
Inducing Receptor Degradation as a Novel Approach to Target CC Chemokine Receptor 2 (CCR2)

 , , , ,
, , , ,
Abstract

1. Introduction
2. Results
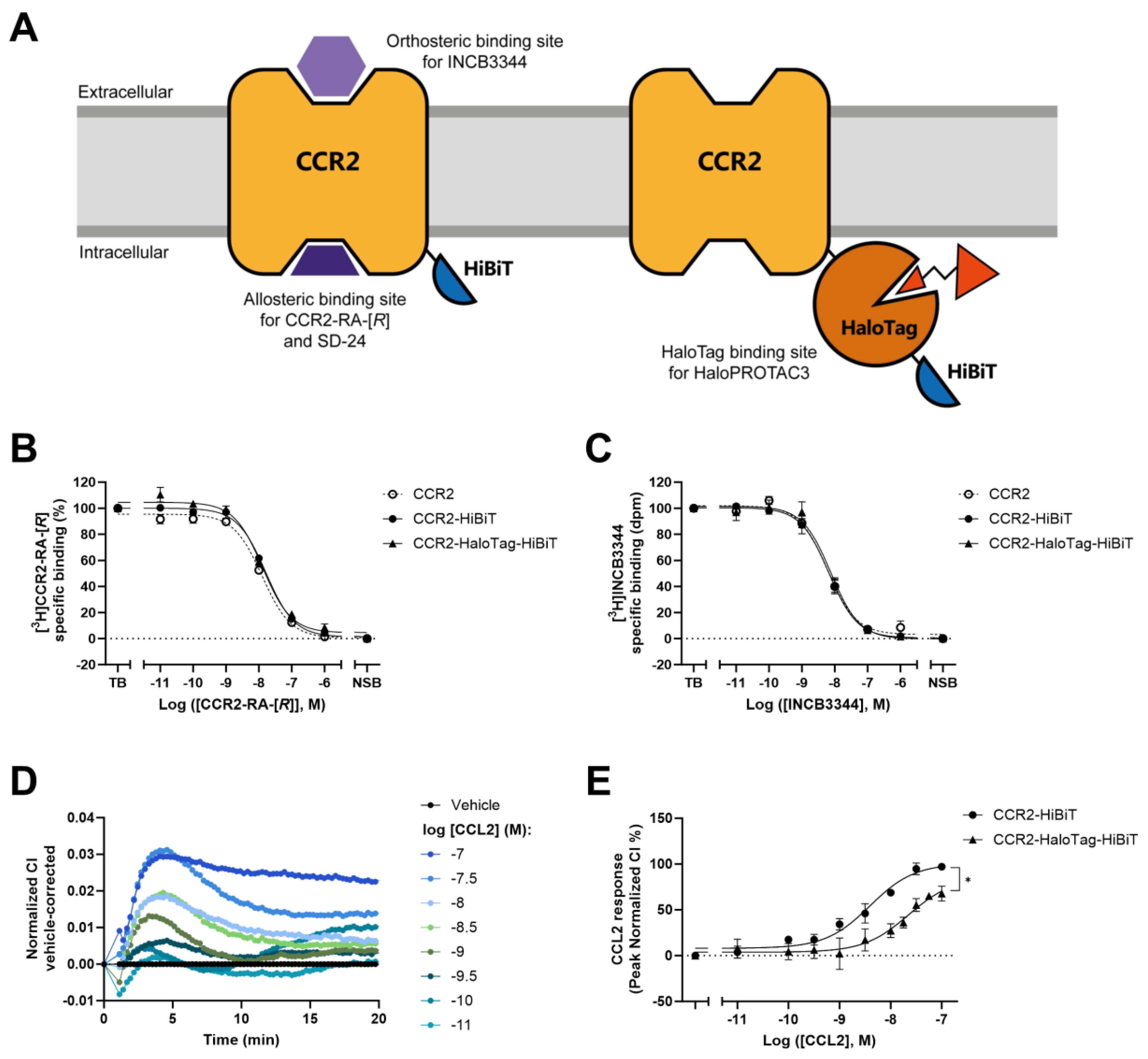
2.1. Insertion of HaloTag7 to CCR2 Alters Receptor Function but Not Ligand Binding
2.2. HaloPROTAC3 Induces Degradation of CCR2-HaloTag-HiBiT
2.3. Real-Time HaloPROTAC3-Induced Degradation of CCR2
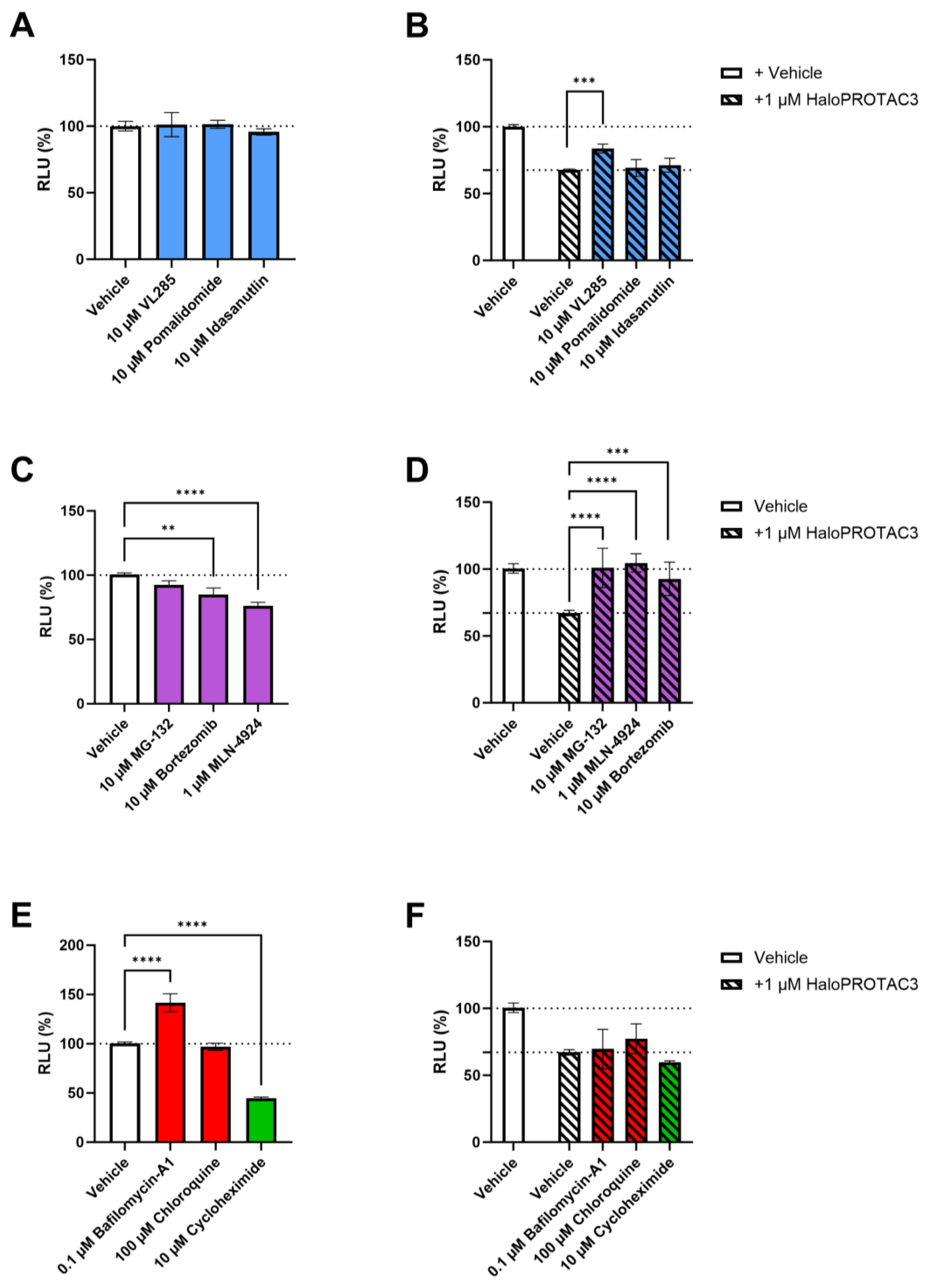
2.4. HaloPROTAC3 Induces Proteasomal Degradation of CCR2-HaloTag-HiBiT
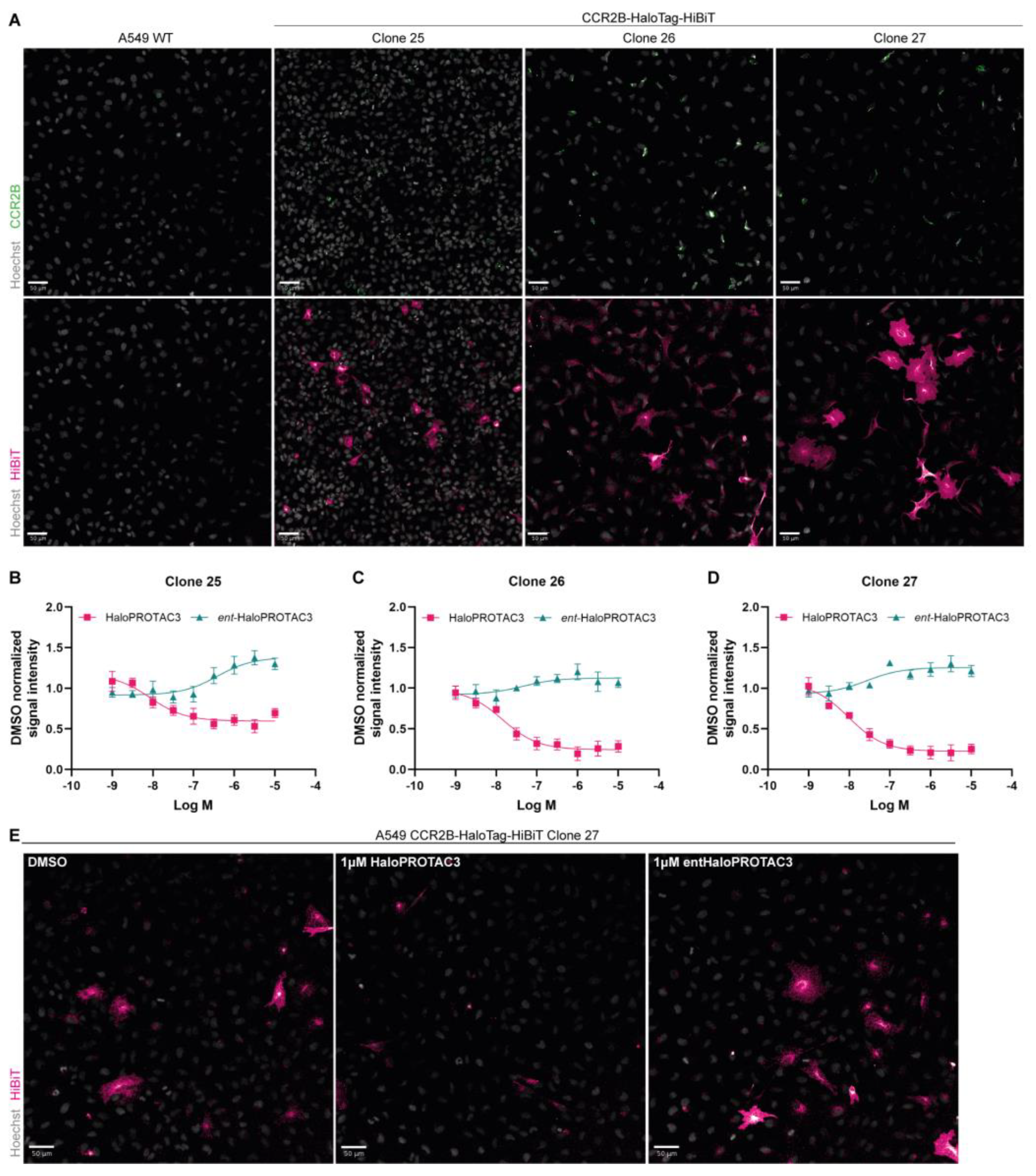
2.5. Immunofluorescence Studies Confirm CCR2-HaloTag-HiBiT Degradation by HaloPROTAC3
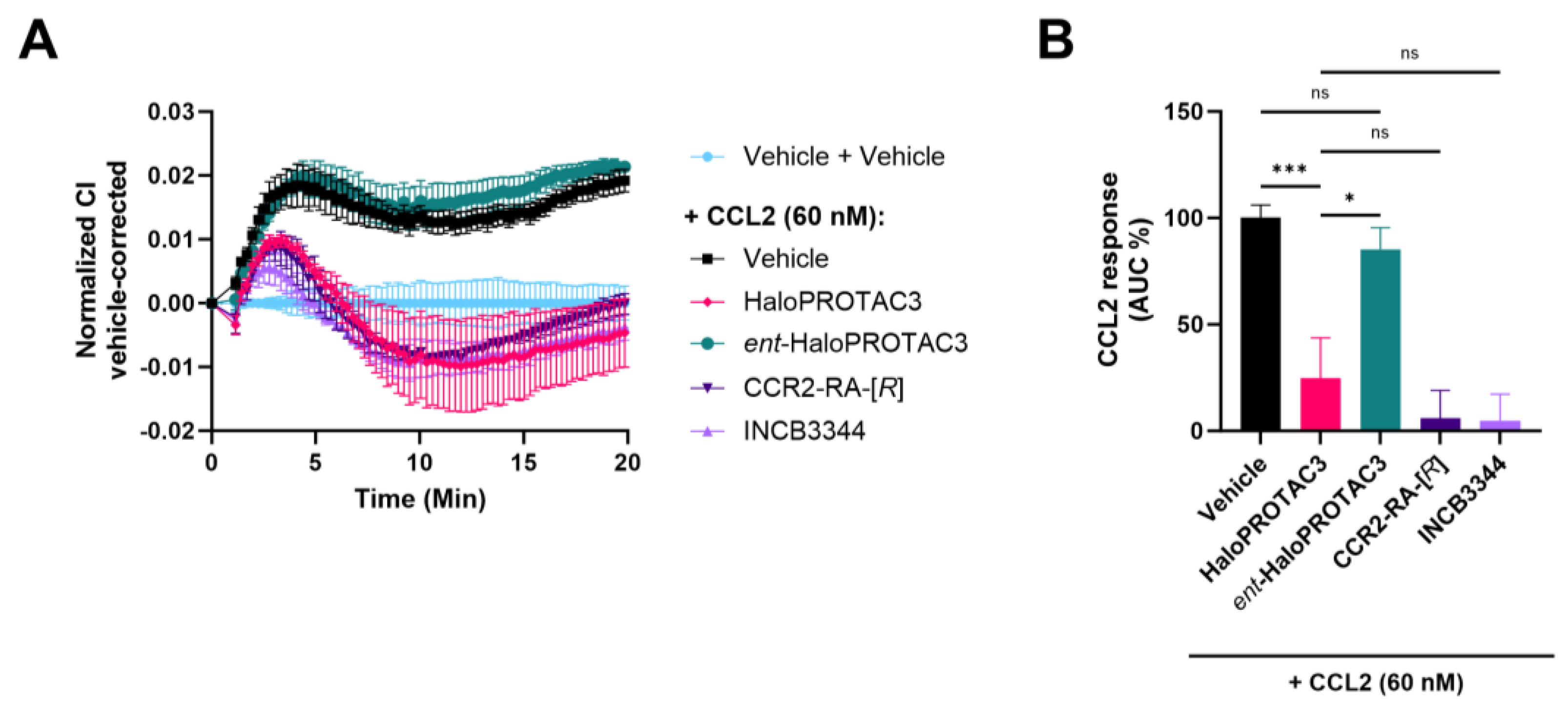
2.6. Degradation of CCR2-HaloTag-HiBiT Translates to Reduced Receptor Activation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Transfection and Lentivirus Generation
4.4. Transduction and Clonal Isolation
4.5. Membrane Preparation
4.6. Radioligand Binding Assays
4.7. Label-Free Phenotypic Whole-Cell Assay (xCELLigence)
4.8. Lytic HiBiT Detection
4.9. Real-Time, Live Cell HiBiT Detection
4.10. Immunofluorescence
4.11. Data Analysis and Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G protein-coupled receptors as targets for approved drugs: How many targets and how many drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Cotarelo, P.; Gomez-Moreira, C.; Criado-Garcia, O.; Sanchez, L.; Rodriguez-Fernandez, J.L. Beyond chemoattraction: Multifunctionality of chemokine receptors in leukocytes. Trends Immunol. 2017, 38, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Luster, A.D. Chemokines and their receptors: Drug targets in immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, L.; Asare, Y.; Bernhagen, J.; Dichgans, M.; Georgakis, M.K. Pharmacological Targeting of the CCL2/CCR2 Axis for Atheroprotection: A Meta-Analysis of Preclinical Studies. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e131–e144. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Bernhagen, J.; Heitman, L.H.; Weber, C.; Dichgans, M. Targeting the CCL2-CCR2 axis for atheroprotection. Eur. Heart J. 2022, 43, 1799–1808. [Google Scholar] [CrossRef]
- Lim, S.Y.; Yuzhalin, A.E.; Gordon-Weeks, A.N.; Muschel, R.J. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 2016, 7, 28697–28710. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the CCL2/CCR2 axis in cancer immunotherapy: One stone, three birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef]
- Alabi, S.B.; Crews, C.M. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 2021, 296, 100647. [Google Scholar] [CrossRef] [PubMed]
- Churcher, I. Protac-induced protein degradation in drug discovery: Breaking the rules or just making new ones? J. Med. Chem. 2018, 61, 444–452. [Google Scholar] [CrossRef]
- Burslem, G.M.; Smith, B.E.; Lai, A.C.; Jaime-Figueroa, S.; McQuaid, D.C.; Bondeson, D.P.; Toure, M.; Dong, H.; Qian, Y.; Wang, J.; et al. The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 2018, 25, 67–77. [Google Scholar] [CrossRef]
- Bensimon, A.; Pizzagalli, M.D.; Kartnig, F.; Dvorak, V.; Essletzbichler, P.; Winter, G.E.; Superti-Furga, G. Targeted Degradation of SLC Transporters Reveals Amenability of Multi-Pass Transmembrane Proteins to Ligand-Induced Proteolysis. Cell Chem. Biol. 2020, 27, 728–739.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lin, Y.; Song, H.; Qin, X.; Yu, Z.; Zhang, Z.; Dong, G.; Li, X.; Shi, X.; Du, L.; et al. First small-molecule PROTACs for G protein-coupled receptors: Inducing α1A-adrenergic receptor degradation. Acta Pharm. Sin. B 2020, 10, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.S.; Rouhimoghadam, M.; Arnatt, C.K.; Filardo, E.J.; Salem, A.K. Proteolytic Targeting Chimeras with Specificity for Plasma Membrane and Intracellular Estrogen Receptors. Mol. Pharm. 2021, 18, 1455–1469. [Google Scholar] [CrossRef]
- Huber, M.E.; Toy, L.; Schmidt, M.F.; Vogt, H.; Budzinski, J.; Wiefhoff, M.F.J.; Merten, N.; Kostenis, E.; Weikert, D.; Schiedel, M. A Chemical Biology Toolbox Targeting the Intracellular Binding Site of CCR9: Fluorescent Ligands, New Drug Leads and PROTACs. Angew. Chem. Int. Ed. Engl. 2022, 61, e202116782. [Google Scholar] [CrossRef] [PubMed]
- Ruffilli, C.; Roth, S.; Rodrigo, M.; Boyd, H.; Zelcer, N.; Moreau, K. Proteolysis Targeting Chimeras (PROTACs): A Perspective on Integral Membrane Protein Degradation. ACS Pharmacol. Transl. Sci. 2022, 5, 849–858. [Google Scholar] [CrossRef] [PubMed]
- Keen, A.C.; Jorg, M.; Halls, M.L. The application of targeted protein degradation technologies to G protein-coupled receptors. Br. J. Pharmacol. 2023, 181, 2351–2358. [Google Scholar] [CrossRef] [PubMed]
- Schapira, M.; Calabrese, M.F.; Bullock, A.N.; Crews, C.M. Targeted protein degradation: Expanding the toolbox. Nat. Rev. Drug Discov. 2019, 18, 949–963. [Google Scholar] [CrossRef]
- Congreve, M.; Oswald, C.; Marshall, F.H. Applying structure-based drug design approaches to allosteric modulators of GPCRs. Trends Pharmacol. Sci. 2017, 38, 837–847. [Google Scholar] [CrossRef]
- Ortiz Zacarias, N.V.; Lenselink, E.B.; IJzerman, A.P.; Handel, T.M.; Heitman, L.H. Intracellular receptor modulation: Novel approach to target GPCRs. Trends Pharmacol. Sci. 2018, 39, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qin, L.; Ortiz Zacarías, N.V.; de Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461. [Google Scholar] [CrossRef]
- Guedeney, N.; Cornu, M.; Schwalen, F.; Kieffer, C.; Voisin-Chiret, A.S. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discov. Today 2023, 28, 103395. [Google Scholar] [CrossRef] [PubMed]
- Kostic, M.; Jones, L.H. Critical Assessment of Targeted Protein Degradation as a Research Tool and Pharmacological Modality. Trends Pharmacol. Sci. 2020, 41, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Yesbolatova, A.; Tominari, Y.; Kanemaki, M.T. Ligand-induced genetic degradation as a tool for target validation. Drug Discov. Today Technol. 2019, 31, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, C.; Magtoto, C.M.; Walker, J.R.; Chua, N.K.; Gabrielyan, A.; Hall, M.; Cobbold, S.A.; Mieruszynski, S.; Brzozowski, M.; Simpson, D.S.; et al. Development of NanoLuc-targeting protein degraders and a universal reporter system to benchmark tag-targeted degradation platforms. Nat. Commun. 2022, 13, 2073. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Raina, K.; Darricarrere, N.; Hines, J.; Gustafson, J.L.; Smith, I.E.; Miah, A.H.; Harling, J.D.; Crews, C.M. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 2015, 10, 1831–1837. [Google Scholar] [CrossRef]
- Nabet, B.; Roberts, J.M.; Buckley, D.L.; Paulk, J.; Dastjerdi, S.; Yang, A.; Leggett, A.L.; Erb, M.A.; Lawlor, M.A.; Souza, A. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 2018, 14, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Bond, A.G.; Craigon, C.; Chan, K.-H.; Testa, A.; Karapetsas, A.; Fasimoye, R.; Macartney, T.; Blow, J.J.; Alessi, D.R.; Ciulli, A. Development of BromoTag: A “bump-and-hole”–PROTAC system to induce potent, rapid, and selective degradation of tagged target proteins. J. Med. Chem. 2021, 64, 15477–15502. [Google Scholar] [CrossRef] [PubMed]
- Caine, E.A.; Mahan, S.D.; Johnson, R.L.; Nieman, A.N.; Lam, N.; Warren, C.R.; Riching, K.M.; Urh, M.; Daniels, D.L. Targeted Protein Degradation Phenotypic Studies Using HaloTag CRISPR/Cas9 Endogenous Tagging Coupled with HaloPROTAC3. Curr. Protoc. Pharmacol. 2020, 91, e81. [Google Scholar] [CrossRef]
- Schwinn, M.K.; Machleidt, T.; Zimmerman, K.; Eggers, C.T.; Dixon, A.S.; Hurst, R.; Hall, M.P.; Encell, L.P.; Binkowski, B.F.; Wood, K.V. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem. Biol. 2018, 13, 467–474. [Google Scholar] [CrossRef]
- Riching, K.M.; Mahan, S.; Corona, C.R.; McDougall, M.; Vasta, J.D.; Robers, M.B.; Urh, M.; Daniels, D.L. Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol. 2018, 13, 2758–2770. [Google Scholar] [CrossRef]
- Daniels, D.L.; Riching, K.M.; Urh, M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discov. Today Technol. 2019, 31, 61–68. [Google Scholar] [CrossRef]
- Riching, K.M.; Mahan, S.D.; Urh, M.; Daniels, D.L. High-throughput cellular profiling of targeted protein degradation compounds using HiBiT CRISPR cell lines. JoVE (J. Vis. Exp.) 2020, e61787. [Google Scholar]
- Zweemer, A.J.; Nederpelt, I.; Vrieling, H.; Hafith, S.; Doornbos, M.L.; de Vries, H.; Abt, J.; Gross, R.; Stamos, D.; Saunders, J.; et al. Multiple binding sites for small-molecule antagonists at the CC chemokine receptor 2. Mol. Pharmacol. 2013, 84, 551–561. [Google Scholar] [CrossRef]
- Ortiz Zacarías, N.V.; van Veldhoven, J.P.D.; Portner, L.; van Spronsen, E.; Ullo, S.; Veenhuizen, M.; van der Velden, W.J.C.; Zweemer, A.J.M.; Kreekel, R.M.; Oenema, K.; et al. Pyrrolone derivatives as intracellular allosteric modulators for chemokine receptors: Selective and dual-targeting inhibitors of CC chemokine receptors 1 and 2. J. Med. Chem. 2018, 61, 9146–9161. [Google Scholar] [CrossRef] [PubMed]
- Doornbos, M.L.J.; Heitman, L.H. Chapter 11—Label-free impedance-based whole cell assay to study GPCR pharmacology. In Methods Cell Biology; Shukla, A.K., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 149, pp. 179–194. [Google Scholar]
- Scott, C.W.; Peters, M.F. Label-free whole-cell assays: Expanding the scope of GPCR screening. Drug Discov. Today 2010, 15, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Röth, S.; Fulcher, L.J.; Sapkota, G.P. Advances in targeted degradation of endogenous proteins. Cell. Mol. Life Sci. 2019, 76, 2761–2777. [Google Scholar] [CrossRef] [PubMed]
- Ohana, R.F.; Encell, L.P.; Zhao, K.; Simpson, D.; Slater, M.R.; Urh, M.; Wood, K.V. HaloTag7: A genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification. Protein Expr. Purif. 2009, 68, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.M.; Glennie, L.; Brewer, A.; Zhao, J.-F.; Crooks, J.; Shpiro, N.; Sapkota, G.P. Target protein localization and its impact on PROTAC-mediated degradation. Cell Chem. Biol. 2022, 29, 1482–1504.e7. [Google Scholar] [CrossRef] [PubMed]
- Neklesa, T.K.; Tae, H.S.; Schneekloth, A.R.; Stulberg, M.J.; Corson, T.W.; Sundberg, T.B.; Raina, K.; Holley, S.A.; Crews, C.M. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 2011, 7, 538–543. [Google Scholar] [CrossRef]
- Bondeson, D.P.; Mullin-Bernstein, Z.; Oliver, S.; Skipper, T.A.; Atack, T.C.; Bick, N.; Ching, M.; Guirguis, A.A.; Kwon, J.; Langan, C.; et al. Systematic profiling of conditional degron tag technologies for target validation studies. Nat. Commun. 2022, 13, 5495. [Google Scholar] [CrossRef]
- Kredel, S.; Wolff, M.; Hobbie, S.; Bieler, M.; Gierschik, P.; Heilker, R. High-content analysis of CCR2 antagonists on human primary monocytes. J. Biomol. Screen. 2011, 16, 683–693. [Google Scholar] [CrossRef]
- Tovell, H.; Testa, A.; Maniaci, C.; Zhou, H.; Prescott, A.R.; Macartney, T.; Ciulli, A.; Alessi, D.R. Rapid and reversible knockdown of endogenously tagged endosomal proteins via an optimized HaloPROTAC degrader. ACS Chem. Biol. 2019, 14, 882–892. [Google Scholar] [CrossRef]
- Ody, B.K.; Zhang, J.; Nelson, S.E.; Xie, Y.; Liu, R.; Dodd, C.J.; Jacobs, S.E.; Whitzel, S.L.; Williams, L.A.; Gozem, S. Synthesis and Evaluation of Cereblon-Recruiting HaloPROTACs. ChemBioChem 2023, 24, e202300498. [Google Scholar] [CrossRef]
- Tomoshige, S.; Hashimoto, Y.; Ishikawa, M. Efficient protein knockdown of HaloTag-fused proteins using hybrid molecules consisting of IAP antagonist and HaloTag ligand. Bioorg. Med. Chem. 2016, 24, 3144–3148. [Google Scholar] [CrossRef] [PubMed]
- Tomoshige, S.; Naito, M.; Hashimoto, Y.; Ishikawa, M. Degradation of HaloTag-fused nuclear proteins using bestatin-HaloTag ligand hybrid molecules. Org. Biomol. Chem. 2015, 13, 9746–9750. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Brownell, J.E.; Sintchak, M.D.; Gavin, J.M.; Liao, H.; Bruzzese, F.J.; Bump, N.J.; Soucy, T.A.; Milhollen, M.A.; Yang, X.; Burkhardt, A.L.; et al. Substrate-Assisted Inhibition of Ubiquitin-like Protein-Activating Enzymes: The NEDD8 E1 Inhibitor MLN4924 Forms a NEDD8-AMP Mimetic In Situ. Mol. Cell 2010, 37, 102–111. [Google Scholar] [CrossRef]
- Bartlett, D.W.; Gilbert, A.M. Translational PK–PD for targeted protein degradation. Chem. Soc. Rev. 2022, 51, 3477–3486. [Google Scholar] [CrossRef] [PubMed]
- Vetma, V.; Perez, L.C.; Eliaš, J.; Stingu, A.; Kombara, A.; Gmaschitz, T.; Braun, N.; Ciftci, T.; Dahmann, G.; Diers, E.; et al. Confounding Factors in Targeted Degradation of Short-Lived Proteins. ACS Chem. Biol. 2024, 19, 1484–1494. [Google Scholar] [CrossRef]
- Sica, A.; Saccani, A.; Borsatti, A.; Power, C.A.; Wells, T.N.; Luini, W.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Bacterial lipopolysaccharide rapidly inhibits expression of C–C chemokine receptors in human monocytes. J. Exp. Med. 1997, 185, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Rahimpour, R.; Ran, L.; Kong, C.; Biragyn, A.; Andrews, J.; Devries, M.; Wang, J.M.; Kelvin, D. Regulation of CCR2 chemokine receptor mRNA stability. J. Leukoc. Biol. 1997, 62, 653–660. [Google Scholar] [CrossRef]
- Kao, S.-H.; Wang, W.-L.; Chen, C.-Y.; Chang, Y.-L.; Wu, Y.-Y.; Wang, Y.-T.; Wang, S.-P.; Nesvizhskii, A.I.; Chen, Y.-J.; Hong, T.-M. Analysis of protein stability by the cycloheximide chase assay. Bio-Protocol 2015, 5, e1374. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Du, Q.; Zhang, H.-G.; Yuan, Y.; Zuo, Y.; Zheng, H. Cycloheximide (CHX) chase assay to examine protein half-life. Bio-Protocol 2023, 13, e4690. [Google Scholar] [CrossRef] [PubMed]
- Merrill, R.A.; Song, J.; Kephart, R.A.; Klomp, A.J.; Noack, C.E.; Strack, S. A robust and economical pulse-chase protocol to measure the turnover of HaloTag fusion proteins. J. Biol. Chem. 2019, 294, 16164–16171. [Google Scholar] [CrossRef] [PubMed]
- Schall, T.J.; Proudfoot, A.E. Overcoming hurdles in developing successful drugs targeting chemokine receptors. Nat. Rev. Immunol. 2011, 11, 355–363. [Google Scholar] [CrossRef]
- Bot, I.; Ortiz Zacarias, N.V.; de Witte, W.E.; de Vries, H.; van Santbrink, P.J.; van der Velden, D.; Kroner, M.J.; van der Berg, D.J.; Stamos, D.; de Lange, E.C.; et al. A novel CCR2 antagonist inhibits atherogenesis in apoE deficient mice by achieving high receptor occupancy. Sci. Rep. 2017, 7, 52. [Google Scholar] [CrossRef]
- Mares, A.; Miah, A.H.; Smith, I.E.D.; Rackham, M.; Thawani, A.R.; Cryan, J.; Haile, P.A.; Votta, B.J.; Beal, A.M.; Capriotti, C.; et al. Extended pharmacodynamic responses observed upon PROTAC-mediated degradation of RIPK2. Commun. Biol. 2020, 3, 140. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pIC50 ± SEM (IC50, nM) 1 | |||
|---|---|---|---|
| Compound | CCR2 2 | CCR2-HiBiT 3 | CCR2-HaloTag-HiBiT 3 |
| CCR2-RA-[R] | 7.9 ± 0.03 (13) | 7.8 ± 0.06 (17) | 7.9 ± 0.08 (13) |
| INCB3344 | 8.2 ± 0.13 (6) | 8.2 ± 0.13 (7) | 8.1 ± 0.10 (8) |
| SD-24 | 8.0 ± 0.06 (10) | 8.3 ± 0.05 (5) | 8.2 ± 0.05 (6) |
| Compound | pDC50/pEC50 ± SEM (DC50/EC50, nM) 1,2 | Dmax ± SEM (%) 1,3 |
|---|---|---|
| HaloPROTAC3 | 7.4 ± 0.01 (42) | 38 ± 5 |
| ent-HaloPROTAC3 | n.a. 4 | −1 ± 4 |
| INCB3344 | 8.4 ± 0.3 (6) | −20 ± 5 |
| CCR2-RA-[R] | 7.8 ± 0.1 (15) | −49 ± 5 |
| SD-24 | 6.8 ± 0.1 (181) | −48 ± 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz Zacarías, N.V.; Röth, S.; Broekhuis, J.D.; van der Es, D.; Moreau, K.; Heitman, L.H. Inducing Receptor Degradation as a Novel Approach to Target CC Chemokine Receptor 2 (CCR2). Int. J. Mol. Sci. 2024, 25, 8984. https://doi.org/10.3390/ijms25168984
Ortiz Zacarías NV, Röth S, Broekhuis JD, van der Es D, Moreau K, Heitman LH. Inducing Receptor Degradation as a Novel Approach to Target CC Chemokine Receptor 2 (CCR2). International Journal of Molecular Sciences. 2024; 25(16):8984. https://doi.org/10.3390/ijms25168984
Chicago/Turabian StyleOrtiz Zacarías, Natalia V., Sascha Röth, Jeremy D. Broekhuis, Daan van der Es, Kevin Moreau, and Laura H. Heitman. 2024. "Inducing Receptor Degradation as a Novel Approach to Target CC Chemokine Receptor 2 (CCR2)" International Journal of Molecular Sciences 25, no. 16: 8984. https://doi.org/10.3390/ijms25168984
APA StyleOrtiz Zacarías, N. V., Röth, S., Broekhuis, J. D., van der Es, D., Moreau, K., & Heitman, L. H. (2024). Inducing Receptor Degradation as a Novel Approach to Target CC Chemokine Receptor 2 (CCR2). International Journal of Molecular Sciences, 25(16), 8984. https://doi.org/10.3390/ijms25168984