Neurobiol. 2020, 154, 79–110. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of Fatty Acid Degradation by Astrocytic Mitochondria Triggers Neuroinflammation and Neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and Functions of Lipid Droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Stelzmann, R.A.; Norman Schnitzlein, H.; Reed Murtagh, F. An English Translation of Alzheimer’s 1907 Paper, “Über Eine Eigenartige Erkankung Der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid-Droplet-Accumulating Microglia Represent a Dysfunctional and Proinflammatory State in the Aging Brain. Nat. Neurosci. 2020, 23, 194–208. [Google Scholar] [CrossRef]
- Ralhan, I.; Chang, C.-L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid Droplets in the Nervous System. J. Cell Biol. 2021, 220, e202102136. [Google Scholar] [CrossRef] [PubMed]
- Feringa, F.M.; van der Kant, R. An inside Job: New Roles for ApoE at the Lipid Droplet. J. Cell Biol. 2024, 223, e202402171. [Google Scholar] [CrossRef]
- Blumenfeld, J.; Yip, O.; Kim, M.J.; Huang, Y. Cell Type-Specific Roles of APOE4 in Alzheimer Disease. Nat. Rev. Neurosci. 2024, 25, 91–110. [Google Scholar] [CrossRef]
- Sienski, G.; Narayan, P.; Bonner, J.M.; Kory, N.; Boland, S.; Arczewska, A.A.; Ralvenius, W.T.; Akay, L.; Lockshin, E.; He, L.; et al. APOE4 Disrupts Intracellular Lipid Homeostasis in Human iPSC-Derived Glia. Sci. Transl. Med. 2021, 13, eaaz4564. [Google Scholar] [CrossRef]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef]
- Windham, I.A.; Powers, A.E.; Ragusa, J.V.; Wallace, E.D.; Zanellati, M.C.; Williams, V.H.; Wagner, C.H.; White, K.K.; Cohen, S. APOE Traffics to Astrocyte Lipid Droplets and Modulates Triglyceride Saturation and Droplet Size. J. Cell Biol. 2024, 223, e202305003. [Google Scholar] [CrossRef]
- Mejhert, N.; Kuruvilla, L.; Gabriel, K.R.; Elliott, S.D.; Guie, M.-A.; Wang, H.; Lai, Z.W.; Lane, E.A.; Christiano, R.; Danial, N.N.; et al. Partitioning of MLX-Family Transcription Factors to Lipid Droplets Regulates Metabolic Gene Expression. Mol. Cell 2020, 77, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of Lipid Droplet Proteomes. Dev. Cell 2018, 44, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-P.; Wang, S.; Zhao, X.; Fang, W.; Wang, Z.; Ye, H.; Wang, M.-J.; Ke, L.; Huang, T.; Lv, P.; et al. Lipid-Accumulated Reactive Astrocytes Promote Disease Progression in Epilepsy. Nat. Neurosci. 2023, 26, 542–554. [Google Scholar] [CrossRef]
- Sayre, N.L.; Sifuentes, M.; Holstein, D.; Cheng, S.-Y.; Zhu, X.; Lechleiter, J.D. Stimulation of Astrocyte Fatty Acid Oxidation by Thyroid Hormone Is Protective against Ischemic Stroke-Induced Damage. J. Cereb. Blood Flow. Metab. 2017, 37, 514–527. [Google Scholar] [CrossRef]
- Polyzos, A.A.; Lee, D.Y.; Datta, R.; Hauser, M.; Budworth, H.; Holt, A.; Mihalik, S.; Goldschmidt, P.; Frankel, K.; Trego, K.; et al. Metabolic Reprogramming in Astrocytes Distinguishes Region-Specific Neuronal Susceptibility in Huntington Mice. Cell Metab. 2019, 29, 1258–1273. [Google Scholar] [CrossRef]
- Farez, M.F.; Quintana, F.J.; Gandhi, R.; Izquierdo, G.; Lucas, M.; Weiner, H.L. Toll-like Receptor 2 and Poly(ADP-Ribose) Polymerase 1 Promote Central Nervous System Neuroinflammation in Progressive EAE. Nat. Immunol. 2009, 10, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.P.; Kanter, J.L.; Johnson, A.M.; Srinagesh, H.K.; Chang, E.-J.; Purdy, T.M.; van Haren, K.; Wikoff, W.R.; Kind, T.; Khademi, M.; et al. Identification of Naturally Occurring Fatty Acids of the Myelin Sheath That Resolve Neuroinflammation. Sci. Transl. Med. 2012, 4, 137ra73. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Yeste, A.; Weiner, H.L.; Covacu, R. Lipids and Lipid-Reactive Antibodies as Biomarkers for Multiple Sclerosis. J. Neuroimmunol. 2012, 248, 53–57. [Google Scholar] [CrossRef]
- Furuhashi, M. Fatty Acid-Binding Protein 4 in Cardiovascular and Metabolic Diseases. J. Atheroscler. Thromb. 2019, 26, 216–232. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Yamamoto, Y.; Sharifi, K.; Kida, H.; Kagawa, Y.; Yasumoto, Y.; Islam, A.; Miyazaki, H.; Shimamoto, C.; Maekawa, M.; et al. Astrocyte-Expressed FABP7 Regulates Dendritic Morphology and Excitatory Synaptic Function of Cortical Neurons. Glia 2016, 64, 48–62. [Google Scholar] [CrossRef]
- Kagawa, Y.; Yasumoto, Y.; Sharifi, K.; Ebrahimi, M.; Islam, A.; Miyazaki, H.; Yamamoto, Y.; Sawada, T.; Kishi, H.; Kobayashi, S.; et al. Fatty Acid-Binding Protein 7 Regulates Function of Caveolae in Astrocytes through Expression of Caveolin-1. Glia 2015, 63, 780–794. [Google Scholar] [CrossRef] [PubMed]
- Killoy, K.M.; Harlan, B.A.; Pehar, M.; Vargas, M.R. FABP7 Upregulation Induces a Neurotoxic Phenotype in Astrocytes. Glia 2020, 68, 2693–2704. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, F.; Nishinaka, T.; Yamashita, T.; Nakamoto, K.; Koyama, Y.; Kasuya, F.; Tokuyama, S. Astrocytes Release Polyunsaturated Fatty Acids by Lipopolysaccharide Stimuli. Biol. Pharm. Bull. 2016, 39, 1100–1106. [Google Scholar] [CrossRef]
- Guttenplan, K.A.; Weigel, M.K.; Prakash, P.; Wijewardhane, P.R.; Hasel, P.; Rufen-Blanchette, U.; Münch, A.E.; Blum, J.A.; Fine, J.; Neal, M.C.; et al. Neurotoxic Reactive Astrocytes Induce Cell Death via Saturated Lipids. Nature 2021, 599, 102–107. [Google Scholar] [CrossRef]
- Wang, H.; Kulas, J.A.; Higginbotham, H.; Kovacs, M.A.; Ferris, H.A.; Hansen, S.B. Regulation of Neuroinflammation by Astrocyte-Derived Cholesterol. BioRxiv 2022. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of Region-Specific Astrocyte Subtypes at Single Cell Resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [PubMed]
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-Nucleus RNA-Seq Identifies Huntington Disease Astrocyte States. Acta Neuropathol. Commun. 2020, 8, 19. [Google Scholar] [CrossRef]
- Valenza, M.; Leoni, V.; Karasinska, J.M.; Petricca, L.; Fan, J.; Carroll, J.; Pouladi, M.A.; Fossale, E.; Nguyen, H.P.; Riess, O.; et al. Cholesterol Defect Is Marked across Multiple Rodent Models of Huntington’s Disease and Is Manifest in Astrocytes. J. Neurosci. 2010, 30, 10844–10850. [Google Scholar] [CrossRef]
- Valenza, M.; Marullo, M.; Di Paolo, E.; Cesana, E.; Zuccato, C.; Biella, G.; Cattaneo, E. Disruption of Astrocyte-Neuron Cholesterol Cross Talk Affects Neuronal Function in Huntington’s Disease. Cell Death Differ. 2015, 22, 690–702. [Google Scholar] [CrossRef]
- Benraiss, A.; Mariani, J.N.; Osipovitch, M.; Cornwell, A.; Windrem, M.S.; Villanueva, C.B.; Chandler-Militello, D.; Goldman, S.A. Cell-Intrinsic Glial Pathology Is Conserved across Human and Murine Models of Huntington’s Disease. Cell Rep. 2021, 36, 109308. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Voskuhl, R.R. Cell Specificity Dictates Similarities in Gene Expression in Multiple Sclerosis, Parkinson’s Disease, and Alzheimer’s Disease. PLoS ONE 2017, 12, e0181349. [Google Scholar] [CrossRef]
- Boisvert, M.M.; Erikson, G.A.; Shokhirev, M.N.; Allen, N.J. The Aging Astrocyte Transcriptome from Multiple Regions of the Mouse Brain. Cell Rep. 2018, 22, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Supplie, L.M.; Düking, T.; Campbell, G.; Diaz, F.; Moraes, C.T.; Götz, M.; Hamprecht, B.; Boretius, S.; Mahad, D.; Nave, K.-A. Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J. Neurosci. 2017, 37, 4231–4242. [Google Scholar] [CrossRef] [PubMed]
- Göbel, J.; Motori, E.; Bergami, M. Spatiotemporal Control of Mitochondrial Network Dynamics in Astroglial Cells. Biochem. Biophys. Res. Commun. 2018, 500, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of Mitochondria from Astrocytes to Neurons after Stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Christie, I.N.; Theparambil, S.M.; Braga, A.; Doronin, M.; Hosford, P.S.; Brazhe, A.; Mascarenhas, A.; Nizari, S.; Hadjihambi, A.; Wells, J.A.; et al. Astrocytes Produce Nitric Oxide via Nitrite Reduction in Mitochondria to Regulate Cerebral Blood Flow during Brain Hypoxia. Cell Rep. 2023, 42, 113514. [Google Scholar] [CrossRef]
- Popov, A.; Brazhe, N.; Morozova, K.; Yashin, K.; Bychkov, M.; Nosova, O.; Sutyagina, O.; Brazhe, A.; Parshina, E.; Li, L.; et al. Mitochondrial Malfunction and Atrophy of Astrocytes in the Aged Human Cerebral Cortex. Nat. Commun. 2023, 14, 8380. [Google Scholar] [CrossRef] [PubMed]
- Fiebig, C.; Keiner, S.; Ebert, B.; Schäffner, I.; Jagasia, R.; Lie, D.C.; Beckervordersandforth, R. Mitochondrial Dysfunction in Astrocytes Impairs the Generation of Reactive Astrocytes and Enhances Neuronal Cell Death in the Cortex Upon Photothrombotic Lesion. Front. Mol. Neurosci. 2019, 12, 40. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Sayre, N.L.; Chen, Y.; Sifuentes, M.; Stoveken, B.; Lechleiter, J.D. Purinergic Receptor Stimulation Decreases Ischemic Brain Damage by Energizing Astrocyte Mitochondria. Adv. Neurobiol. 2014, 11, 121–150. [Google Scholar] [CrossRef]
- Cao, X.; Li, L.-P.; Wang, Q.; Wu, Q.; Hu, H.-H.; Zhang, M.; Fang, Y.-Y.; Zhang, J.; Li, S.-J.; Xiong, W.-C.; et al. Astrocyte-Derived ATP Modulates Depressive-like Behaviors. Nat. Med. 2013, 19, 773–777. [Google Scholar] [CrossRef]
- Viana, J.F.; Machado, J.L.; Abreu, D.S.; Veiga, A.; Barsanti, S.; Tavares, G.; Martins, M.; Sardinha, V.M.; Guerra-Gomes, S.; Domingos, C.; et al. Astrocyte Structural Heterogeneity in the Mouse Hippocampus. Glia 2023, 71, 1667–1682. [Google Scholar] [CrossRef] [PubMed]
- Calì, C.; Tauffenberger, A.; Magistretti, P. The Strategic Location of Glycogen and Lactate: From Body Energy Reserve to Brain Plasticity. Front. Cell Neurosci. 2019, 13, 82. [Google Scholar] [CrossRef] [PubMed]
- Troidl, J.; Calì, C.; Gröller, E.; Pfister, H.; Hadwiger, M.; Beyer, J. Barrio: Customizable Spatial Neighborhood Analysis and Comparison for Nanoscale Brain Structures. Comput. Graph. Forum 2022, 41, 183–194. [Google Scholar] [CrossRef]
- Agus, M.; Boges, D.; Gagnon, N.; Magistretti, P.J.; Hadwiger, M.; Calí, C. GLAM: Glycogen-Derived Lactate Absorption Map for Visual Analysis of Dense and Sparse Surface Reconstructions of Rodent Brain Structures on Desktop Systems and Virtual Environments. Comput. Graph. 2018, 74, 85–98. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calì, C.; Cantando, I.; Veloz Castillo, M.F.; Gonzalez, L.; Bezzi, P. Metabolic Reprogramming of Astrocytes in Pathological Conditions: Implications for Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 8922. https://doi.org/10.3390/ijms25168922
Calì C, Cantando I, Veloz Castillo MF, Gonzalez L, Bezzi P. Metabolic Reprogramming of Astrocytes in Pathological Conditions: Implications for Neurodegenerative Diseases. International Journal of Molecular Sciences. 2024; 25(16):8922. https://doi.org/10.3390/ijms25168922
Chicago/Turabian StyleCalì, Corrado, Iva Cantando, Maria Fernanda Veloz Castillo, Laurine Gonzalez, and Paola Bezzi. 2024. "Metabolic Reprogramming of Astrocytes in Pathological Conditions: Implications for Neurodegenerative Diseases" International Journal of Molecular Sciences 25, no. 16: 8922. https://doi.org/10.3390/ijms25168922
APA StyleCalì, C., Cantando, I., Veloz Castillo, M. F., Gonzalez, L., & Bezzi, P. (2024). Metabolic Reprogramming of Astrocytes in Pathological Conditions: Implications for Neurodegenerative Diseases. International Journal of Molecular Sciences, 25(16), 8922. https://doi.org/10.3390/ijms25168922