
Chronic Neuronal Hyperexcitation Exacerbates Tau Propagation in a Mouse Model of Tauopathy

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
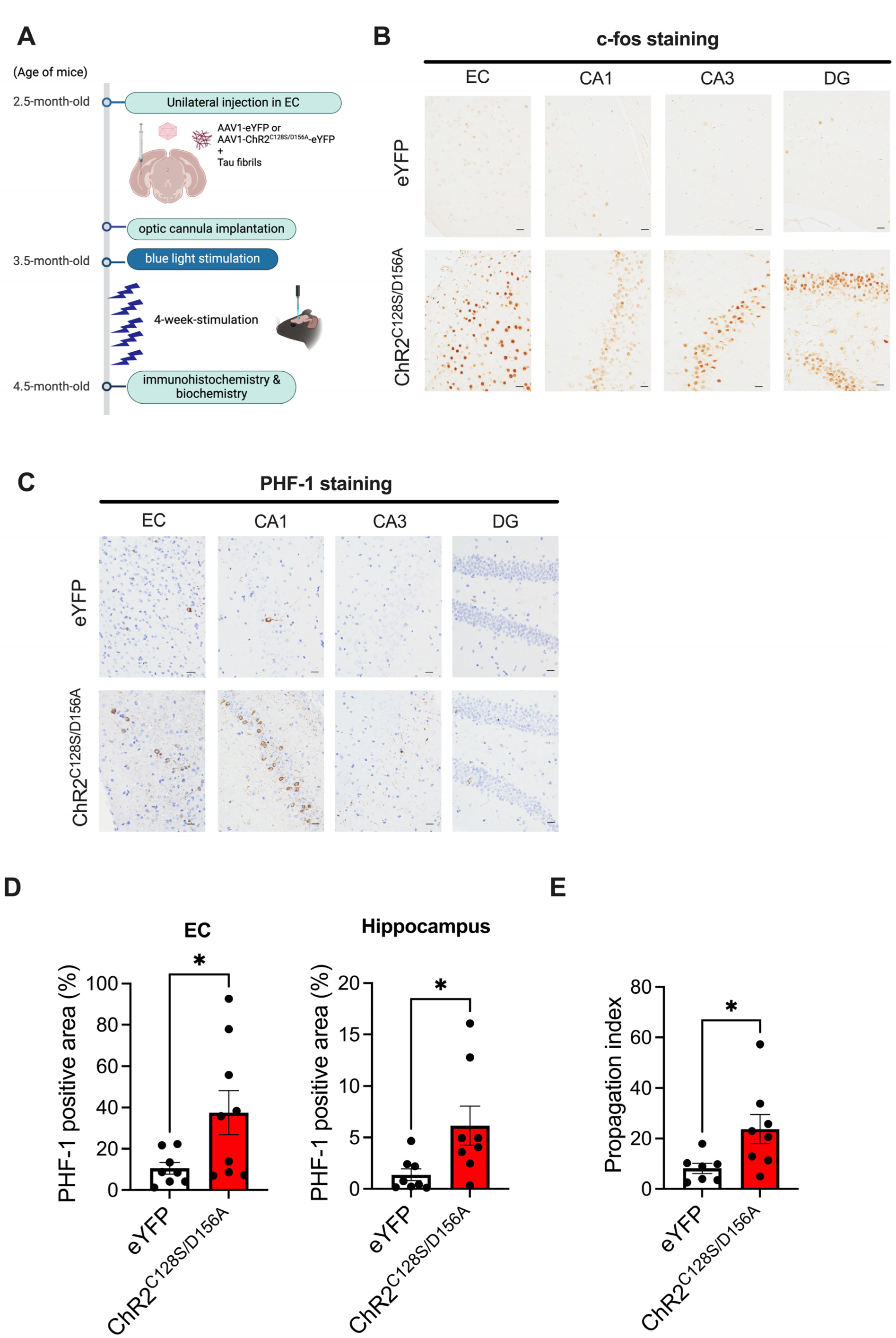
2.1. Tau Pathology Propagation from EC to Hippocampus Following Tau Fibril Microinjection
2.2. Immunohistochemical Analysis of the Effects of Chronic Neuronal Stimulation on Tau Propagation
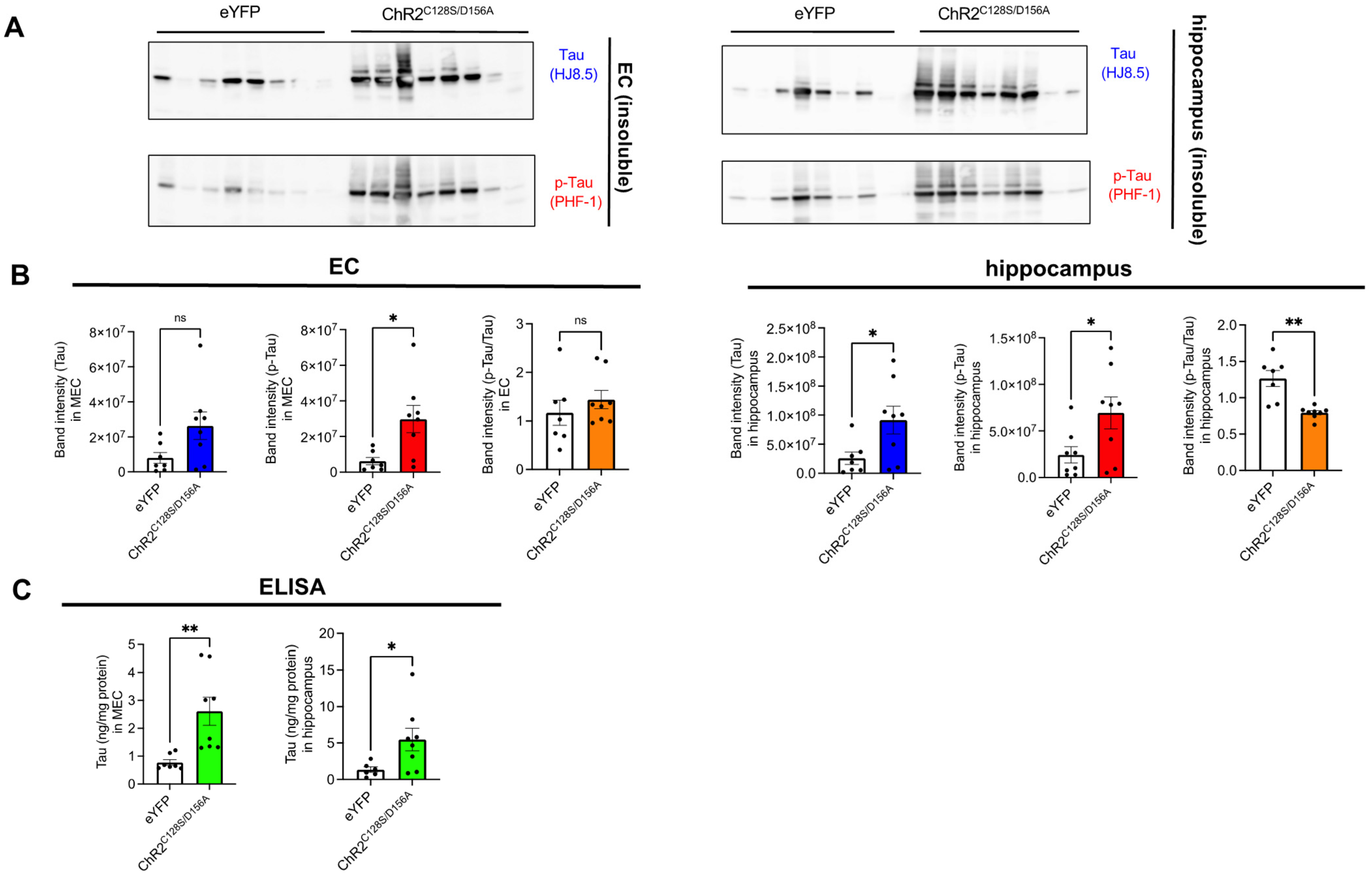
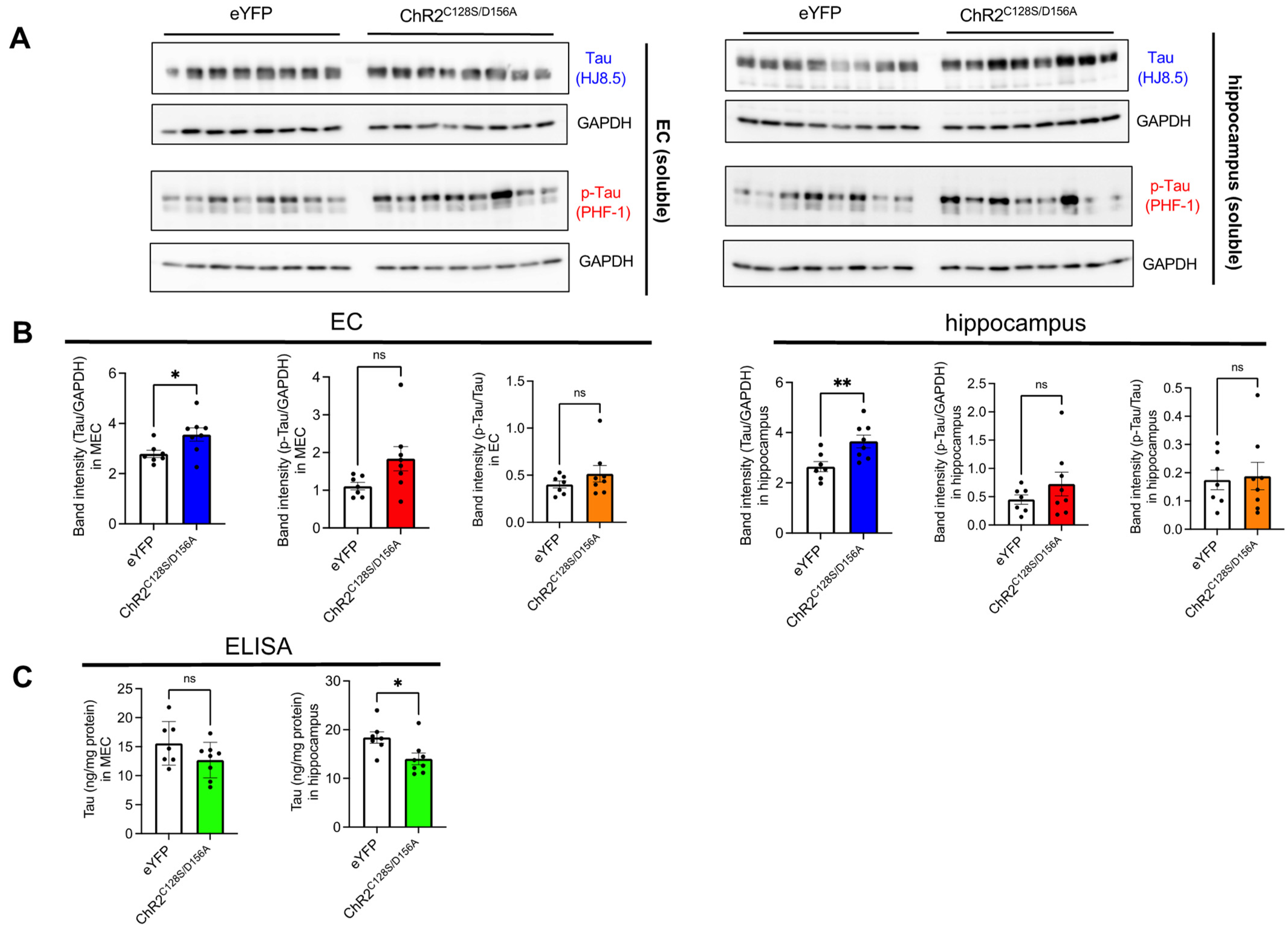
2.3. Biochemical Analysis of the Effects of Chronic Neuronal Stimulation on Tau Propagation
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Recombinant Tau Purification and Fibrilization
4.3. Stereotaxic Injection of AAV and Tau Fibrils
4.4. Optogenetics
4.5. Immunohistochemistry
4.6. Biochemistry
4.7. Immunoblots
4.8. Image Analysis
4.9. Human-Specific Tau ELISA
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Braak, E. Staging of Alzheimer’s Disease-Related Neurofibrillary Changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hoenig, M.C.; Bischof, G.N.; Seemiller, J.; Hammes, J.; Kukolja, J.; Onur, Ö.A.; Jessen, F.; Fliessbach, K.; Neumaier, B.; Fink, G.R.; et al. Networks of Tau Distribution in Alzheimer’s Disease. Brain 2018, 141, 568–581. [Google Scholar] [CrossRef] [PubMed]
- La Joie, R.; Visani, A.V.; Baker, S.L.; Brown, J.A.; Bourakova, V.; Cha, J.; Chaudhary, K.; Edwards, L.; Iaccarino, L.; Janabi, M.; et al. Prospective Longitudinal Atrophy in Alzheimer’s Disease Correlates with the Intensity and Topography of Baseline Tau-PET. Sci. Transl. Med. 2020, 12, eaau5732. [Google Scholar] [CrossRef] [PubMed]
- Ossenkoppele, R.; Smith, R.; Mattsson-Carlgren, N.; Groot, C.; Leuzy, A.; Strandberg, O.; Palmqvist, S.; Olsson, T.; Jögi, J.; Stormrud, E.; et al. Accuracy of Tau Positron Emission Tomography as a Prognostic Marker in Preclinical and Prodromal Alzheimer Disease: A Head-to-Head Comparison against Amyloid Positron Emission Tomography and Magnetic Resonance Imaging. JAMA Neurol. 2021, 78, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and Spreading of Tauopathy in Transgenic Mouse Brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Iba, M.; Guo, J.L.; McBride, J.D.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Synthetic Tau Fibrils Mediate Transmission of Neurofibrillary Tangles in a Transgenic Mouse Model of Alzheimer’s-Like Tauopathy. J. Neurosci. 2013, 33, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Stancu, I.C.; Vasconcelos, B.; Ris, L.; Wang, P.; Villers, A.; Peeraer, E.; Buist, A.; Terwel, D.; Baatsen, P.; Oyelami, T.; et al. Templated Misfolding of Tau by Prion-like Seeding along Neuronal Connections Impairs Neuronal Network Function and Associated Behavioral Outcomes in Tau Transgenic Mice. Acta Neuropathol. 2015, 129, 875–894. [Google Scholar] [CrossRef] [PubMed]
- Schoonhoven, D.N.; Coomans, E.M.; Millán, A.P.; Van Nifterick, A.M.; Visser, D.; Ossenkoppele, R.; Tuncel, H.; Van Der Flier, W.M.; Golla, S.S.V.; Scheltens, P.; et al. Tau Protein Spreads through Functionally Connected Neurons in Alzheimer’s Disease: A Combined MEG/PET Study. Brain 2023, 146, 4040–4054. [Google Scholar] [CrossRef] [PubMed]
- Montal, V.; Diez, I.; Kim, C.M.; Orwig, W.; Bueichekú, E.; Gutiérrez-Zúñiga, R.; Bejanin, A.; Pegueroles, J.; Dols-Icardo, O.; Vannini, P.; et al. Network Tau Spreading Is Vulnerable to the Expression Gradients of APOE and Glutamatergic-Related Genes. Sci. Transl. Med. 2022, 14, eabn7273. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Corjuc, B.T.; Oakley, D.H.; Nobuhara, C.K.; Bannon, R.N.; Chase, A.; Commins, C.; Gonzalez, J.A.; Dooley, P.M.; Frosch, M.P.; et al. Synaptic Tau Seeding Precedes Tau Pathology in Human Alzheimer’s Disease Brain. Front. Neurosci. 2018, 12, 267. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Davies, C.; Sirisi, S.; Lee, J.E.; Simzer, E.M.; Tzioras, M.; Querol-Vilaseca, M.; Sánchez-Aced, É.; Chang, Y.Y.; Holt, K.; et al. Synaptic Oligomeric Tau in Alzheimer’s Disease—A Potential Culprit in the Spread of Tau Pathology through the Brain. Neuron 2023, 111, 2170–2183.e6. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Salat, D.H.; Greve, D.N.; Chua, E.F.; Rand-Giovannetti, E.; Rentz, D.M.; Bertram, L.; Mullin, K.; Tanzi, R.E.; Blacker, D.; et al. Increased Hippocampal Activation in Mild Cognitive Impairment Compared to Normal Aging and AD. Neurology 2005, 65, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.S.; Yousem, D.M.; Cristinzio, C.; Kusevic, I.; Yassa, M.A.; Caffo, B.S.; Zeger, S.L. Familial Risk for Alzheimer’s Disease Alters FMRI Activation Patterns. Brain 2006, 129, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological Release of Endogenous Tau Is Stimulated by Neuronal Activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M.; et al. Neuronal Activity Regulates Extracellular Tau in Vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal Activity Enhances Tau Propagation and Tau Pathology in Vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical Excitation/Inhibition Balance in Information Processing and Social Dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tanei, Z.; Hashimoto, T.; Wakabayashi, T.; Okuno, H.; Naka, Y.; Yizhar, O.; Fenno, L.E.; Fukayama, M.; Bito, H.; et al. Chronic Optogenetic Activation Augments Aβ Pathology in a Mouse Model of Alzheimer Disease. Cell Rep. 2015, 11, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.M.; Wells, J.A.; Ma, D.; Wallis, L.; Park, D.; Llewellyn, S.K.; Ahmed, Z.; Lythgoe, M.F.; Harrison, I.F. Glymphatic Inhibition Exacerbates Tau Propagation in an Alzheimer’s Disease Model. Alzheimer’s Res. Ther. 2024, 16, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. LRP1 Is a Master Regulator of Tau Uptake and Spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Wegmann, S.; Maury, E.A.; Kirk, M.J.; Saqran, L.; Roe, A.; DeVos, S.L.; Nicholls, S.; Fan, Z.; Takeda, S.; Cagsal-Getkin, O.; et al. Removing Endogenous Tau Does Not Prevent Tau Propagation yet Reduces Its Neurotoxicity. EMBO J. 2015, 34, 3028–3041. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.K.; Gentzel, R.; Usenovic, M.; Gretzula, C.; Ware, C.; Parmentier-Batteur, S.; Schachter, J.B.; Zariwala, H.A. Pharmacogenetic Neuronal Stimulation Increases Human Tau Pathology and Trans-Synaptic Spread of Tau to Distal Brain Regions in Mice. Neurobiol. Dis. 2018, 118, 161–176. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The Release and Trans-Synaptic Transmission of Tau via Exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Yamada, K.; Satake, K.; Nishida, I.; Heuberger, M.; Kuwahara, T.; Iwatsubo, T. Seeding Activity-Based Detection Uncovers the Different Release Mechanisms of Seed-Competent Tau Versus Inert Tau via Lysosomal Exocytosis. Front. Neurosci. 2019, 13, 1258. [Google Scholar] [CrossRef] [PubMed]
- Padamsey, Z.; McGuinness, L.; Bardo, S.J.; Reinhart, M.; Tong, R.; Hedegaard, A.; Hart, M.L.; Emptage, N.J. Activity-Dependent Exocytosis of Lysosomes Regulates the Structural Plasticity of Dendritic Spines. Neuron 2017, 93, 132–146. [Google Scholar] [CrossRef]
- Fauré, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes Are Released by Cultured Cortical Neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Cheung, G.; Jupp, O.J.; Cousin, M.A. Activity-Dependent Bulk Endocytosis and Clathrin-Dependent Endocytosis Replenish Specific Synaptic Vesicle Pools in Central Nerve Terminals. J. Neurosci. 2010, 30, 8151–8161. [Google Scholar] [CrossRef] [PubMed]
- Kawles, A.; Minogue, G.; Zouridakis, A.; Keszycki, R.; Gill, N.; Nassif, C.; Coventry, C.; Zhang, H.; Rogalski, E.; Flanagan, M.E.; et al. Differential Vulnerability of the Dentate Gyrus to Tauopathies in Dementias. Acta Neuropathol. Commun. 2023, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Polydoro, M.; Suárez-Calvet, M.; William, C.; Adamowicz, D.H.; Kopeikina, K.J.; Pitstick, R.; Sahara, N.; Ashe, K.H.; Carlson, G.A.; et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [Google Scholar] [CrossRef]
- Delpech, J.C.; Pathak, D.; Varghese, M.; Kalavai, S.V.; Hays, E.C.; Hof, P.R.; Evan Johnson, W.; Ikezu, S.; Medalla, M.; Luebke, J.I.; et al. Wolframin-1-Expressing Neurons in the Entorhinal Cortex Propagate Tau to CA1 Neurons and Impair Hippocampal Memory in Mice. Sci. Transl. Med. 2021, 13, eabe8455. [Google Scholar] [CrossRef]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic Activity in Alzheimer’s Disease: Causes and Clinical Relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Pollanen, M.S.; Onzivua, S.; Robertson, J.; McKeever, P.M.; Olawa, F.; Kitara, D.L.; Fong, A. Nodding Syndrome in Uganda Is a Tauopathy. Acta Neuropathol. 2018, 136, 691–697. [Google Scholar] [CrossRef]
- Thom, M.; Liu, J.Y.W.; Thompson, P.; Phadke, R.; Narkiewicz, M.; Martinian, L.; Marsdon, D.; Koepp, M.; Caboclo, L.; Catarino, C.B.; et al. Neurofibrillary Tangle Pathology and Braak Staging in Chronic Epilepsy in Relation to Traumatic Brain Injury and Hippocampal Sclerosis: A Post-Mortem Study. Brain 2011, 134, 2969–2981. [Google Scholar] [CrossRef]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense Reduction of Tau in Adult Mice Protects against Seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef]
- Gomez-Murcia, V.; Sandau, U.; Ferry, B.; Parrot, S.; Laurent, C.; Basquin, M.; Buée, L.; Boison, D.; Blum, D. Hyperexcitability and Seizures in the THY-Tau22 Mouse Model of Tauopathy. Neurobiol. Aging 2020, 94, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Yamada, K.; Nishiyama, R.; Hashimoto, T.; Nishida, I.; Abe, Y.; Yasui, M.; Iwatsubo, T. Glymphatic System Clears Extracellular Tau and Protects from Tau Aggregation and Neurodegeneration. J. Exp. Med. 2022, 219, e20211275. [Google Scholar] [CrossRef]
- Yamada, K.; Patel, T.K.; Hochgräfe, K.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Mandelkow, E.-M.; Holtzman, D.M. Analysis of in Vivo Turnover of Tau in a Mouse Model of Tauopathy. Mol. Neurodegener. 2015, 10, 55. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishida, I.; Yamada, K.; Sakamoto, A.; Wakabayashi, T.; Iwatsubo, T. Chronic Neuronal Hyperexcitation Exacerbates Tau Propagation in a Mouse Model of Tauopathy. Int. J. Mol. Sci. 2024, 25, 9004. https://doi.org/10.3390/ijms25169004
Nishida I, Yamada K, Sakamoto A, Wakabayashi T, Iwatsubo T. Chronic Neuronal Hyperexcitation Exacerbates Tau Propagation in a Mouse Model of Tauopathy. International Journal of Molecular Sciences. 2024; 25(16):9004. https://doi.org/10.3390/ijms25169004
Chicago/Turabian StyleNishida, Itaru, Kaoru Yamada, Asami Sakamoto, Tomoko Wakabayashi, and Takeshi Iwatsubo. 2024. "Chronic Neuronal Hyperexcitation Exacerbates Tau Propagation in a Mouse Model of Tauopathy" International Journal of Molecular Sciences 25, no. 16: 9004. https://doi.org/10.3390/ijms25169004
APA StyleNishida, I., Yamada, K., Sakamoto, A., Wakabayashi, T., & Iwatsubo, T. (2024). Chronic Neuronal Hyperexcitation Exacerbates Tau Propagation in a Mouse Model of Tauopathy. International Journal of Molecular Sciences, 25(16), 9004. https://doi.org/10.3390/ijms25169004