

Cellular and Molecular Genetic Mechanisms of Lung Fibrosis Development and the Role of Vitamin D: A Review


Abstract
:1. Introduction
2. Key Pathogenetic Mechanisms and Cell Populations in the Development of Pulmonary Fibrosis
2.1. Alveolar Epithelium
2.2. Cells of the Stromal Microenvironment—Fibroblasts, Myofibroblasts, and Vascular Endothelium
2.3. Immunocompetent Cells
2.4. Cell Aging and Apoptosis
3. Effects of Vitamin D and Its Analogues Implemented through Ligand-Associated Activation of Vitamin D Receptors
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Full Name |
| EMT | Epithelial–mesenchymal transition |
| FGFR | Fibroblast growth factor receptor |
| IFN-γ | Interferon gamma |
| IL | Interleukin |
| IPF | Idiopathic pulmonary fibrosis |
| LXR | Liver X-receptor |
| M1 | Macrophage type 1 |
| M2 | Macrophage type 2 |
| miRNA | Micro RNA |
| MMP | Metalloproteinase |
| PAI | Plasminogen activator inhibitor |
| PDGFR | Platelet-derived growth factor receptor |
| RAS | Renin–angiotensin system |
| RXR | Retinoic acid receptors |
| SASP | Senescence-associated secretory phenotype |
| TGF-β | Transforming growth factor beta |
| Th1 | T-helper type 1 |
| Th17 | T-helper type 17 |
| Th2 | T-helper type 2 |
| Th9 | T-helper type 9 |
| TNF-α | Tumor necrosis factor alpha |
| Treg | Regulatory T-cell |
| VDR | Vitamin D receptor |
| VEGFR | Vascular endothelial growth factor receptor |
| Vit D | Vitamin D |
| α-SMA | Alpha-smooth muscle actin |
| γδT cells | Gamma delta T-cell |
References
- Mei, Q.; Liu, Z.; Zuo, H.; Yang, Z.; Qu, J. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Front. Pharmacol. 2022, 12, 797292. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic Pulmonary Fibrosis: Current and Future Treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Cox, I.A.; Campbell, J.A.; Xia, Q.; Otahal, P.; De Graaff, B.; Corte, T.J.; Teoh, A.K.Y.; Walters, E.H.; Palmer, A.J. Mortality and Survival in Idiopathic Pulmonary Fibrosis: A Systematic Review and Meta-Analysis. ERJ Open Res. 2022, 8, 00591–2021. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Chen, S.Y.; Yeh, W.S.; Maroni, B.; Li, Q.; Lee, Y.C.; Collard, H.R. Idiopathic Pulmonary Fibrosis in US Medicare Beneficiaries Aged 65 Years and Older: Incidence, Prevalence, and Survival, 2001–2011. Lancet Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Kaul, B.; Lee, J.S.; Zhang, N.; Vittinghoff, E.; Sarmiento, K.; Collard, H.R.; Whooley, M.A. Epidemiology of Idiopathic Pulmonary Fibrosis among U.S. Veterans, 2010–2019. Ann. Am. Thorac. Soc. 2022, 19, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Maher, T.M.; Bendstrup, E.; Dron, L.; Langley, J.; Smith, G.; Khalid, J.M.; Patel, H.; Kreuter, M. Global Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Respir. Res. 2021, 22, 197. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Šterclová, M.; Mogulkoc, N.; Lewandowska, K.; Müller, V.; Hájková, M.; Kramer, M.R.; Jovanović, D.; Tekavec-Trkanjec, J.; Studnicka, M.; et al. The European MultiPartner IPF registry (EMPIRE): Validating Long-Term Prognostic Factors in Idiopathic Pulmonary Fibrosis. Respir. Res. 2020, 21, 11. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L.; Luppi, F.; Ferrara, G.; Mura, M. Immunomodulatory Treatment of Interstitial Lung Disease. Ther. Adv. Respir. Dis. 2022, 16, 175346662211170. [Google Scholar] [CrossRef]
- Kubo, H.; Nakayama, K.; Yanai, M.; Suzuki, T.; Yamaya, M.; Watanabe, M.; Sasaki, H. Anticoagulant Therapy for Idiopathic Pulmonary Fibrosis. Chest 2005, 128, 1475–1482. [Google Scholar] [CrossRef]
- Li, S.; Pan, Y.; Xin, W.; Yan, C. The Potential Benefit of Endothelin Receptor Antagonists’ Therapy in Idiopathic Pulmonary Fibrosis: A Meta-Analysis of Results from Randomized Controlled Trials. Medicine 2022, 101, e29981. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Brown, K.K.; Costabel, U.; Cottin, V.; Du Bois, R.M.; Lasky, J.A.; Thomeer, M.; Utz, J.P.; Khandker, R.K.; McDermott, L.; et al. Treatment of Idiopathic Pulmonary Fibrosis with Etanercept: An Exploratory, Placebo-Controlled Trial. Am. J. Respir. Crit. Care Med. 2008, 178, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G. Idiopathic Pulmonary Fibrosis: Lessons from Clinical Trials over the Past 25 Years. Eur. Respir. J. 2017, 50, 1701209. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Wang, Z.; Zhu, H.; Liu, W.; Zhao, M.; Jiang, X.; Zhao, J.; Ren, C.; Zhang, Y.; Luo, L. Research Progress on Drugs Targeting the TGF-β Signaling Pathway in Fibrotic Diseases. Immunol. Res. 2022, 70, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Alkhatib, A.; Kolls, J.K.; Kondoh, Y.; Lasky, J.A. Pharmacotherapy and Adjunctive Treatment for Idiopathic Pulmonary Fibrosis (IPF). J. Thorac. Dis. 2019, 11, S1740–S1754. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in Patients with Progressive Fibrotic Interstitial Lung Diseases Other than Idiopathic Pulmonary Fibrosis (RELIEF): A Double-Blind, Randomised, Placebo-Controlled, Phase 2b Trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Alonzi, V.; D’Alessandro, M.; Bergantini, L.; Pordon, E.; Guerrieri, M.; Refini, R.M.; Sestini, P.; Bargagli, E. The Effectiveness of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis, Familial Pulmonary Fibrosis and Progressive Fibrosing Interstitial Lung Diseases: A Real-World Study. Biomedicines 2022, 10, 1973. [Google Scholar] [CrossRef]
- Santos, G.; Fabiano, A.; Mota, P.C.; Rodrigues, I.; Carvalho, D.; Melo, N.; Novais-Bastos, H.; Alexandre, A.T.; Moura, C.S.; Guimarães, S.; et al. The Impact of Nintedanib and Pirfenidone on Lung Function and Survival in Patients with Idiopathic Pulmonary Fibrosis in Real-Life Setting. Pulm. Pharmacol. Ther. 2023, 83, 102261. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (CAPACITY): Two Randomised Trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Costabel, U.; Albera, C.; Lancaster, L.H.; Lin, C.-Y.; Hormel, P.; Hulter, H.N.; Noble, P.W. An Open-Label Study of the Long-Term Safety of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (RECAP). Respiration 2017, 94, 408–415. [Google Scholar] [CrossRef]
- Raghu, G.; Mouded, M.; Chambers, D.C.; Martinez, F.J.; Richeldi, L.; Lancaster, L.H.; Hamblin, M.J.; Gibson, K.F.; Rosas, I.O.; Prasse, A.; et al. A Phase IIb Randomized Clinical Study of an Anti-α v β 6 Monoclonal Antibody in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 206, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.; Cottin, V.; Ramaswamy, M.; Wuyts, W.A.; Jenkins, R.G.; Scholand, M.B.; Kreuter, M.; Valenzuela, C.; Ryerson, C.J.; Goldin, J.; et al. Bexotegrast in Patients with Idiopathic Pulmonary Fibrosis: The INTEGRIS-IPF Study. Am. J. Respir. Crit. Care Med. 2024, 210, 424–434. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in Progressive Fibrosing Interstitial Lung Diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Richeldi, L.; Fernández Pérez, E.R.; De Salvo, M.C.; Silva, R.S.; Song, J.W.; Ogura, T.; Xu, Z.J.; Belloli, E.A.; Zhang, X.; et al. Pamrevlumab for Idiopathic Pulmonary Fibrosis: The ZEPHYRUS-1 Randomized Clinical Trial. JAMA 2024, 332, 380–389. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Jarzebska, N.; Karetnikova, E.S.; Markov, A.G.; Kasper, M.; Rodionov, R.N.; Spieth, P.M. Scarred Lung. An Update on Radiation-Induced Pulmonary Fibrosis. Front. Med. 2021, 7, 585756. [Google Scholar] [CrossRef] [PubMed]
- Bellou, V.; Belbasis, L.; Evangelou, E. Tobacco Smoking and Risk for Pulmonary Fibrosis. Chest 2021, 160, 983–993. [Google Scholar] [CrossRef] [PubMed]
- Bae, W.; Lee, C.-H.; Lee, J.; Kim, Y.W.; Han, K.; Choi, S.M. Impact of Smoking on the Development of Idiopathic Pulmonary Fibrosis: Results from a Nationwide Population-Based Cohort Study. Thorax 2022, 77, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.-Y.; Kim, H.; Bae, Y.; Song, J.W. Smoking Status and Clinical Outcome in Idiopathic Pulmonary Fibrosis: A Nationwide Study. Respir. Res. 2024, 25, 191. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Pathogenesis and Management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef]
- Michalski, J.E.; Schwartz, D.A. Genetic Risk Factors for Idiopathic Pulmonary Fibrosis: Insights into Immunopathogenesis. J. Inflamm. Res. 2021, 13, 1305–1318. [Google Scholar] [CrossRef] [PubMed]
- McElroy, A.N.; Invernizzi, R.; Laskowska, J.W.; O’Neill, A.; Doroudian, M.; Moghoofei, M.; Mostafaei, S.; Li, F.; Przybylski, A.A.; O’Dwyer, D.N.; et al. Candidate Role for Toll-like Receptor 3 L412F Polymorphism and Infection in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2022, 205, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Maher, T.M. The Role of Infection in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Eur. Respir. Rev. 2013, 22, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Thannickal, V.J. The Aging Lung and Idiopathic Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Yang, I.V.; Schwartz, D.A. Epigenetics of Idiopathic Pulmonary Fibrosis. Transl. Res. 2015, 165, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Perera, U.E.; Derseh, H.B.; Dewage, S.N.V.; Stent, A.; Wijayarathna, R.; Snibson, K.J. Evaluation of microRNA Expression in a Sheep Model for Lung Fibrosis. BMC Genom. 2021, 22, 827. [Google Scholar] [CrossRef] [PubMed]
- Cadena-Suárez, A.R.; Hernández-Hernández, H.A.; Alvarado-Vásquez, N.; Rangel-Escareño, C.; Sommer, B.; Negrete-García, M.C. Role of MicroRNAs in Signaling Pathways Associated with the Pathogenesis of Idiopathic Pulmonary Fibrosis: A Focus on Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2022, 23, 6613. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. miR-21 Mediates Fibrogenic Activation of Pulmonary Fibroblasts and Lung Fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Moimas, S.; Salton, F.; Kosmider, B.; Ring, N.; Volpe, M.C.; Bahmed, K.; Braga, L.; Rehman, M.; Vodret, S.; Graziani, M.L.; et al. miR-200 Family Members Reduce Senescence and Restore Idiopathic Pulmonary Fibrosis Type II Alveolar Epithelial Cell Transdifferentiation. ERJ Open Res. 2019, 5, 00138–2019. [Google Scholar] [CrossRef]
- Guiot, J.; Henket, M.; Remacle, C.; Cambier, M.; Struman, I.; Winandy, M.; Moermans, C.; Louis, E.; Malaise, M.; Ribbens, C.; et al. Systematic Review of Overlapping microRNA Patterns in COVID-19 and Idiopathic Pulmonary Fibrosis. Respir. Res. 2023, 24, 112. [Google Scholar] [CrossRef]
- Yan, L.; Su, Y.; Hsia, I.; Xu, Y.; Vincent-Chong, V.K.; Mojica, W.; Seshadri, M.; Zhao, R.; Wu, Y. Delivery of Anti-microRNA-21 by Lung-Targeted Liposomes for Pulmonary Fibrosis Treatment. Mol. Ther.-Nucleic Acids 2023, 32, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef]
- Selman, M. Role of Epithelial Cells in Idiopathic Pulmonary Fibrosis: From Innocent Targets to Serial Killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-Cell RNA-Seq Reveals Ectopic and Aberrant Lung-Resident Cell Populations in Idiopathic Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human Alveolar Type 2 Epithelium Transdifferentiates into Metaplastic KRT5+ Basal Cells. Nat. Cell Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef]
- Goldmann, T.; Zissel, G.; Watz, H.; Drömann, D.; Reck, M.; Kugler, C.; Rabe, K.F.; Marwitz, S. Human Alveolar Epithelial Cells Type II Are Capable of TGFβ-Dependent Epithelial-Mesenchymal-Transition and Collagen-Synthesis. Respir. Res. 2018, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Homps-Legrand, M.; Crestani, B.; Mailleux, A.A. Origins of Pathological Myofibroblasts in Lung Fibrosis: Insights from Lineage Tracing Mouse Models in the Single-Cell RNA Sequencing Era. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2023, 324, L737–L746. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Y. TGF β1: Gentlemanly Orchestrator in Idiopathic Pulmonary Fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef]
- Moore, M.W.; Herzog, E.L. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 199–208. [Google Scholar] [CrossRef]
- Decaris, M.L.; Schaub, J.R.; Chen, C.; Cha, J.; Lee, G.G.; Rexhepaj, M.; Ho, S.S.; Rao, V.; Marlow, M.M.; Kotak, P.; et al. Dual Inhibition of Avβ6 and Avβ1 Reduces Fibrogenesis in Lung Tissue Explants from Patients with IPF. Respir. Res. 2021, 22, 265. [Google Scholar] [CrossRef]
- Bahram Yazdroudi, F.; Malek, A. Optimal Controlling of Anti-TGF-β and Anti-PDGF Medicines for Preventing Pulmonary Fibrosis. Sci. Rep. 2023, 13, 15073. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.C. Procoagulant Signalling Mechanisms in Lung Inflammation and Fibrosis: Novel Opportunities for Pharmacological Intervention? Br. J. Pharmacol. 2008, 153, S367–S378. [Google Scholar] [CrossRef]
- May, J.; Mitchell, J.A.; Jenkins, R.G. Beyond Epithelial Damage: Vascular and Endothelial Contributions to Idiopathic Pulmonary Fibrosis. J. Clin. Investig. 2023, 133, e172058. [Google Scholar] [CrossRef]
- Murray, L.A.; Habiel, D.M.; Hohmann, M.; Camelo, A.; Shang, H.; Zhou, Y.; Coelho, A.L.; Peng, X.; Gulati, M.; Crestani, B.; et al. Antifibrotic Role of Vascular Endothelial Growth Factor in Pulmonary Fibrosis. JCI Insight 2017, 2, e92192. [Google Scholar] [CrossRef]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial–Mesenchymal Transition in Bleomycin-Induced Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef]
- Xaubet, A.; Agustí, C.; Luburich, P.; Barberá, J.A.; Carrión, M.; Ayuso, M.C.; Roca, J.; Rodriguez-Roisin, R. Interleukin-8 Expression in Bronchoalveolar Lavage Cells in the Evaluation of Alveolitis in Idiopathic Pulmonary Fibrosis. Respir. Med. 1998, 92, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, J.; Zhang, C.; Zhang, X.; Gao, P. Neutrophils Modulate Fibrogenesis in Chronic Pulmonary Diseases. Front. Med. 2021, 8, 616200. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Mikroulis, D.; Konstantinidis, T.; Sivridis, E.; Koffa, M.; Giatromanolaki, A.; Boumpas, D.T.; et al. Neutrophil Extracellular Traps Promote Differentiation and Function of Fibroblasts. J. Pathol. 2014, 233, 294–307. [Google Scholar] [CrossRef]
- Pokhreal, D.; Crestani, B.; Helou, D.G. Macrophage Implication in IPF: Updates on Immune, Epigenetic, and Metabolic Pathways. Cells 2023, 12, 2193. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, Functional, and Plasticity Features of Classical and Alternatively Activated Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Wu, G.; Xiong, W.; Gu, W.; Wang, C.-Y. Macrophages: Friend or Foe in Idiopathic Pulmonary Fibrosis? Respir. Res. 2018, 19, 170. [Google Scholar] [CrossRef]
- Zhang, Z.; Tian, H.; Liu, H.; Xie, R. The Role of Macrophage-Derived TGF-Β1 on SiO2-Induced Pulmonary Fibrosis: A Review. Toxicol. Ind. Health 2021, 37, 240–250. [Google Scholar] [CrossRef]
- Osorio-Valencia, S.; Zhou, B. Roles of Macrophages and Endothelial Cells and Their Crosstalk in Acute Lung Injury. Biomedicines 2024, 12, 632. [Google Scholar] [CrossRef]
- Deng, L.; Jian, Z.; Xu, T.; Li, F.; Deng, H.; Zhou, Y.; Lai, S.; Xu, Z.; Zhu, L. Macrophage Polarization: An Important Candidate Regulator for Lung Diseases. Molecules 2023, 28, 2379. [Google Scholar] [CrossRef]
- Kass, D.J.; Yu, G.; Loh, K.S.; Savir, A.; Borczuk, A.; Kahloon, R.; Juan-Guardela, B.; Deiuliis, G.; Tedrow, J.; Choi, J.; et al. Cytokine-Like Factor 1 Gene Expression Is Enriched in Idiopathic Pulmonary Fibrosis and Drives the Accumulation of CD4+ T Cells in Murine Lungs. Am. J. Pathol. 2012, 180, 1963–1978. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.A.; McKenzie, A.N.J. TH2 Cell Development and Function. Nat. Rev. Immunol. 2018, 18, 121–133. [Google Scholar] [CrossRef]
- Deng, L.; Huang, T.; Zhang, L. Correction to: T Cells in Idiopathic Pulmonary Fibrosis: Crucial but Controversial. Cell Death Discov. 2023, 9, 74. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yi, X.; Zhong, H.; Ruan, W.; Huang, D. Effect and Mechanism of Imbalance via Th9 Cells and Th17/Treg Cells in Inflammatory and Fibrotic Phases of Pulmonary Fibrosis in Mice. Biotechnol. Genet. Eng. Rev. 2023, 1–11. [Google Scholar] [CrossRef]
- Segawa, S.; Goto, D.; Iizuka, A.; Kaneko, S.; Yokosawa, M.; Kondo, Y.; Matsumoto, I.; Sumida, T. The Regulatory Role of Interferon-γ Producing Gamma Delta T Cells via the Suppression of T Helper 17 Cell Activity in Bleomycin-Induced Pulmonary Fibrosis. Clin. Exp. Immunol. 2016, 185, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.-J.; Wu, S.-H.; Xuan, Y.-H.; Yan, G. Role of IL-17 Family Cytokines in the Progression of IPF from Inflammation to Fibrosis. Mil. Med. Res. 2022, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Ramani, K.; Biswas, P.S. Interleukin-17: Friend or Foe in Organ Fibrosis. Cytokine 2019, 120, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Li, Z.; Yang, H.-Z.; Liu, H.; Wang, J.-P.; Ma, Y.-G.; Wang, X.-X.; Liu, H.-Z.; Sun, W.; Hu, Z.-W. Blocking IL-17A Promotes the Resolution of Pulmonary Inflammation and Fibrosis Via TGF-Β1–Dependent and–Independent Mechanisms. J. Immunol. 2011, 187, 3003–3014. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y. Biology of Interleukin-17 and Its Pathophysiological Significance in Sepsis. Front. Immunol. 2020, 11, 1558. [Google Scholar] [CrossRef]
- Cipolla, E.; Fisher, A.J.; Gu, H.; Mickler, E.A.; Agarwal, M.; Wilke, C.A.; Kim, K.K.; Moore, B.B.; Vittal, R. IL-17A Deficiency Mitigates Bleomycin-induced Complement Activation during Lung Fibrosis. FASEB J. 2017, 31, 5543–5556. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1β–Mediated Pulmonary Fibrosis Is IL-17A Dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Martinu, T.; McManigle, W.C.; Kelly, F.L.; Nelson, M.E.; Sun, J.; Zhang, H.L.; Kolls, J.K.; Gowdy, K.M.; Palmer, S.M. IL-17A Contributes to Lung Fibrosis in a Model of Chronic Pulmonary Graft-versus-Host Disease. Transplantation 2019, 103, 2264–2274. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, R.J.J.; Knight, D.A.; Richards, C.D.; Prêle, C.M.; Lau, H.L.; Jarnicki, A.G.; Jones, J.; Bozinovski, S.; Vlahos, R.; Thiem, S.; et al. Genetic Partitioning of Interleukin-6 Signalling in Mice Dissociates Stat3 from Smad3-mediated Lung Fibrosis. EMBO Mol. Med. 2012, 4, 939–951. [Google Scholar] [CrossRef]
- Roman, J.; Chiba, H. B Cells in Idiopathic Pulmonary Fibrosis: Targeting Immune Cells with Antifibrotic Agents. Am. J. Respir. Cell Mol. Biol. 2021, 64, 652–654. [Google Scholar] [CrossRef]
- Prêle, C.M.; Miles, T.; Pearce, D.R.; O’Donoghue, R.J.; Grainge, C.; Barrett, L.; Birnie, K.; Lucas, A.D.; Baltic, S.; Ernst, M.; et al. Plasma Cell but Not CD20-Mediated B-Cell Depletion Protects from Bleomycin-Induced Lung Fibrosis. Eur. Respir. J. 2022, 60, 2101469. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Gonzalez, F.; Faner, R.; Rojas, M.; Agustí, A.; Serrano, M.; Sellarés, J. Cellular Senescence in Lung Fibrosis. Int. J. Mol. Sci. 2021, 22, 7012. [Google Scholar] [CrossRef]
- Zhu, M.; Ding, Q.; Lin, Z.; Chen, X.; Chen, S.; Zhu, Y. New Insights of Epigenetics in Vascular and Cellular Senescence. J. Transl. Intern. Med. 2021, 9, 239–248. [Google Scholar] [CrossRef]
- Shreeya, T.; Ansari, M.S.; Kumar, P.; Saifi, M.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Senescence: A DNA Damage Response and Its Role in Aging and Neurodegenerative Diseases. Front. Aging 2024, 4, 1292053. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lu, Q.; Liu, X. Advances in Cellular Senescence in Idiopathic Pulmonary Fibrosis (Review). Exp. Ther. Med. 2023, 25, 145. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.P.; Misso, N.L.A.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse Effects of Interleukin-6 on Apoptosis of Fibroblasts from Pulmonary Fibrosis and Normal Lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef]
- Ng, B.; Cook, S.A.; Schafer, S. Interleukin-11 Signaling Underlies Fibrosis, Parenchymal Dysfunction, and Chronic Inflammation of the Airway. Exp. Mol. Med. 2020, 52, 1871–1878. [Google Scholar] [CrossRef]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef]
- Rana, T.; Jiang, C.; Banerjee, S.; Yi, N.; Zmijewski, J.W.; Liu, G.; Liu, R.-M. PAI-1 Regulation of P53 Expression and Senescence in Type II Alveolar Epithelial Cells. Cells 2023, 12, 2008. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Z. Fibroblast Senescence in Idiopathic Pulmonary Fibrosis. Front. Cell Dev. Biol. 2020, 8, 593283. [Google Scholar] [CrossRef]
- Yanai, H.; Shteinberg, A.; Porat, Z.; Budovsky, A.; Braiman, A.; Zeische, R.; Fraifeld, V.E. Cellular Senescence-like Features of Lung Fibroblasts Derived from Idiopathic Pulmonary Fibrosis Patients. Aging 2015, 7, 664–672. [Google Scholar] [CrossRef]
- Raghu, G.; Weycker, D.; Edelsberg, J.; Bradford, W.Z.; Oster, G. Incidence and Prevalence of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2006, 174, 810–816. [Google Scholar] [CrossRef]
- Hashimoto, M.; Asai, A.; Kawagishi, H.; Mikawa, R.; Iwashita, Y.; Kanayama, K.; Sugimoto, K.; Sato, T.; Maruyama, M.; Sugimoto, M. Elimination of p19ARF-Expressing Cells Enhances Pulmonary Function in Mice. JCI Insight 2016, 1, e87732. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Korfei, M.; Mutze, K.; Klee, S.; Skronska-Wasek, W.; Alsafadi, H.N.; Ota, C.; Costa, R.; Schiller, H.B.; Lindner, M.; et al. Senolytic Drugs Target Alveolar Epithelial Cell Function and Attenuate Experimental Lung Fibrosis Ex Vivo. Eur. Respir. J. 2017, 50, 1602367. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in Idiopathic Pulmonary Fibrosis: Results from a First-in-Human, Open-Label, Pilot Study. eBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, A.; Kellogg, D.; Justice, J.; Goros, M.; Gelfond, J.; Pascual, R.; Hashmi, S.; Masternak, M.; Prata, L.; LeBrasseur, N.; et al. Senolytics Dasatinib and Quercetin in Idiopathic Pulmonary Fibrosis: Results of a Phase I, Single-Blind, Single-Center, Randomized, Placebo-Controlled Pilot Trial on Feasibility and Tolerability. eBioMedicine 2023, 90, 104481. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J. Evolving Concepts of Apoptosis in Idiopathic Pulmonary Fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef]
- Hanson, K.M.; Hernady, E.B.; Reed, C.K.; Johnston, C.J.; Groves, A.M.; Finkelstein, J.N. Apoptosis Resistance in Fibroblasts Precedes Progressive Scarring in Pulmonary Fibrosis and Is Partially Mediated by Toll-Like Receptor 4 Activation. Toxicol. Sci. 2019, 170, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Zhang, K.; Jiang, S.; Fu, L.; Shi, Y.; Tan, R.; Cui, J.; Zhou, Y. P53: A Key Protein That Regulates Pulmonary Fibrosis. Oxidative Med. Cell. Longev. 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Kuwano, K.; Hagimoto, N.; Nakanishi, Y. The Role of Apoptosis in Pulmonary Fibrosis. Histol. Histopathol. 2004, 19, 867–881. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Harokopos, V.; Paparountas, T.; Oikonomou, N.; Chatziioannou, A.; Vilaras, G.; Tsiambas, E.; Karameris, A.; Bouros, D.; Aidinis, V. Comparative Expression Profiling in Pulmonary Fibrosis Suggests a Role of Hypoxia-Inducible Factor-1α in Disease Pathogenesis. Am. J. Respir. Crit. Care Med. 2007, 176, 1108–1119. [Google Scholar] [CrossRef]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A.; Hogaboam, C.M. Quercetin Enhances Ligand-Induced Apoptosis in Senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef]
- Jiang, C.; Liu, G.; Cai, L.; Deshane, J.; Antony, V.; Thannickal, V.J.; Liu, R.-M. Divergent Regulation of Alveolar Type 2 Cell and Fibroblast Apoptosis by Plasminogen Activator Inhibitor 1 in Lung Fibrosis. Am. J. Pathol. 2021, 191, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Disayabutr, S.; Kim, E.K.; Cha, S.-I.; Green, G.; Naikawadi, R.P.; Jones, K.D.; Golden, J.A.; Schroeder, A.; Matthay, M.A.; Kukreja, J.; et al. miR-34 miRNAs Regulate Cellular Senescence in Type II Alveolar Epithelial Cells of Patients with Idiopathic Pulmonary Fibrosis. PLoS ONE 2016, 11, e0158367. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Ge, J.; Xie, N.; Banerjee, S.; Zhou, Y.; Liu, R.-M.; Thannickal, V.J.; Liu, G. miR-34a Promotes Fibrosis in Aged Lungs by Inducing Alveolarepithelial Dysfunctions. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 312, L415–L424. [Google Scholar] [CrossRef]
- Pludowski, P.; Grant, W.B.; Konstantynowicz, J.; Holick, M.F. Editorial: Classic and Pleiotropic Actions of Vitamin D. Front. Endocrinol. 2019, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.-G.; Kerlan, V.; Desailloud, R. Non-Classical Effects of Vitamin D: Non-Bone Effects of Vitamin D. Ann. D’endocrinologie 2021, 82, 43–51. [Google Scholar] [CrossRef]
- Carlberg, C. Vitamin D and Its Target Genes. Nutrients 2022, 14, 1354. [Google Scholar] [CrossRef]
- Adams, J.S.; Singer, F.R.; Gacad, M.A.; Sharma, O.P.; Hayes, M.J.; Vouros, P.; Holick, M.F. Isolation and Structural Identification of 1,25-Dihydroxyvitamin D3 Produced by Cultured Alveolar Macrophages in Sarcoidosis. J. Clin. Endocrinol. Metab. 1985, 60, 960–966. [Google Scholar] [CrossRef]
- Menezes, R.J.; Cheney, R.T.; Husain, A.; Tretiakova, M.; Loewen, G.; Johnson, C.S.; Jayaprakash, V.; Moysich, K.B.; Salgia, R.; Reid, M.E. Vitamin D Receptor Expression in Normal, Premalignant, and Malignant Human Lung Tissue. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1104–1110. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Arora, J. Two Lineages of Immune Cells That Differentially Express the Vitamin D Receptor. J. Steroid Biochem. Mol. Biol. 2023, 228, 106253. [Google Scholar] [CrossRef]
- Slominski, A.T.; Kim, T.-K.; Li, W.; Postlethwaite, A.; Tieu, E.W.; Tang, E.K.Y.; Tuckey, R.C. Detection of Novel CYP11A1-Derived Secosteroids in the Human Epidermis and Serum and Pig Adrenal Gland. Sci. Rep. 2015, 5, 14875. [Google Scholar] [CrossRef]
- Lin, Z.; Marepally, S.R.; Goh, E.S.Y.; Cheng, C.Y.S.; Janjetovic, Z.; Kim, T.-K.; Miller, D.D.; Postlethwaite, A.E.; Slominski, A.T.; Tuckey, R.C.; et al. Investigation of 20S-Hydroxyvitamin D3 Analogs and Their 1α-OH Derivatives as Potent Vitamin D Receptor Agonists with Anti-Inflammatory Activities. Sci. Rep. 2018, 8, 1478. [Google Scholar] [CrossRef]
- Wang, M.; Ramirez, J.; Han, J.; Jia, Y.; Domenico, J.; Seibold, M.A.; Hagman, J.R.; Gelfand, E.W. The Steroidogenic Enzyme Cyp11a1 Is Essential for Development of Peanut-Induced Intestinal Anaphylaxis. J. Allergy Clin. Immunol. 2013, 132, 1174–1183.e8. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Hobrath, J.V.; Oak, A.S.W.; Tang, E.K.Y.; Tieu, E.W.; Li, W.; Tuckey, R.C.; Jetten, A.M. Endogenously Produced Nonclassical Vitamin D Hydroxy-Metabolites Act as “Biased” Agonists on VDR and Inverse Agonists on RORα and RORγ. J. Steroid Biochem. Mol. Biol. 2017, 173, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Kim, T.-K.; Qayyum, S.; Song, Y.; Janjetovic, Z.; Oak, A.S.W.; Slominski, R.M.; Raman, C.; Stefan, J.; Mier-Aguilar, C.A.; et al. Vitamin D and Lumisterol Derivatives Can Act on Liver X Receptors (LXRs). Sci. Rep. 2021, 11, 8002. [Google Scholar] [CrossRef]
- Ito, I.; Waku, T.; Aoki, M.; Abe, R.; Nagai, Y.; Watanabe, T.; Nakajima, Y.; Ohkido, I.; Yokoyama, K.; Miyachi, H.; et al. A Nonclassical Vitamin D Receptor Pathway Suppresses Renal Fibrosis. J. Clin. Investig. 2013, 123, 4579–4594. [Google Scholar] [CrossRef]
- Zhu, W.; Ding, Q.; Wang, L.; Xu, G.; Diao, Y.; Qu, S.; Chen, S.; Shi, Y. Vitamin D3 Alleviates Pulmonary Fibrosis by Regulating the MAPK Pathway via Targeting PSAT1 Expression In Vivo and In Vitro. Int. Immunopharmacol. 2021, 101, 108212. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D Receptor Inhibits Nuclear Factor κB Activation by Interacting with IκB Kinase β Protein. J. Biol. Chem. 2013, 288, 19450–19458. [Google Scholar] [CrossRef]
- Lange, C.M.; Gouttenoire, J.; Duong, F.H.T.; Morikawa, K.; Heim, M.H.; Moradpour, D. Vitamin D Receptor and Jak–STAT Signaling Crosstalk Results in Calcitriol-Mediated Increase of Hepatocellular Response to IFN-α. J. Immunol. 2014, 192, 6037–6044. [Google Scholar] [CrossRef]
- Matsuda, S.; Kitagishi, Y. Peroxisome Proliferator-Activated Receptor and Vitamin D Receptor Signaling Pathways in Cancer Cells. Cancers 2013, 5, 1261–1270. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Hu, X.; Qi, X.; Zhu, R.; Li, C.; Zhu, Y.; Yin, S.; Cheng, L.; Zhu, R. Vitamin D Ameliorates Angiotensin II-Induced Human Endothelial Progenitor Cell Injury via the PPAR-γ/HO-1 Pathway. J. Vasc. Res. 2019, 56, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Nieuwland, A.J.; Kokje, V.B.; Koning, O.H.; Hamming, J.F.; Szuhai, K.; Claas, F.H.J.; Lindeman, J.H.N. Activation of the Vitamin D Receptor Selectively Interferes with Calcineurin-Mediated Inflammation: A Clinical Evaluation in the Abdominal Aortic Aneurysm. Lab. Investig. 2016, 96, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, W.; Yang, Y.; Wei, S.; Xue, L.; Tao, S. Pharmaceutic Application of Vitamin D3 on Particle-Induced Fibrotic Effects through Induction of Nrf2 Signals. Toxicol. Res. 2020, 9, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Nie, H.; Ge, X.; Du, J.; Liu, W.; Li, X.; Sun, Y.; Wei, X.; Xun, Z.; Li, Y.C. Vitamin D Suppresses Bleomycin-Induced Pulmonary Fibrosis by Targeting the Local Renin–Angiotensin System in the Lung. Sci. Rep. 2021, 11, 16525. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, T.; Yao, L.; Xing, Y.; Zhao, X.; Fu, J.; Xue, X. Chronic Vitamin D Deficiency Induces Lung Fibrosis through Activation of the Renin-Angiotensin System. Sci. Rep. 2017, 7, 3312. [Google Scholar] [CrossRef]
- Boonstra, A.; Barrat, F.J.; Crain, C.; Heath, V.L.; Savelkoul, H.F.J.; O’Garra, A. 1α,25-Dihydroxyvitamin D3 Has a Direct Effect on Naive CD4+ T Cells to Enhance the Development of Th2 Cells. J. Immunol. 2001, 167, 4974–4980. [Google Scholar] [CrossRef]
- Fisher, S.A.; Rahimzadeh, M.; Brierley, C.; Gration, B.; Doree, C.; Kimber, C.E.; Plaza Cajide, A.; Lamikanra, A.A.; Roberts, D.J. The Role of Vitamin D in Increasing Circulating T Regulatory Cell Numbers and Modulating T Regulatory Cell Phenotypes in Patients with Inflammatory Disease or in Healthy Volunteers: A Systematic Review. PLoS ONE 2019, 14, e0222313. [Google Scholar] [CrossRef]
- Barron, L.; Wynn, T.A. Fibrosis Is Regulated by Th2 and Th17 Responses and by Dynamic Interactions between Fibroblasts and Macrophages. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 300, G723–G728. [Google Scholar] [CrossRef]
- Joshi, S.; Pantalena, L.-C.; Liu, X.K.; Gaffen, S.L.; Liu, H.; Rohowsky-Kochan, C.; Ichiyama, K.; Yoshimura, A.; Steinman, L.; Christakos, S.; et al. 1,25-Dihydroxyvitamin D 3 Ameliorates Th17 Autoimmunity via Transcriptional Modulation of Interleukin-17A. Mol. Cell. Biol. 2011, 31, 3653–3669. [Google Scholar] [CrossRef]
- Han, H.; Chung, S.I.; Park, H.J.; Oh, E.Y.; Kim, S.-R.; Park, K.H.; Lee, J.-H.; Park, J.-W. Obesity-Induced Vitamin D Deficiency Contributes to Lung Fibrosis and Airway Hyperresponsiveness. Am. J. Respir. Cell Mol. Biol. 2021, 64, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.M.; Wongtrakool, C.; Welch, T.; Steinmeyer, A.; Zügel, U.; Roman, J. Vitamin D Inhibition of Pro-Fibrotic Effects of Transforming Growth Factor Β1 in Lung Fibroblasts and Epithelial Cells. J. Steroid Biochem. Mol. Biol. 2010, 118, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhan, J.; Ji, H.; Xu, Y.; Xu, Q.; Zhu, X.; Liu, Y. Fibroblast Upregulation of Vitamin D Receptor Represents a Self-Protective Response to Limit Fibroblast Proliferation and Activation during Pulmonary Fibrosis. Antioxidants 2023, 12, 1634. [Google Scholar] [CrossRef] [PubMed]
- Guijarro, T.; Magro-Lopez, E.; Manso, J.; Garcia-Martinez, R.; Fernandez-Aceñero, M.J.; Liste, I.; Zambrano, A. Detrimental Pro-Senescence Effects of Vitamin D on Lung Fibrosis. Mol. Med. 2018, 24, 64. [Google Scholar] [CrossRef] [PubMed]
- Magro-Lopez, E.; Chamorro-Herrero, I.; Zambrano, A. Effects of Hypocalcemic Vitamin D Analogs in the Expression of DNA Damage Induced in Minilungs from hESCs: Implications for Lung Fibrosis. Int. J. Mol. Sci. 2022, 23, 4921. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, Y.; Xue, L.; Li, B.; Zhang, Z. 1α,25-Dihydroxyvitamin D3 Attenuates TGF-β-Induced Pro-Fibrotic Effects in Human Lung Epithelial Cells through Inhibition of Epithelial–Mesenchymal Transition. Nutrients 2017, 9, 980. [Google Scholar] [CrossRef] [PubMed]
- Sari, E.; Oztay, F.; Tasci, A.E. Vitamin D Modulates E-Cadherin Turnover by Regulating TGF-β and Wnt Signalings during EMT-Mediated Myofibroblast Differentiation in A459 Cells. J. Steroid Biochem. Mol. Biol. 2020, 202, 105723. [Google Scholar] [CrossRef] [PubMed]
- Izbicki, G.; Segel, M.J.; Christensen, T.G.; Conner, M.W.; Breuer, R. Time Course of Bleomycin-induced Lung Fibrosis. Int. J. Exp. Path. 2002, 83, 111–119. [Google Scholar] [CrossRef]
- Gul, A.; Yang, F.; Xie, C.; Du, W.; Mohammadtursun, N.; Wang, B.; Le, J.; Dong, J. Pulmonary Fibrosis Model of Mice Induced by Different Administration Methods of Bleomycin. BMC Pulm. Med. 2023, 23, 91. [Google Scholar] [CrossRef]
- Robbe, A.; Tassin, A.; Carpentier, J.; Declèves, A.-E.; Mekinda Ngono, Z.L.; Nonclercq, D.; Legrand, A. Intratracheal Bleomycin Aerosolization: The Best Route of Administration for a Scalable and Homogeneous Pulmonary Fibrosis Rat Model? BioMed Res. Int. 2015, 2015, 198418. [Google Scholar] [CrossRef]
- Abidi, A.; Bahri, S.; Ben Khamsa, S.; Legrand, A. A Comparative Study of Intratracheal and Aerosolization Instillations of Bleomycin Inducing Experimental Lung Fibrosis in Rat. Toxicol. Mech. Methods 2019, 29, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, X.; Fang, X.; Liang, A.; Yu, Z.; Gu, P.; Zeng, Y.; He, J.; Zhu, H.; Li, S.; et al. Preventive Effects of Vitamin D Treatment on Bleomycin-Induced Pulmonary Fibrosis. Sci. Rep. 2015, 5, 17638. [Google Scholar] [CrossRef] [PubMed]
- Tzilas, V.; Bouros, E.; Barbayianni, I.; Karampitsakos, T.; Kourtidou, S.; Ntassiou, M.; Ninou, I.; Aidinis, V.; Bouros, D.; Tzouvelekis, A. Vitamin D Prevents Experimental Lung Fibrosis and Predicts Survival in Patients with Idiopathic Pulmonary Fibrosis. Pulm. Pharmacol. Ther. 2019, 55, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Zhou, F.; Mao, H.; Xie, Z.; Jin, Y. Vitamin D and Idiopathic Pulmonary Fibrosis: A Two-Sample Mendelian Randomization Study. BMC Pulm. Med. 2023, 23, 309. [Google Scholar] [CrossRef] [PubMed]
- Seuter, S.; Virtanen, J.K.; Nurmi, T.; Pihlajamäki, J.; Mursu, J.; Voutilainen, S.; Tuomainen, T.-P.; Neme, A.; Carlberg, C. Molecular Evaluation of Vitamin D Responsiveness of Healthy Young Adults. J. Steroid Biochem. Mol. Biol. 2017, 174, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Haq, A. The Concept of the Personal Vitamin D Response Index. J. Steroid Biochem. Mol. Biol. 2018, 175, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Martineau, A.R.; Jolliffe, D.A.; Hooper, R.L.; Greenberg, L.; Aloia, J.F.; Bergman, P.; Dubnov-Raz, G.; Esposito, S.; Ganmaa, D.; Ginde, A.A.; et al. Vitamin D Supplementation to Prevent Acute Respiratory Tract Infections: Systematic Review and Meta-Analysis of Individual Participant Data. BMJ 2017, 356, i6583. [Google Scholar] [CrossRef] [PubMed]
- Janssens, W.; Bouillon, R.; Claes, B.; Carremans, C.; Lehouck, A.; Buysschaert, I.; Coolen, J.; Mathieu, C.; Decramer, M.; Lambrechts, D. Vitamin D Deficiency Is Highly Prevalent in COPD and Correlates with Variants in the Vitamin D-Binding Gene. Thorax 2010, 65, 215–220. [Google Scholar] [CrossRef]
- Yavari, M.; Mousavi, S.A.J.; Janani, L.; Feizy, Z.; Vafa, M. Effects of Supplementation of Vitamins D, C and E on Idiopathic Pulmonary Fibrosis (IPF): A Clinical Trial. Clin. Nutr. ESPEN 2022, 49, 295–300. [Google Scholar] [CrossRef]
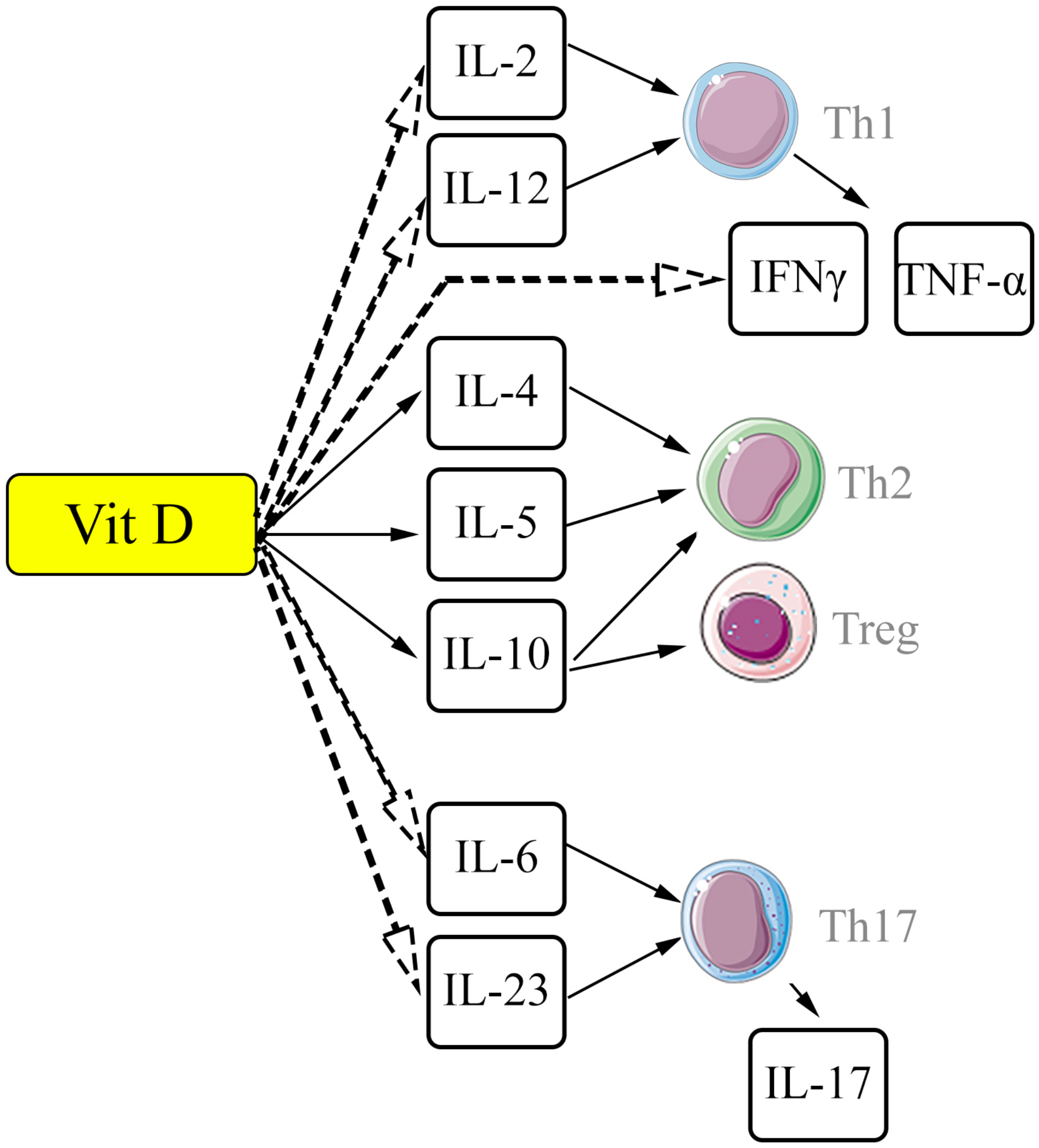


{kind=link}
{kind=link}
| Drug | Target | Study Design, Phase, and Number of Participants | Summary of Main Findings | Name and Status of Clinical Trial (NCT) |
|---|---|---|---|---|
| Pirfenidone | TGF-β | Randomized, double-blind, placebo-controlled, parallel-group study; Phase 3; in total, 779 patients with IPF | Positive dose-dependent effect with reduced decline in FVC vs. placebo and favorable benefit risk profile [18] | CAPACITY, completed (NCT00287729, NCT00287716) |
| Randomized, double-blind, placebo-controlled, parallel-group study; Phase 3; 555 patients with IPF | Reduced disease progression, as reflected by lung function, exercise tolerance, and progression-free survival [19] | ASCEND, completed (NCT01366209) | ||
| Open-label extension study; Phase 3; 1058 patients with IPF | Mean change in percent predicted FVC from baseline at 180 weeks was −9.6%; median on-treatment survival from the first pirfenidone dose was 77.2 months [20] | RECAP, completed (NCT00662038) | ||
| BG00011 (anti-αvβ6 monoclonal antibody) | TGF-β/TGF-β receptors—integrins αV/β6 | Randomized, double-blind, placebo-controlled, parallel-group study; Phase 2; 109 patients with IPF | Early trial termination due to imbalance in adverse events and lack of clinical benefit [21] | SPIRIT, terminated (NCT03573505) |
| PLN-74809 (Bexotegrast) | TGF-β/TGF-β receptors—integrins αV/β1, αV/β6 | Randomized, double-blind, dose-ranging, placebo-controlled, parallel-group study; Phase 2a; 120 patients with IPF | Dose-dependent antifibrotic effect (reduction in FVC decline over 12 weeks vs. placebo) [22] | INTEGRIS-IPF, completed (NCT04396756) |
| Nintedanib | Tyrosine kinases (VEGFR 1–3, FGFR 1–3, PDGFR α, β) | Randomized, double-blind, dose-ranging, placebo-controlled, parallel-group study; Phase 3; 663 patients with progressive fibrosing interstitial lung disease | Slower rate of progression of the interstitial lung disease (reduction in FVC decline) [23] | INBUILD, completed (NCT02999178) |
| Pamrevlumab | CTGF | Randomized, double-blind, placebo-controlled, parallel-group study; Phase 3; in total, 728 patients with IPF | Termination of a planned open-label extension study of pamrevlumab as well as the ongoing ZEPHYRUS-2 trial due to a lack of effectiveness [24] | ZEPHYRUS-1,2, terminated (NCT03955146, NCT04419558) |
| Signaling Pathway | Signaling Pathway Inductor | The Implemented Cascade of Reactions | Effect of Vit D Complex/VDR |
|---|---|---|---|
| SMAD | TGF-β/receptors TGF-β; angiotensin II | Regulation of cellular cycle, differentiation (in particular of myofibroblasts), immune reactions | Suppression by reducing TGF-β expression and nuclear translocation of SMAD components |
| MARK | TGF-β, growth factors, lipopolysaccharides, etc. | Regulation of cellular proliferation and differentiation, inflammatory response, and elimination of cells by apoptosis | Suppression by enhancing the expression of proteinase (MAPK phosphatase-1) followed by inhibition of p38 MAPK |
| NF-κβ | Proinflammatory cytokines (IL-1 and TNF-α), lipopolysaccharides, and growth factors | Regulation of non-specific and adaptive immunity and inflammatory response | Suppression by inhibiting Ikkß kinase and preventing activation of NF-kß, as well as by suppressing nuclear translocation of signaling pathway components |
| JAK/STAT | Cytokines (IL-2, IL-6, IL-10, IL-12, IFN-α, etc.) and growth factors (EGF) | Regulation of cellular proliferation, differentiation, and inflammatory response | Suppression of binding of transcription factor STAT3 to the promoter (ATF6) |
| PPAR-α/γ | Unsaturated fatty acids, eicosanoids, etc. | Regulation of metabolism of fatty acids and energy balance, regulation of immune response, and cellular cycle | Competitive interaction due to binding to RXR and suppression of the PPAR-α/γ promoter |
| NFAT | Ca2+ | Regulation of cellular cycle and inflammatory response | Blocking of NFAT components of Runx1 transcription factor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enzel, D.; Kriventsov, M.; Sataieva, T.; Malygina, V. Cellular and Molecular Genetic Mechanisms of Lung Fibrosis Development and the Role of Vitamin D: A Review. Int. J. Mol. Sci. 2024, 25, 8946. https://doi.org/10.3390/ijms25168946
Enzel D, Kriventsov M, Sataieva T, Malygina V. Cellular and Molecular Genetic Mechanisms of Lung Fibrosis Development and the Role of Vitamin D: A Review. International Journal of Molecular Sciences. 2024; 25(16):8946. https://doi.org/10.3390/ijms25168946
Chicago/Turabian StyleEnzel, Darya, Maxim Kriventsov, Tatiana Sataieva, and Veronika Malygina. 2024. "Cellular and Molecular Genetic Mechanisms of Lung Fibrosis Development and the Role of Vitamin D: A Review" International Journal of Molecular Sciences 25, no. 16: 8946. https://doi.org/10.3390/ijms25168946
APA StyleEnzel, D., Kriventsov, M., Sataieva, T., & Malygina, V. (2024). Cellular and Molecular Genetic Mechanisms of Lung Fibrosis Development and the Role of Vitamin D: A Review. International Journal of Molecular Sciences, 25(16), 8946. https://doi.org/10.3390/ijms25168946