

Generic Reporter Sets for Colorimetric Multiplex dPCR Demonstrated with 6-Plex SNP Quantification Panels

 , , ,
, , ,
Abstract
1. Introduction

2. Results
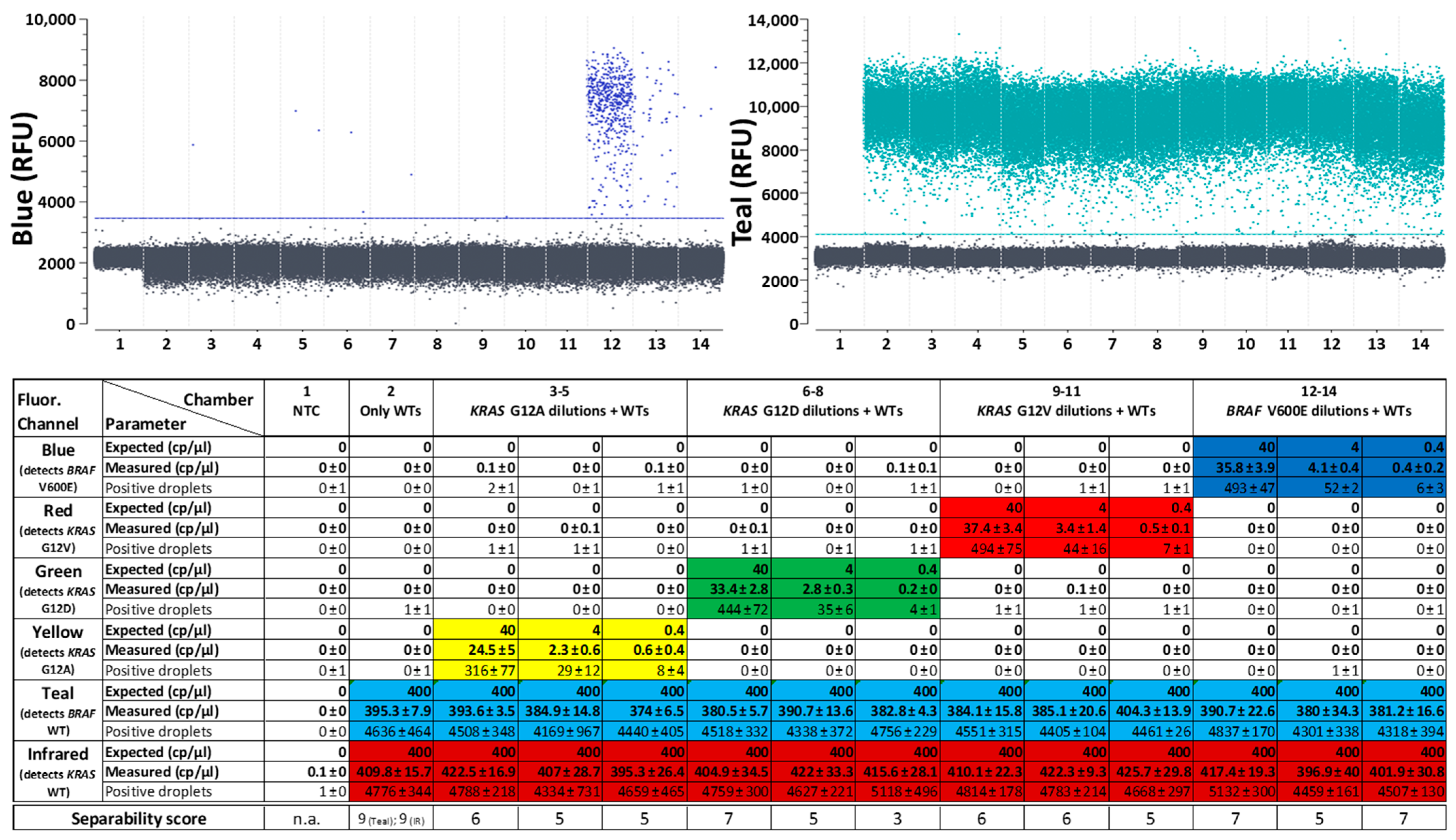
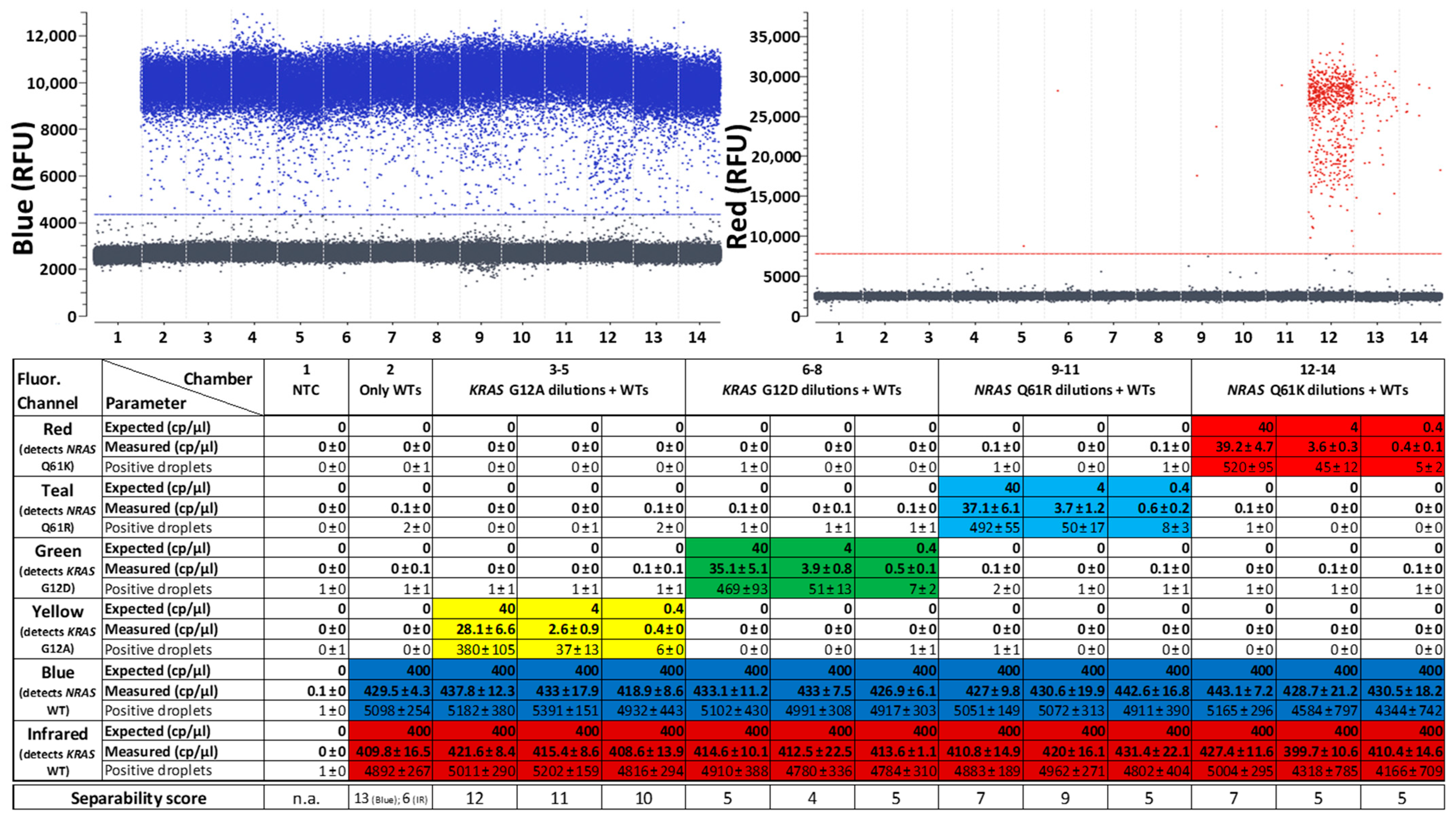
2.1. Characterization of a Fluorogenic Generic Reporter Set for Quantification of 6-Plex SNP Detection Panels
2.2. Development of a New Generic Reporter Set

3. Discussion
3.1. Impact of the Developed Generic Reporter Sets
- Efficient oligonucleotide design: One generic reporter set can be used for different colorimetric multiplex assays. When using the same dPCR platform, no further optimization steps are needed. For other platforms, generic reporter set optimization is necessary only once.
- Higher degree of freedom and deeper insight in assay design: The differentiability and quantifiability of the respective targets used is independent of the fluorescence signal generation, since signal generation is standardized by the generic reporter set. Alternative, TaqManTM-based approaches exhibit a higher dependency of fluorescence signal generation on the respective target-specific probe sequence. For example, guanine bases are known to quench the fluorescence of several fluorescent dyes when in close proximity [36,37]. If this type of quenching is attempted to be avoided by changing the length of the probe, shortening the probe often makes it less specific and extending it reduces the efficiency of the quencher, which is usually located at the other end of the TaqManTM probe [38]. When using generic reporter sets instead, if individual targets do not yield proper fluorescence signals, the problem lies not in the generic signal generation but in the decoupled target detection, which is why fewer parameters need to be checked during troubleshooting.
- Good absolute quantifiability: Quantification results show little difference between expected and measured copy numbers, as has already been shown by Schlenker et al. [20]. dPCR in general has a better quantitative resolution than standard qPCR [39] which is resolved via comparing threshold cycle numbers that can be dependent on many factors as inhibitors [40,41] and the applied detection system [42]. With the presented assay, SNP mutations can still be reliably quantified in presence of at least 1000-fold excess of the corresponding WT (plus another WT) in the same reaction. It can be assumed that even higher WT excesses do not present a problem for mutation target detection, since the detection of mutations in the presence or absence of the highly concentrated WTs did not show any noticeable differences, while many of the mutation target-positive droplets must already have contained WT targets when adding them in 1000-fold concentrations.
- High specificity: Despite the simultaneous detection of different SNPs, the assays detected the correct targets, differentiating reliably between single base mismatches [20]. In all tested reactions, the number of false positives was so low that it was never within the 95% confidence interval for a positive result.
3.2. Strategies to Reduce Signal Crosstalk
3.3. Parameters for Evaluation of dPCR Result Quality
3.4. Workflow for the Generic Creation of New Reporter Sets

4. Materials and Methods
4.1. Reagents and Devices
4.2. Mediator Extension Assays and Digital PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Producer | Final Concentration |
|---|---|---|
| Buffer A—naica® (multiplex) PCR MIX 10 X | Stilla Technologies | 1× |
| Buffer B—naica® (multiplex) PCR MIX | Stilla Technologies | 4% |
| H2O | Qiagen (Hilden, Germany) | - |
| Reporter | biomers.net | 0.4 µM |
| Mediator probe | biomers.net | 1.2 µM |
| Primers | biomers.net | 0.5–2 µM (depending on assay) |
| Target DNA | Integrated DNA Technologies | variable |
| Herring sperm DNA | Promega | 2.4 µg/mL |
| Temperature [°C] | Duration [min:s] | Cycles | |
|---|---|---|---|
| Partitioning | 40 | 12:00 | - |
| Initial denaturation | 95 | 3:00 | - |
| Amplification | 95 | 0:15 | 60 |
| 58 | 0:60 | ||
| Pressure release | 25 | 33:00 | - |
| Component | Producer | Final Concentration |
|---|---|---|
| Buffer A—naica® multiplex PCR MIX 10 X | Stilla Technologies | 1× |
| Buffer B—naica® multiplex PCR MIX | Stilla Technologies | 4% |
| H2O | Qiagen | - |
| Reporter | biomers.net | 0.1 µM |
| Mediator | biomers.net | 0.15 µM |
| Temperature [°C] | Duration [min:s] | Cycles | |
|---|---|---|---|
| Partitioning | 40 | 12:00 | - |
| Initial denaturation | 95 | 5:00 | - |
| Amplification | 95 | 0:10 | 20 |
| 60 | 0:30 | ||
| Pressure release | 25 | 33:00 | - |
4.3. Data Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olmedillas-López, S.; Olivera-Salazar, R.; García-Arranz, M.; García-Olmo, D. Current and Emerging Applications of Droplet Digital PCR in Oncology: An Updated Review. Mol. Diagn. Ther. 2022, 26, 61–87. [Google Scholar] [CrossRef]
- Fang, W.; Liu, X.; Maiga, M.; Cao, W.; Mu, Y.; Yan, Q.; Zhu, Q. Digital PCR for Single-Cell Analysis. Biosensors 2024, 14, 64. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef]
- Hou, Y.; Chen, S.; Zheng, Y.; Zheng, X.; Lin, J.-M. Droplet-based digital PCR (ddPCR) and its applications. TrAC Trends Anal. Chem. 2023, 158, 116897. [Google Scholar] [CrossRef]
- Whale, A.S.; Huggett, J.F.; Tzonev, S. Fundamentals of multiplexing with digital PCR. Biomol. Detect. Quantif. 2016, 10, 15–23. [Google Scholar] [CrossRef]
- Tan, L.L.; Loganathan, N.; Agarwalla, S.; Yang, C.; Yuan, W.; Zeng, J.; Wu, R.; Wang, W.; Duraiswamy, S. Current commercial dPCR platforms: Technology and market review. Crit. Rev. Biotechnol. 2022, 43, 433–464. [Google Scholar] [CrossRef]
- Lai, J.H.; Won Keum, J.; Lee, H.G.; Molaei, M.; Blair, E.J.; Li, S.; Soliman, J.W.; Raol, V.K.; Barker, C.L.; Fodor, S.P.A.; et al. New realm of precision multiplexing enabled by massively-parallel single molecule UltraPCR. biorXiv 2023. [Google Scholar] [CrossRef]
- Zhao, Y.; Xia, Q.; Yin, Y.; Wang, Z. Comparison of Droplet Digital PCR and Quantitative PCR Assays for Quantitative Detection of Xanthomonas citri Subsp. citri. PLoS ONE 2016, 11, e0159004. [Google Scholar] [CrossRef] [PubMed]
- Cusenza, V.Y.; Bisagni, A.; Rinaldini, M.; Cattani, C.; Frazzi, R. Copy Number Variation and Rearrangements Assessment in Cancer: Comparison of Droplet Digital PCR with the Current Approaches. Int. J. Mol. Sci. 2021, 22, 4732. [Google Scholar] [CrossRef]
- Lievens, A.; Jacchia, S.; Kagkli, D.; Savini, C.; Querci, M. Measuring Digital PCR Quality: Performance Parameters and Their Optimization. PLoS ONE 2016, 11, e0153317. [Google Scholar] [CrossRef] [PubMed]
- Van Heuverswyn, F.; Karczmarczyk, M.; Schimmel, H.; Trapmann, S.; Emons, H. Influence of primer and probe chemistry and amplification target on reverse transcription digital PCR quantification of viral RNA. Biomol. Detect. Quantif. 2016, 9, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Hays, A.; Wissel, M.; Colletti, K.; Soon, R.; Azadeh, M.; Smith, J.; Doddareddy, R.; Chalfant, M.; Adamowicz, W.; Ramaswamy, S.S.; et al. Recommendations for Method Development and Validation of qPCR and dPCR Assays in Support of Cell and Gene Therapy Drug Development. AAPS J. 2024, 26, 24. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.A.; Huang, C.; Yamamoto, F.; Sahoo, M.K.; Le, Q.-T.; Pinsky, B.A. Comparison of Real-Time PCR and Digital PCR for Detection of Plasma Epstein-Barr Virus DNA in Nasopharyngeal Carcinoma. J. Mol. Diagn. 2023, 25, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, B.; De Reu, K.; De Zutter, L.; Verstraete, K.; Heyndrickx, M.; Van Coillie, E. Comparison of Droplet Digital PCR and qPCR for the Quantification of Shiga Toxin-Producing Escherichia coli in Bovine Feces. Toxins 2016, 8, 157. [Google Scholar] [CrossRef]
- Voegel, T.M.; Larrabee, M.M.; Nelson, L.M. Development of droplet digital PCR assays to quantify genes involved in nitrification and denitrification, comparison with quantitative real-time PCR and validation of assays in vineyard soil. Can. J. Microbiol. 2021, 67, 174–187. [Google Scholar] [CrossRef]
- Hu, G.; Huang, K.; Zhou, W.; Wang, R.; Zhao, W.; Zou, H.; Li, W.; Wu, S.; Li, M.; Wang, G. Comparison of droplet digital PCR and real-time quantitative PCR for quantitative detection of the parasitic ciliate Ichthyophthirius multifiliis in the water environment. J. Fish Dis. 2023, 46, 357–367. [Google Scholar] [CrossRef]
- Wadle, S.; Lehnert, M.; Schuler, F.; Köppel, R.; Serr, A.; Zengerle, R.; von Stetten, F. Simplified development of multiplex real-time PCR through master mix augmented by universal fluorogenic reporters. Biotechniques 2016, 61, 123–128. [Google Scholar] [CrossRef]
- Faltin, B.; Wadle, S.; Roth, G.; Zengerle, R.; von Stetten, F. Mediator Probe PCR: A Novel Approach for Detection of Real-Time PCR Based on Label-Free Primary Probes and Standardized Secondary Universal Fluorogenic Reporters. Clin. Chem. 2012, 58, 1546–1556. [Google Scholar] [CrossRef]
- Lehnert, M.; Kipf, E.; Schlenker, F.; Borst, N.; Zengerle, R.; Von Stetten, F. Fluorescence signal-to-noise optimisation for real-time PCR using universal reporter oligonucleotides. Anal. Methods 2018, 10, 3444–3454. [Google Scholar] [CrossRef]
- Schlenker, F.; Kipf, E.; Deuter, M.; Höffkes, I.; Lehnert, M.; Zengerle, R.; von Stetten, F.; Scherer, F.; Wehrle, J.; von Bubnoff, N.; et al. Stringent Base Specific and Optimization-Free Multiplex Mediator Probe ddPCR for the Quantification of Point Mutations in Circulating Tumor DNA. Cancers 2021, 13, 5742. [Google Scholar] [CrossRef]
- Roth, G.; Faltin, B.; von Stetten, F.; Wadle, S. Bifunctional Oligonucleotide Probe for Universal Real Time Multianalyte Detection. WO/2013/079307, 6 June 2013. [Google Scholar]
- Schlenker, F.; Kipf, E.; Borst, N.; Hutzenlaub, T.; Zengerle, R.; Von Stetten, F.; Juelg, P. Virtual Fluorescence Color Channels by Selective Photobleaching in Digital PCR Applied to the Quantification of KRAS Point Mutations. Anal. Chem. 2021, 93, 10538–10545. [Google Scholar] [CrossRef] [PubMed]
- Wadle, S.; Rubenwolf, S.; Lehnert, M.; Faltin, B.; Weidmann, M.; Hufert, F.; Zengerle, R.; von Stetten, F. Mediator Probe PCR: Detection of Real-Time PCR by Label-Free Probes and a Universal Fluorogenic Reporter. In Quantitative Real-Time PCR; Methods in Molecular Biology: Totowa, NJ, USA, 2014; Volume 1160, pp. 55–73. [Google Scholar]
- Lyamichev, V.; Brow, M.A.D.; Dahlberg, J.E. Structure-Specific Endonucleolytic Cleavage of Nucleic Acids by Eubacterial DNA Polymerases. Science 1993, 260, 778–783. [Google Scholar] [CrossRef]
- Bożyk, A.; Krawczyk, P.; Reszka, K.; Krukowska, K.; Kolak, A.; Mańdziuk, S.; Wojas-Krawczyk, K.; Ramlau, R.; Milanowski, J. Correlation between KRAS, NRAS and BRAF mutations and tumor localizations in patients with primary and metastatic colorectal cancer. Arch. Med. Sci. 2021, 18, 1221–1230. [Google Scholar] [CrossRef]
- Heppt, M.V.; Siepmann, T.; Engel, J.; Schubert-Fritschle, G.; Eckel, R.; Mirlach, L.; Kirchner, T.; Jung, A.; Gesierich, A.; Ruzicka, T.; et al. Prognostic significance of BRAF and NRAS mutations in melanoma: A German study from routine care. BMC Cancer 2017, 17, 536. [Google Scholar] [CrossRef]
- Dogan, S.; Shen, R.; Ang, D.C.; Johnson, M.L.; D’Angelo, S.P.; Paik, P.K.; Brzostowski, E.B.; Riely, G.J.; Kris, M.G.; Zakowski, M.F.; et al. Molecular Epidemiology of EGFR and KRAS Mutations in 3,026 Lung Adenocarcinomas: Higher Susceptibility of Women to Smoking-Related KRAS -Mutant Cancers. Clin. Cancer Res. 2012, 18, 6169–6177. [Google Scholar] [CrossRef]
- Sanchez-Ibarra, H.E.; Jiang, X.; Gallegos-Gonzalez, E.Y.; Cavazos-González, A.C.; Chen, Y.; Morcos, F.; Barrera-Saldaña, H.A. KRAS, NRAS, and BRAF mutation prevalence, clinicopathological association, and their application in a predictive model in Mexican patients with metastatic colorectal cancer: A retrospective cohort study. PLoS ONE 2020, 15, e0235490. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Hussung, S.; Follo, M.; Klar, R.F.U.; Michalczyk, S.; Fritsch, K.; Nollmann, F.; Hipp, J.; Duyster, J.; Scherer, F.; von Bubnoff, N.; et al. Development and Clinical Validation of Discriminatory Multitarget Digital Droplet PCR Assays for the Detection of Hot Spot KRAS and NRAS Mutations in Cell-Free DNA. J. Mol. Diagn. 2020, 22, 943–956. [Google Scholar] [CrossRef]
- Palmirotta, R.; Lovero, D.; Cafforio, P.; Felici, C.; Silvestris, F. Liquid biopsy of cancer: A multimodal diagnostic tool in clinical oncology. Ther. Adv. Med. Oncol. 2018, 10, 1758835918794630. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, S.; Markl, A.M.; Neugebauer, M.; Krauth, S.J.; Borst, N.; von Stetten, F.; Lehnert, M. Reporter emission multiplexing in digital PCRs (REM-dPCRs): Direct quantification of multiple target sequences per detection channel by population specific reporters. Analyst 2023, 148, 5243–5254. [Google Scholar] [CrossRef]
- Stilla Technologies Guidelines for 6-Color Multiplex Assay Design for Optimized Performance with Crystal Digital PCRTM. Available online: https://www.stillatechnologies.com/digital-pcr/naica-system-analysis/guidelines-for-6-color-multiplex-assay-design-for-optimized-performance-with-crystal-digital-pcr/ (accessed on 27 March 2024).
- Stilla Technologies Stilla FAQ: What Size Are the Droplets Generated in the Sapphire Chips? Available online: https://www.stillatechnologies.com/digital-pcr/naica-system-test-faqs/what-size-are-the-droplets-generated-in-the-sapphire-chips/ (accessed on 2 April 2024).
- Schwaederlé, M.C.; Patel, S.P.; Husain, H.; Ikeda, M.; Lanman, R.B.; Banks, K.C.; Talasaz, A.; Bazhenova, L.; Kurzrock, R. Utility of Genomic Assessment of Blood-Derived Circulating Tumor DNA (ctDNA) in Patients with Advanced Lung Adenocarcinoma. Clin. Cancer Res. 2017, 23, 5101–5111. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.A.M.; Schulz, A.; Sauer, M.H.M. Nucleobase-Specific Quenching of Fluorescent Dyes. 1. Nucleobase One-Electron Redox Potentials and Their Correlation with Static and Dynamic Quenching Efficiencies. J. Phys. Chem. 1996, 100, 5541–5553. [Google Scholar] [CrossRef]
- Marras, S.A.E.; Kramer, F.R.; Tyagi, S. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 2002, 30, e122. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Rodríguez, M.; Córdoba, J.J.; Andrade, M.J. Design of Primers and Probes for Quantitative Real-Time PCR Methods. In PCR Primer Design; Methods in Molecular Biology: Totowa, NJ, USA, 2015; pp. 31–56. [Google Scholar]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Benito, L.T. Low Input or Inhibition? Quantitative Approaches to Detect qPCR Inhibitors. BioEcho Life Sci. GmbH BLOG. 2022. Available online: https://www.bioecho.com/blog/low-input-or-inhibition-quantitative-approaches-to-detect-qpcr-inhibitors (accessed on 16 July 2024).
- Scott, G.; Evens, N.; Porter, J.; Walker, D.I. The Inhibition and Variability of Two Different RT-qPCR Assays Used for Quantifying SARS-CoV-2 RNA in Wastewater. Food Environ. Virol. 2023, 15, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, J.M.; Lorenz, P.; Tuomi, J.M.; Hecker, M.; van den Hoff, M.J.B. Fluorescent-increase kinetics of different fluorescent reporters used for qPCR depend on monitoring chemistry, targeted sequence, type of DNA input and PCR efficiency. Microchim. Acta 2014, 181, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.A.; Touré, M.B.; Sanogo, D.; Shaffer, J.G.; Doumbia, S.O.; Krogstad, D.J. Template copy number and the sensitivity of quantitative PCR for Plasmodium falciparum in asymptomatic individuals. Malar. J. 2020, 19, 295. [Google Scholar] [CrossRef]
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Catherine Muenker, M.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nat. Microbiol. 2020, 5, 1299–1305. [Google Scholar] [CrossRef]
- Chen, A.K.; Cheng, Z.; Behlke, M.A.; Tsourkas, A. Assessing the Sensitivity of Commercially Available Fluorophores to the Intracellular Environment. Anal. Chem. 2008, 80, 7437–7444. [Google Scholar] [CrossRef]
- ATTO-TEC GmbH. Determination of Fluorescence Quantum Yield. Available online: https://www.atto-tec.com/fluorescence/determination-of-fluorescence-quantum-yield/?language=en (accessed on 15 July 2024).
- Bio-Rad Laboratories, I. Droplet Digital PCR Applications Guide (Bulletin 6407 Ver B). The Resource Is a Guide by Bio-Rad That Can Be Accessed Online. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf (accessed on 15 July 2024).
- Universitätsklinikum Freiburg Digital PCR. Available online: https://www.uniklinik-freiburg.de/medizin1/forschung/lighthouse-core-facility/nucleic-acid-analysis/dpcr.html (accessed on 8 April 2024).
- Chao, L.-C.; Tsai, H.-Y.; Li, C.-R.; Huang, K.-C.; Lin, Y.-H.; Shieh, D.-B. Fluorescence Signal-to-Noise Ratio Enhanced by Off-Plane Excitation for Quantitative PCR Device. In Proceedings of the 2020 IEEE International Symposium on Medical Measurements and Applications (MeMeA), Bari, Italy, 1 June 2020; pp. 1–6. [Google Scholar]
- Mehndiratta, M.; Palanichamy, J.K.; Ramalingam, P.; Pal, A.; Das, P.; Sinha, S.; Chattopadhyay, P. Fluorescence acquisition during hybridization phase in quantitative real-time PCR improves specificity and signal-to-noise ratio. Biotechniques 2008, 45, 625–634. [Google Scholar] [CrossRef]
- Wilding, T.A.; Stoeck, T.; Morrissey, B.J.; Carvalho, S.F.; Coulson, M.W. Maximising signal-to-noise ratios in environmental DNA-based monitoring. Sci. Total Environ. 2023, 858, 159735. [Google Scholar] [CrossRef] [PubMed]
- Sint, D.; Raso, L.; Traugott, M. Advances in multiplex PCR: Balancing primer efficiencies and improving detection success. Methods Ecol. Evol. 2012, 3, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, V.; Rutkowski, A.J.; Meuser, E.; Carr, T.H.; Harrington, E.A.; Barrett, J.C. Optimisation of robust singleplex and multiplex droplet digital PCR assays for high confidence mutation detection in circulating tumour DNA. Sci. Rep. 2019, 9, 12620. [Google Scholar] [CrossRef] [PubMed]
- Nour, A.M.A.; Pfaffl, M.W. MIQE qPCR & dPCR How to Apply the MIQE Guidelines; bioMCC: Freising, Germany, 2022; ISBN 9783000488061. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neugebauer, M.; Calabrese, S.; Müller, S.; Truong, T.-T.; Juelg, P.; Borst, N.; Hutzenlaub, T.; Dazert, E.; Bubnoff, N.C.C.v.; von Stetten, F.; et al. Generic Reporter Sets for Colorimetric Multiplex dPCR Demonstrated with 6-Plex SNP Quantification Panels. Int. J. Mol. Sci. 2024, 25, 8968. https://doi.org/10.3390/ijms25168968
Neugebauer M, Calabrese S, Müller S, Truong T-T, Juelg P, Borst N, Hutzenlaub T, Dazert E, Bubnoff NCCv, von Stetten F, et al. Generic Reporter Sets for Colorimetric Multiplex dPCR Demonstrated with 6-Plex SNP Quantification Panels. International Journal of Molecular Sciences. 2024; 25(16):8968. https://doi.org/10.3390/ijms25168968
Chicago/Turabian StyleNeugebauer, Maximilian, Silvia Calabrese, Sarah Müller, Truong-Tu Truong, Peter Juelg, Nadine Borst, Tobias Hutzenlaub, Eva Dazert, Nikolas Christian Cornelius von Bubnoff, Felix von Stetten, and et al. 2024. "Generic Reporter Sets for Colorimetric Multiplex dPCR Demonstrated with 6-Plex SNP Quantification Panels" International Journal of Molecular Sciences 25, no. 16: 8968. https://doi.org/10.3390/ijms25168968
APA StyleNeugebauer, M., Calabrese, S., Müller, S., Truong, T.-T., Juelg, P., Borst, N., Hutzenlaub, T., Dazert, E., Bubnoff, N. C. C. v., von Stetten, F., & Lehnert, M. (2024). Generic Reporter Sets for Colorimetric Multiplex dPCR Demonstrated with 6-Plex SNP Quantification Panels. International Journal of Molecular Sciences, 25(16), 8968. https://doi.org/10.3390/ijms25168968