
Activation of G Protein-Coupled Estrogen Receptor (GPER) Negatively Modulates Cardiac Excitation–Contraction Coupling (ECC) through the PI3K/NOS/NO Pathway

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
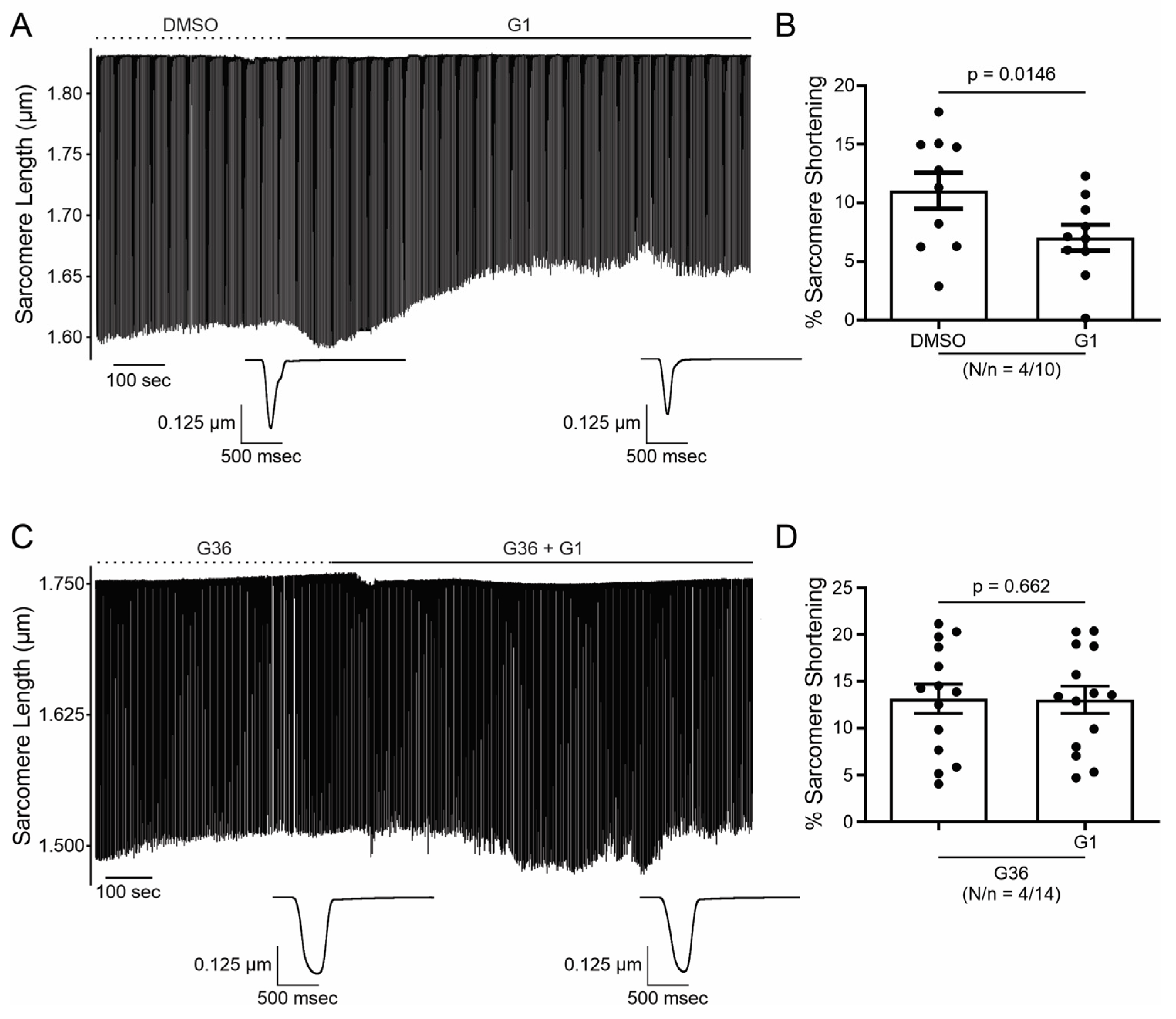
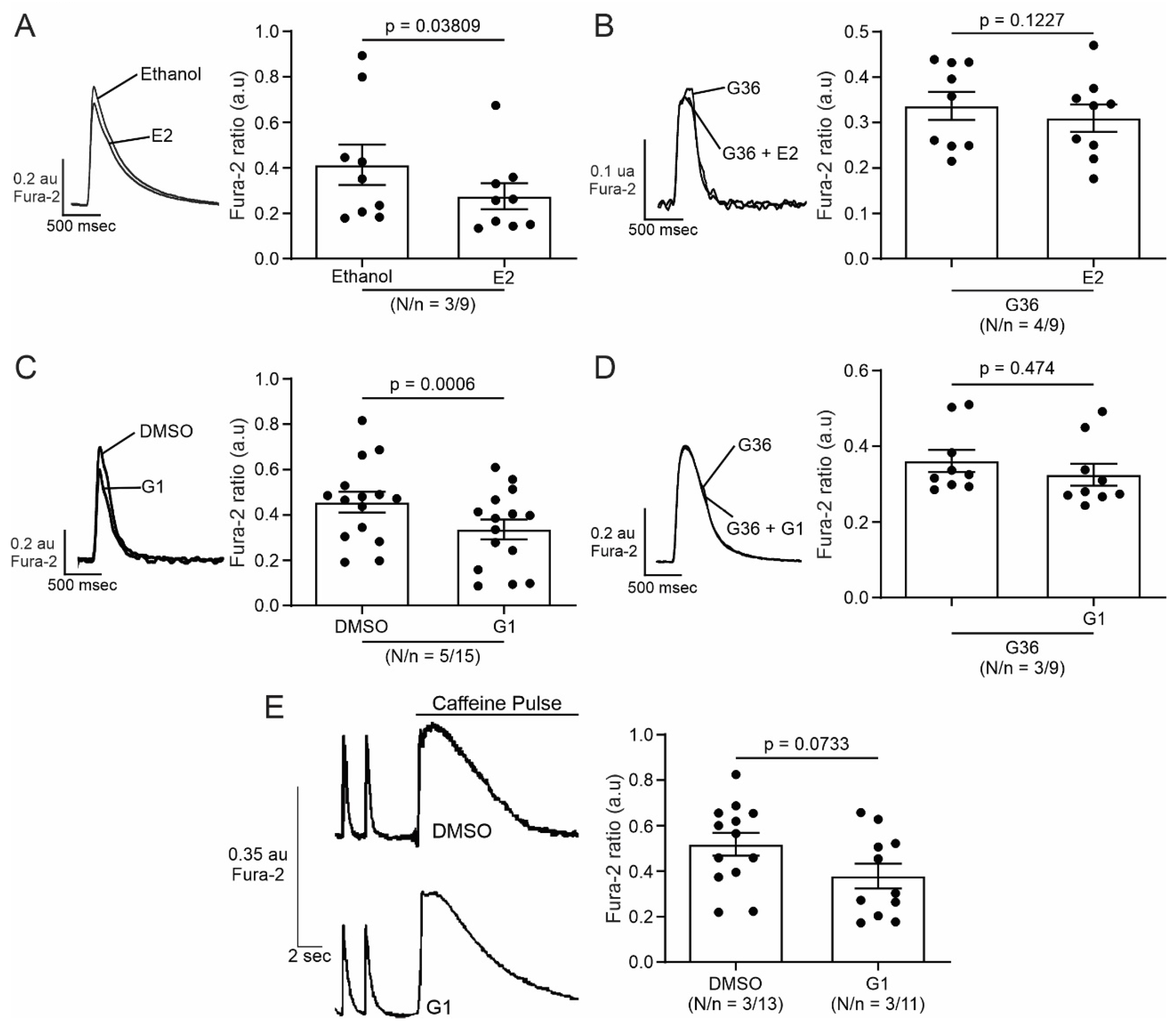
2.1. E2 and G1 Reduce Cardiomyocyte Contractility through GPER Signaling
2.2. Activation of GPER Decreases L-Type Calcium Current (ICaL) in Cardiac Myocytes
2.3. PI3K and NOS Are Involved in the GPER-Triggered Intracellular Pathway
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Ventricular Myocyte Isolation
4.3. Measure of Calcium Transient and Cell Shortening
4.4. Patch Clamp Recording
4.5. Nitric Oxide Production
4.6. Treatments
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kvingedal, A.M.; Smeland, E.B. A novel putative G-protein-coupled receptor expressed in lung, heart and lymphoid tissue. FEBS Lett. 1997, 407, 59–62. [Google Scholar] [CrossRef]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Arterburn, J.B. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol. Rev. 2015, 67, 505–540. [Google Scholar] [CrossRef]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef]
- Frump, A.L.; Albrecht, M.; Yakubov, B.; Breuils-Bonnet, S.; Nadeau, V.; Tremblay, E.; Potus, F.; Omura, J.; Cook, T.; Fisher, A.; et al. 17beta-Estradiol and estrogen receptor alpha protect right ventricular function in pulmonary hypertension via BMPR2 and apelin. J. Clin. Investig. 2021, 131, 129433. [Google Scholar] [CrossRef]
- Mihai, M.C.; Popa, M.A.; Suica, V.I.; Antohe, F.; Jackson, E.K.; Simionescu, M.; Dubey, R.K. Mechanism of 17beta-estradiol stimulated integration of human mesenchymal stem cells in heart tissue. J. Mol. Cell. Cardiol. 2019, 133, 115–124. [Google Scholar] [CrossRef]
- Filice, E.; Recchia, A.G.; Pellegrino, D.; Angelone, T.; Maggiolini, M.; Cerra, M.C. A new membrane G protein-coupled receptor (GPR30) is involved in the cardiac effects of 17beta-estradiol in the male rat. J. Physiol. Pharmacol. 2009, 60, 3–10. [Google Scholar] [PubMed]
- Holm, A.; Hellstrand, P.; Olde, B.; Svensson, D.; Leeb-Lundberg, L.M.; Nilsson, B.O. The G protein-coupled estrogen receptor 1 (GPER1/GPR30) agonist G-1 regulates vascular smooth muscle cell Ca2+ handling. J. Vasc. Res. 2013, 50, 421–429. [Google Scholar] [CrossRef]
- Deschamps, A.M.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef]
- Yu, X.; Ma, H.; Barman, S.A.; Liu, A.T.; Sellers, M.; Stallone, J.N.; Prossnitz, E.R.; White, R.E.; Han, G. Activation of G protein-coupled estrogen receptor induces endothelium-independent relaxation of coronary artery smooth muscle. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E882–E888. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Li, F.; Klussmann, E.; Stallone, J.N.; Han, G. G protein-coupled estrogen receptor 1 mediates relaxation of coronary arteries via cAMP/PKA-dependent activation of MLCP. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E398–E407. [Google Scholar] [CrossRef]
- Lindsey, S.H.; Liu, L.; Chappell, M.C. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids 2014, 81, 99–102. [Google Scholar] [CrossRef]
- De Giusti, V.C.; Orlowski, A.; Ciancio, M.C.; Espejo, M.S.; Gonano, L.A.; Caldiz, C.I.; Vila Petroff, M.G.; Villa-Abrille, M.C.; Aiello, E.A. Aldosterone stimulates the cardiac sodium/bicarbonate cotransporter via activation of the g protein-coupled receptor gpr30. J. Mol. Cell. Cardiol. 2015, 89, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.H.; Cohen, J.A.; Brosnihan, K.B.; Gallagher, P.E.; Chappell, M.C. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology 2009, 150, 3753–3758. [Google Scholar] [CrossRef]
- Jessup, J.A.; Lindsey, S.H.; Wang, H.; Chappell, M.C.; Groban, L. Attenuation of salt-induced cardiac remodeling and diastolic dysfunction by the GPER agonist G-1 in female mRen2.Lewis rats. PLoS ONE 2010, 5, e15433. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jessup, J.A.; Lin, M.S.; Chagas, C.; Lindsey, S.H.; Groban, L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc. Res. 2012, 94, 96–104. [Google Scholar] [CrossRef]
- Kang, S.; Liu, Y.; Sun, D.; Zhou, C.; Liu, A.; Xu, C.; Hao, Y.; Li, D.; Yan, C.; Sun, H. Chronic activation of the G protein-coupled receptor 30 with agonist G-1 attenuates heart failure. PLoS ONE 2012, 7, e48185. [Google Scholar] [CrossRef]
- Weil, B.R.; Manukyan, M.C.; Herrmann, J.L.; Wang, Y.; Abarbanell, A.M.; Poynter, J.A.; Meldrum, D.R. Signaling via GPR30 protects the myocardium from ischemia/reperfusion injury. Surgery 2010, 148, 436–443. [Google Scholar] [CrossRef]
- Kabir, M.E.; Singh, H.; Lu, R.; Olde, B.; Leeb-Lundberg, L.M.; Bopassa, J.C. G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3beta Pathway after Ischemia/Reperfusion. PLoS ONE 2015, 10, e0135988. [Google Scholar] [CrossRef]
- Feng, Y.; Madungwe, N.B.; da Cruz Junho, C.V.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 2017, 174, 4329–4344. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, A.M.; Gonzalez Arbelaez, L.F.; Ciocci Pardo, A.; Mosca, S.; Lofeudo, J.M.; Velez Rueda, J.O.; Aiello, E.A.; De Giusti, V.C. Chronic GPER activation prevents ischemia/reperfusion injury in ovariectomized rats. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130060. [Google Scholar] [CrossRef] [PubMed]
- Moreyra, A.E.; Gelpi, R.J.; Mosca, S.M.; Cingolani, H.E. Chronic administration of nicardipine attenuates myocardial stunning in isolated rabbit hearts. J. Mol. Cell. Cardiol. 1994, 26, 979–984. [Google Scholar] [CrossRef] [PubMed]
- de Cingolani, G.E.; Mosca, S.M.; Moreyra, A.E.; Cingolani, H.E. Chronic nifedipine treatment diminishes cardiac inotropic response to nifedifine: Functional upregulation of dihydropyridine receptors. J. Cardiovasc. Pharmacol. 1996, 27, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Pardo, A.C.; Diaz Zegarra, L.A.; Gonzalez Arbelaez, L.F.; Ibanez, A.M.; Diaz, R.G.; Aiello, E.A.; Mosca, S.M. Cardioprotective effects of N-methylacetazolamide mediated by inhibition of L-type Ca(2+) channel current. Biochim. Biophys. Acta Gen. Subj. 2022, 1866, 130098. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.M.; Colecraft, H.M. L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc. Res. 2013, 98, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Kamp, T.J.; Hell, J.W. Regulation of cardiac L-type calcium channels by protein kinase A and protein kinase C. Circ. Res. 2000, 87, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Carl, S.L.; Felix, K.; Caswell, A.H.; Brandt, N.R.; Ball, W.J., Jr.; Vaghy, P.L.; Meissner, G.; Ferguson, D.G. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J. Cell Biol. 1995, 129, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Hool, L.C. Elucidating the role of the L-type calcium channel in excitability and energetics in the heart: The ISHR 2020 Research Achievement Award Lecture. J. Mol. Cell. Cardiol. 2022, 172, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Spinale, F.G. L-type calcium channel abundance and function with cardiac hypertrophy and failure: A review. J. Mol. Cell. Cardiol. 1998, 30, 1899–1916. [Google Scholar] [CrossRef]
- Grohe, C.; Kahlert, S.; Lobbert, K.; Meyer, R.; Linz, K.W.; Karas, R.H.; Vetter, H. Modulation of hypertensive heart disease by estrogen. Steroids 1996, 61, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.; Linz, K.W.; Surges, R.; Meinardus, S.; Vees, J.; Hoffmann, A.; Windholz, O.; Grohe, C. Rapid modulation of L-type calcium current by acutely applied oestrogens in isolated cardiac myocytes from human, guinea-pig and rat. Exp. Physiol. 1998, 83, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Firth, J.M.; Francis, A.J.; Alvarez-Laviada, A.; MacLeod, K.T. Effect of ovariectomy on intracellular Ca2+ regulation in guinea pig cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H1031–H1043. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, N.D.; Krust, A.; Collins, P.; MacLeod, K.T. Genomic deletion of estrogen receptors ERalpha and ERbeta does not alter estrogen-mediated inhibition of Ca2+ influx and contraction in murine cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2421–H2427. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, V.; Wauson, E.; Christian, D.; Clayton, S.; Giles, J.; Tran, Q.K. Regulation of beta adrenoceptor-mediated myocardial contraction and calcium dynamics by the G protein-coupled estrogen receptor 1. Biochem. Pharmacol. 2020, 171, 113727. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, A.; De Giusti, V.C.; Ciancio, M.C.; Espejo, M.S.; Aiello, E.A. The cardiac electrogenic sodium/bicarbonate cotransporter (NBCe1) is activated by aldosterone through the G protein-coupled receptor 30 (GPR 30). Channels 2016, 10, 428–434. [Google Scholar] [CrossRef]
- Rocca, C.; Femmino, S.; Aquila, G.; Granieri, M.C.; De Francesco, E.M.; Pasqua, T.; Rigiracciolo, D.C.; Fortini, F.; Cerra, M.C.; Maggiolini, M.; et al. Notch1 Mediates Preconditioning Protection Induced by GPER in Normotensive and Hypertensive Female Rat Hearts. Front. Physiol. 2018, 9, 521. [Google Scholar] [CrossRef] [PubMed]
- Carnes, C.A.; Janssen, P.M.; Ruehr, M.L.; Nakayama, H.; Nakayama, T.; Haase, H.; Bauer, J.A.; Chung, M.K.; Fearon, I.M.; Gillinov, A.M.; et al. Atrial glutathione content, calcium current, and contractility. J. Biol. Chem. 2007, 282, 28063–28073. [Google Scholar] [CrossRef]
- Sun, J.; Picht, E.; Ginsburg, K.S.; Bers, D.M.; Steenbergen, C.; Murphy, E. Hypercontractile female hearts exhibit increased S-nitrosylation of the L-type Ca2+ channel alpha1 subunit and reduced ischemia/reperfusion injury. Circ. Res. 2006, 98, 403–411. [Google Scholar] [CrossRef]
- Wang, Y.; Youm, J.B.; Jin, C.Z.; Shin, D.H.; Zhao, Z.H.; Seo, E.Y.; Jang, J.H.; Kim, S.J.; Jin, Z.H.; Zhang, Y.H. Modulation of L-type Ca2+ channel activity by neuronal nitric oxide synthase and myofilament Ca2+ sensitivity in cardiac myocytes from hypertensive rat. Cell Calcium 2015, 58, 264–274. [Google Scholar] [CrossRef]
- Burkard, N.; Rokita, A.G.; Kaufmann, S.G.; Hallhuber, M.; Wu, R.; Hu, K.; Hofmann, U.; Bonz, A.; Frantz, S.; Cartwright, E.J.; et al. Conditional neuronal nitric oxide synthase overexpression impairs myocardial contractility. Circ. Res. 2007, 100, e32–e44. [Google Scholar] [CrossRef]
- Gonzalez, D.R.; Treuer, A.; Sun, Q.A.; Stamler, J.S.; Hare, J.M. S-Nitrosylation of cardiac ion channels. J. Cardiovasc. Pharmacol. 2009, 54, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.J.; Firth, J.M.; Sanchez-Alonso, J.L.; Gorelik, J.; MacLeod, K.T. GPER limits adverse changes to Ca2+ signalling and arrhythmogenic activity in ovariectomised guinea pig cardiomyocytes. Front. Physiol. 2022, 13, 1023755. [Google Scholar] [CrossRef] [PubMed]
- Marchese, A.; Paing, M.M.; Temple, B.R.; Trejo, J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 601–629. [Google Scholar] [CrossRef] [PubMed]
- Scita, G.; Di Fiore, P.P. The endocytic matrix. Nature 2010, 463, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Jean-Alphonse, F.; Hanyaloglu, A.C. Regulation of GPCR signal networks via membrane trafficking. Mol. Cell. Endocrinol. 2011, 331, 205–214. [Google Scholar] [CrossRef]
- Qin, W.; Cao, L.; Massey, I.Y. Role of PI3K/Akt signaling pathway in cardiac fibrosis. Mol. Cell. Biochem. 2021, 476, 4045–4059. [Google Scholar] [CrossRef] [PubMed]
- Aiello, E.A.; Petroff, M.G.; Mattiazzi, A.R.; Cingolani, H.E. Evidence for an electrogenic Na+-HCO3− symport in rat cardiac myocytes. J. Physiol. 1998, 512 Pt 1, 137–148. [Google Scholar] [CrossRef]
- Espejo, M.S.; Aiello, I.; Sepulveda, M.; Vila Petroff, M.G.; Aiello, E.A.; De Giusti, V.C. The reduced myofilament responsiveness to calcium contributes to the negative force-frequency relationship in rat cardiomyocytes: Role of reactive oxygen species and p-38 map kinase. Pflugers Arch. 2017, 469, 1663–1673. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diaz-Zegarra, L.A.; Espejo, M.S.; Ibañez, A.M.; Rando, M.E.; Pagola, L.E.; De Giusti, V.C.; Aiello, E.A. Activation of G Protein-Coupled Estrogen Receptor (GPER) Negatively Modulates Cardiac Excitation–Contraction Coupling (ECC) through the PI3K/NOS/NO Pathway. Int. J. Mol. Sci. 2024, 25, 8993. https://doi.org/10.3390/ijms25168993
Diaz-Zegarra LA, Espejo MS, Ibañez AM, Rando ME, Pagola LE, De Giusti VC, Aiello EA. Activation of G Protein-Coupled Estrogen Receptor (GPER) Negatively Modulates Cardiac Excitation–Contraction Coupling (ECC) through the PI3K/NOS/NO Pathway. International Journal of Molecular Sciences. 2024; 25(16):8993. https://doi.org/10.3390/ijms25168993
Chicago/Turabian StyleDiaz-Zegarra, Leandro A., María S. Espejo, Alejandro M. Ibañez, Mónica E. Rando, Lucia E. Pagola, Verónica C. De Giusti, and Ernesto A. Aiello. 2024. "Activation of G Protein-Coupled Estrogen Receptor (GPER) Negatively Modulates Cardiac Excitation–Contraction Coupling (ECC) through the PI3K/NOS/NO Pathway" International Journal of Molecular Sciences 25, no. 16: 8993. https://doi.org/10.3390/ijms25168993