
Discovery of Novel Amino Acids (Analogues)-Substituted Thiophene[3,2-d]pyrimidine Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Design, Synthesis, and Biological Evaluation
 , , , and
, , , and
Abstract
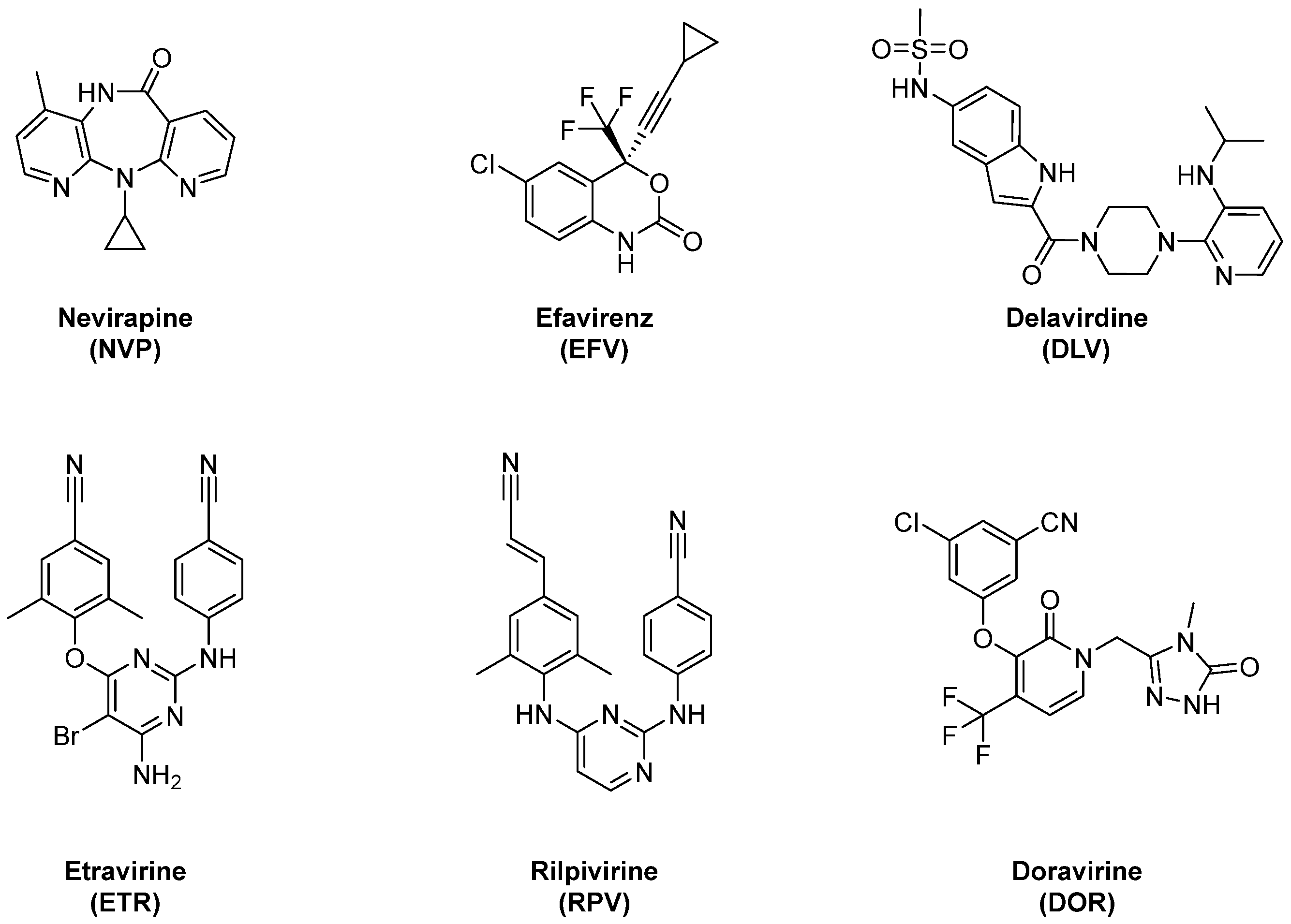
:1. Introduction
2. Results and Discussion
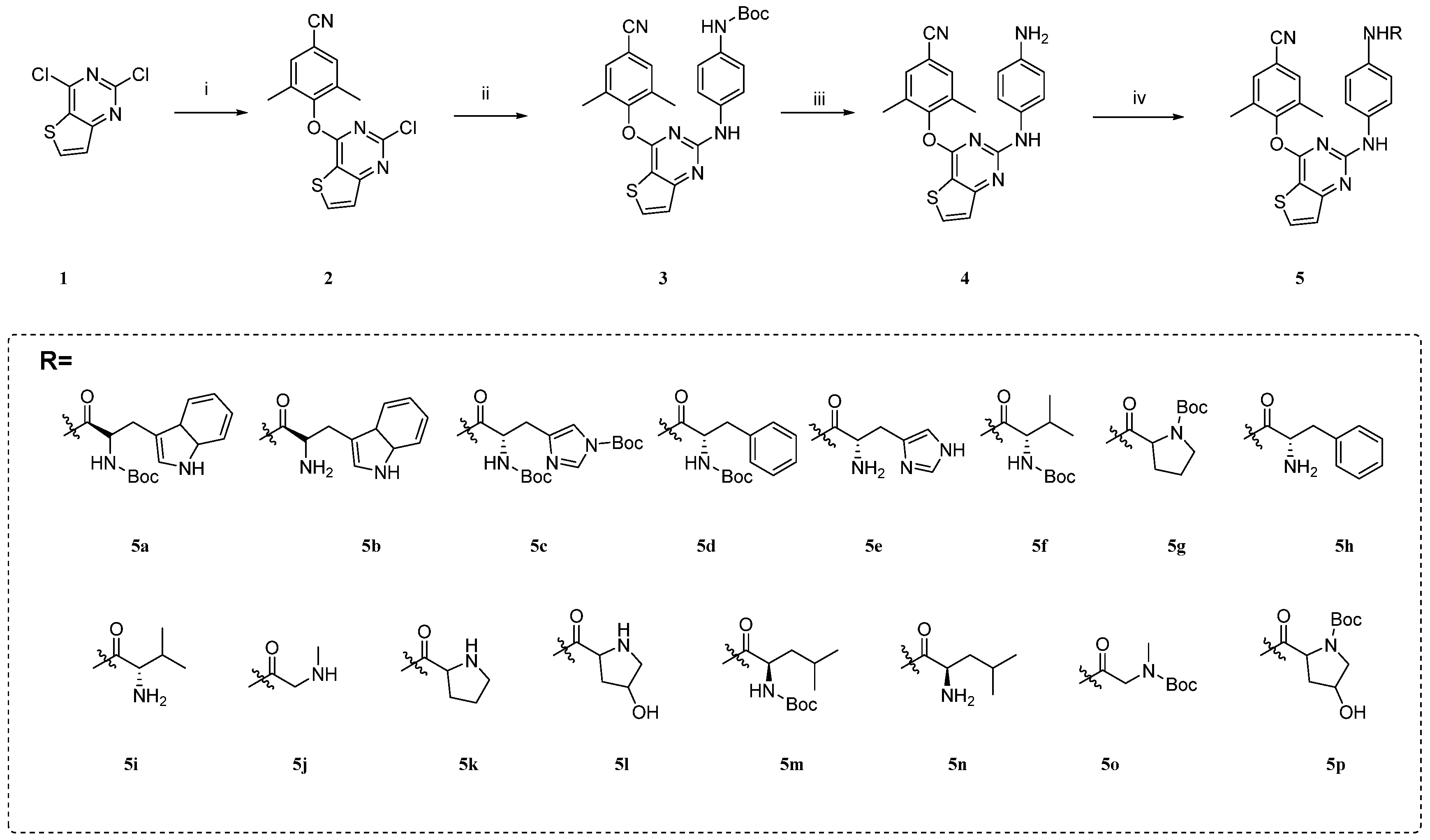
2.1. Chemistry
2.2. Anti-HIV Activity Evaluation
2.3. Inhibition of HIV-1 RT
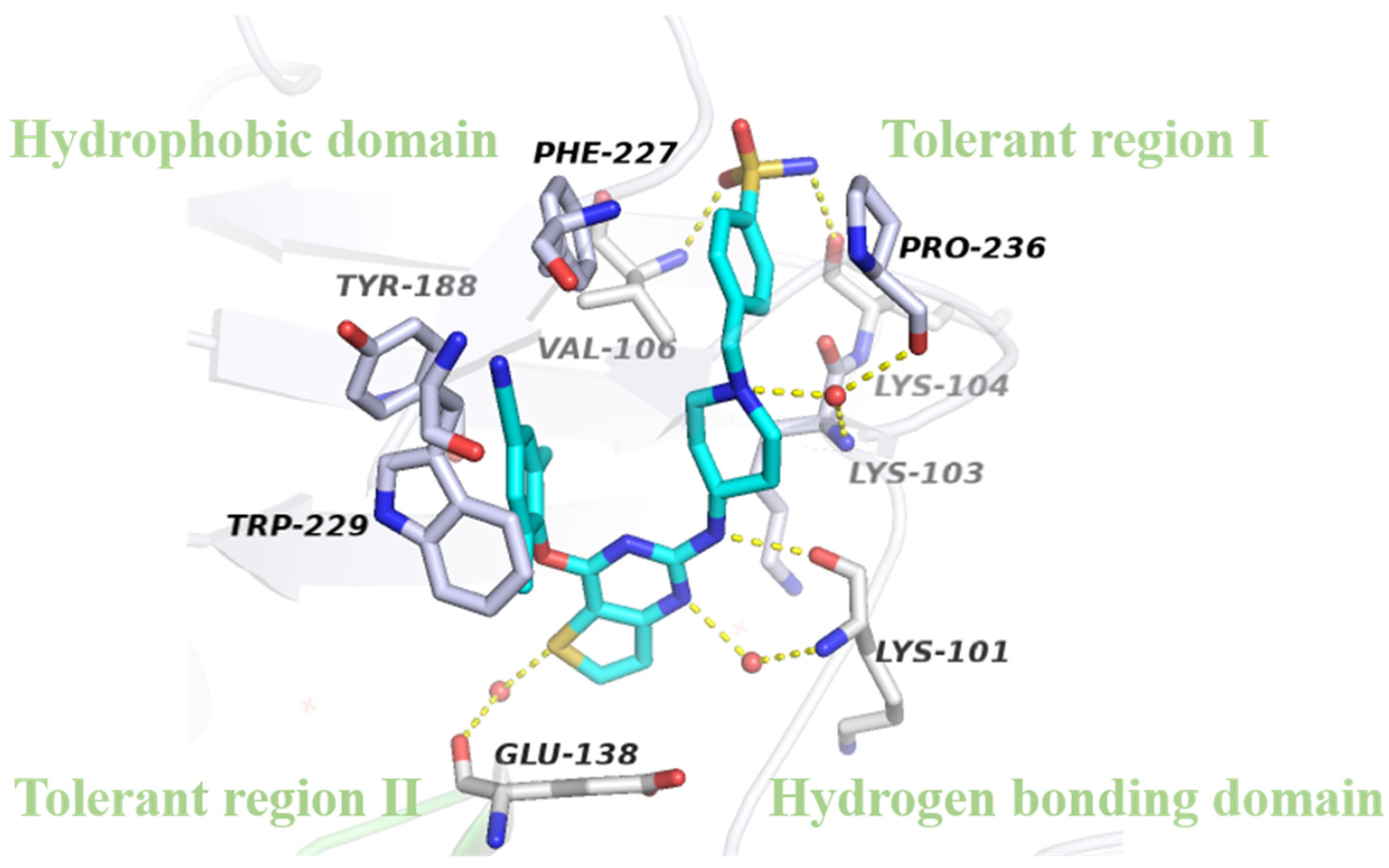
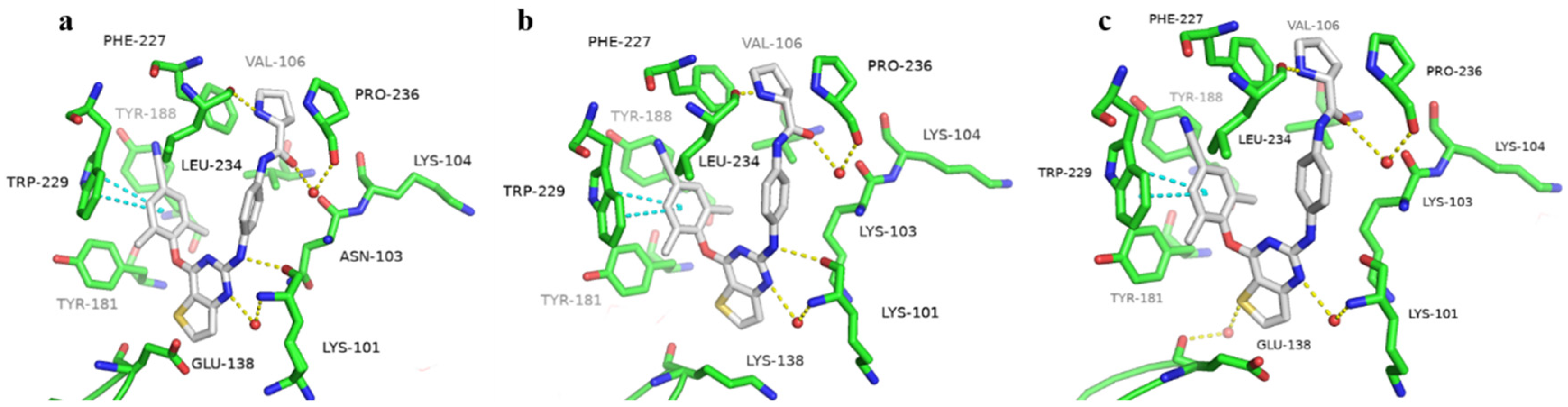
2.4. Molecular Modeling Analysis
3. Methods and Materials
3.1. Chemistry
3.2. In Vitro Anti-HIV Assay
3.3. HIV-1 RT Inhibition Assay
3.4. Molecular Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- HIV Data and Statistics. 2023. Available online: https://www.who.int/teams/global-hiv-hepatitis-and-stis-programmes/hiv/strategic-information/hiv-data-and-statistics (accessed on 1 July 2024).
- Martín-Alonso, S.; Kang, D.; Martínez Del Río, J.; Luczkowiak, J.; Frutos-Beltrán, E.; Zhang, L.; Cheng, X.; Liu, X.; Zhan, P.; Menéndez-Arias, L. Novel RNase H Inhibitors Blocking RNA-directed Strand Displacement DNA Synthesis by HIV-1 Reverse Transcriptase. J. Mol. Biol. 2022, 434, 167507. [Google Scholar] [CrossRef] [PubMed]
- Battini, L.; Bollini, M. Challenges and approaches in the discovery of human immunodeficiency virus type-1 non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev. 2019, 39, 1235–1273. [Google Scholar] [CrossRef]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem. Rev. 2021, 121, 3271–3296. [Google Scholar] [CrossRef]
- Bertoletti, N.; Chan, A.H.; Schinazi, R.F.; Anderson, K.S. Post-Catalytic Complexes with Emtricitabine or Stavudine and HIV-1 Reverse Transcriptase Reveal New Mechanistic Insights for Nucleotide Incorporation and Drug Resistance. Molecules 2020, 25, 4868. [Google Scholar] [CrossRef]
- Li, G.; Wang, Y.; De Clercq, E. Approved HIV reverse transcriptase inhibitors in the past decade. Acta Pharm. Sin. B 2022, 12, 1567–1590. [Google Scholar] [CrossRef] [PubMed]
- Agneskog, E.; Nowak, P.; Källander, C.F.; Sönnerborg, A. Evaluation of etravirine resistance in clinical samples by a simple phenotypic test. J. Med. Virol. 2013, 85, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, Z.; Julias, J.G.; Boyer, P.L.; Kewalramani, V.N.; Hughes, S.H. The level of reverse transcriptase (RT) in human immunodeficiency virus type 1 particles affects susceptibility to nonnucleoside RT inhibitors but not to lamivudine. J. Virol. 2006, 80, 2578–2581. [Google Scholar] [CrossRef]
- Ding, L.; Zhuang, C.; Chen, F. Druggability modification strategies of the diarylpyrimidine-type non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev. 2021, 41, 1255–1290. [Google Scholar] [CrossRef]
- Singh, A.K.; Martinez, S.E.; Gu, W.; Nguyen, H.; Schols, D.; Herdewijn, P.; De Jonghe, S.; Das, K. Sliding of HIV-1 reverse transcriptase over DNA creates a transient P pocket - targeting P-pocket by fragment screening. Nat. Commun. 2021, 12, 7127. [Google Scholar] [CrossRef]
- Gu, S.X.; Zhu, Y.Y.; Wang, C.; Wang, H.F.; Liu, G.Y.; Cao, S.; Huang, L. Recent discoveries in HIV-1 reverse transcriptase inhibitors. Curr. Opin. Pharmacol. 2020, 54, 166–172. [Google Scholar] [CrossRef]
- Zhao, L.M.; Pannecouque, C.; Clercq, E.; Wang, S.; Chen, F.E. Structure-based design of novel heterocycle-substituted ATDP analogs as non-nucleoside reverse transcriptase inhibitors with improved selectivity and solubility. Acta Pharm. Sin. B 2023, 13, 4906–4917. [Google Scholar] [CrossRef]
- Ambrose, Z.; Herman, B.D.; Sheen, C.W.; Zelina, S.; Moore, K.L.; Tachedjian, G.; Nissley, D.V.; Sluis-Cremer, N. The human immunodeficiency virus type 1 nonnucleoside reverse transcriptase inhibitor resistance mutation I132M confers hypersensitivity to nucleoside analogs. J. Virol. 2009, 83, 3826–3833. [Google Scholar] [CrossRef]
- Olson, A.; Bannert, N.; Sönnerborg, A.; de Mendoza, C.; Price, M.; Zangerle, R.; Chaix, M.L.; Prins, M.; Kran, A.B.; Gill, J.; et al. Temporal trends of transmitted HIV drug resistance in a multinational seroconversion cohort. AIDS 2018, 32, 161–169. [Google Scholar] [CrossRef]
- Vanangamudi, M.; Kurup, S.; Namasivayam, V. Non-nucleoside reverse transcriptase inhibitors (NNRTIs): A brief overview of clinically approved drugs and combination regimens. Curr. Opin. Pharmacol. 2020, 54, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Beyrer, C.; Pozniak, A. HIV Drug Resistance—An Emerging Threat to Epidemic Control. N. Engl. J. Med. 2017, 377, 1605–1607. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Li, J.F.; Morris, L.; Martinson, N.; Gray, G.; McIntyre, J.; Heneine, W. Emergence of drug-resistant HIV-1 after intrapartum administration of single-dose nevirapine is substantially underestimated. J. Infect. Dis. 2005, 192, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Ruiz, F.X.; Feng, D.; Pilch, A.; Zhao, T.; Wei, F.; Wang, Z.; Sun, Y.; Fang, Z.; De Clercq, E.; et al. Discovery and Characterization of Fluorine-Substituted Diarylpyrimidine Derivatives as Novel HIV-1 NNRTIs with Highly Improved Resistance Profiles and Low Activity for the hERG Ion Channel. J. Med. Chem. 2020, 63, 1298–1312. [Google Scholar] [CrossRef]
- Chow, S.Y.; Nelson, A. Embarking on a Chemical Space Odyssey. J. Med. Chem. 2017, 60, 3591–3593. [Google Scholar] [CrossRef]
- Pauwels, R.; Balzarini, J.; Baba, M.; Snoeck, R.; Schols, D.; Herdewijn, P.; Desmyter, J.; De Clercq, E. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 1988, 20, 309–321. [Google Scholar] [CrossRef]
- Pannecouque, C.; Daelemans, D.; De Clercq, E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: Revisited 20 years later. Nat. Protoc. 2008, 3, 427–434. [Google Scholar] [CrossRef]
- Suzuki, K.; Craddock, B.P.; Okamoto, N.; Kano, T.; Steigbigel, R.T. Poly A-linked colorimetric microtiter plate assay for HIV reverse transcriptase. J. Virol. Methods 1993, 44, 189–198. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compds | R | a EC50 (μM) | b CC50 (μM) | c SI | ||
|---|---|---|---|---|---|---|
| IIIB | RES056 | IIIB | RES056 | |||
| 5a | 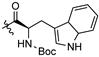 | 0.940 ± 0.278 | >185.66 | >185.66 | >197 | - |
| 5b |  | 0.089 ± 0.043 | >22.401 | 22.394 ± 0.560 | 250 | <1 |
| 5c | 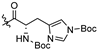 | 0.672 ± 0.648 | >19.607 | 19.601 ± 0.720 | 29 | <1 |
| 5d | 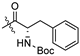 | 0.771 ± 0.236 | >197.086 | >197.086 | >256 | - |
| 5e | 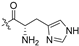 | 0.052 ± 0.022 | ≥4.884 | 25.105 ± 2.301 | 484 | ≤5 |
| 5f | 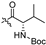 | 0.848 ± 0.247 | >213.223 | >213.223 | >251 | - |
| 5g |  | 0.924 ± 0.242 | >190.630 | 190.630 | 206 | <1 |
| 5h | 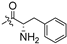 | 0.055 ± 0.012 | 8.474 ± 0.483 | 23.709 ± 0.521 | 429 | 3 |
| 5i |  | 0.042 ± 0.008 | ≥6.623 | 6.535 ± 1.343 | 157 | ≤1 |
| 5j |  | 0.242 ± 0.065 | >17.272 | 17.280 ± 11.530 | 72 | <1 |
| 5k | 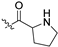 | 0.054 ± 0.018 | 7.530 ± 0.280 | 27.901 ± 0.910 | 513 | 4 |
| 5l |  | 0.059 ± 0.010 | >26.765 | 26.765 ± 1.071 | 451 | <1 |
| 5m | 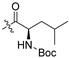 | 9.865 ± 1.182 | >208.247 | >208.247 | >21 | - |
| 5n | 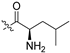 | 0.062 ± 0.019 | 5.502 ± 1.358 | 24.330 ± 4.280 | 391 | 4 |
| 5o | 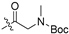 | 0.863 ± 0.253 | - | 5.844 ± 1.180 | 7 | - |
| 5p |  | 2.585 ± 0.520 | - | 22.532 ± 0.391 | 9 | - |
| NVP | 0.236 ± 0.114 | >15.031 | >15.031 | >64 | - | |
| ETR | 0.003 ± 0.001 | 0.040 ± 0.011 | >4.608 | >1387 | >116 | |
| EFV | 0.006 ± 0.002 | 0.698 ± 0.449 | >6.349 | >1053 | >9 | |
| Compds | a EC50 (μM) | |||||
|---|---|---|---|---|---|---|
| L100I | K103N | Y181C | Y188L | E138K | F227L/V106A | |
| 5e | 0.902 ± 0.311 | 0.035 ± 0.009 | 2.585 ± 0.477 | 2.258 ± 0.733 | 0.260 ± 0.075 | 1.465 ± 0.979 |
| 5h | 1.833 ± 0.208 | 0.073 ± 0.016 | 2.724 ± 0.422 | 2.391 ± 0.352 | 0.679 ± 0.569 | 2.113 ± 0.293 |
| 5i | 1.199 ± 0.218 | 0.045 ± 0.010 | >6.541 | 6.907 ± 0.944 | 0.247 ± 0.107 | 3.032 ± 1.755 |
| 5k | 0.601 ± 0.261 | 0.031 ± 0.004 | 1.847 ± 0.621 | 2.355 ± 0.057 | 0.094 ± 0.022 | 1.755 ± 0.686 |
| 5l | 1.481 ± 0.545 | 0.072 ± 0.018 | 2.595 ± 0.225 | 2.741 ± 0.103 | 0.314 ± 0.095 | 2.192 ± 0.045 |
| 5n | 2.124 ± 0.455 | 0.131 ± 0.004 | 2.157 ± 0.057 | 2.111 ± 0.288 | 0.231 ± 0.154 | 2.031 ± 1.184 |
| NVP | 1.741 ± 0.879 | 4.091 ± 3.708 | 8.035 ± 1.675 | 9.868 ± 2.474 | 0.181 ± 0.067 | 7.499 ± 2.590 |
| ETR | 0.006 ± 0.8002 | 0.003 ± 0.001 | 0.015 ± 0.005 | 0.021 ± 0.008 | 0.009 ± 0.004 | 0.009 ± 0.004 |
| EFV | 0.054 ± 0.036 | 0.132 ± 0.050 | 0.008 ± 0.001 | 0.280 ± 0.091 | 0.010 ± 0.005 | 0.329 ± 0.228 |
| Compds | IC50 (μM) | Compds | a IC50 (μM) |
|---|---|---|---|
| 5e | 0.927 ± 0.064 | 5n | 2.390 ± 0.086 |
| 5h | 1.535 ± 0.119 | NVP | 0.735 ± 0.152 |
| 5i | 1.351 ± 0.148 | EFV | 0.012 ± 0.002 |
| 5k | 1.124 ± 0.074 | ETR | 0.018 ± 0.002 |
| 5l | 1.708 ± 0.298 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuo, Z.; Wang, Z.; Jing, L.; Zhang, T.; Ge, A.; Zhou, Z.; Liu, Y.; Li, X.; De Clercq, E.; Pannecouque, C.; et al. Discovery of Novel Amino Acids (Analogues)-Substituted Thiophene[3,2-d]pyrimidine Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Design, Synthesis, and Biological Evaluation. Int. J. Mol. Sci. 2024, 25, 9028. https://doi.org/10.3390/ijms25169028
Zhuo Z, Wang Z, Jing L, Zhang T, Ge A, Zhou Z, Liu Y, Li X, De Clercq E, Pannecouque C, et al. Discovery of Novel Amino Acids (Analogues)-Substituted Thiophene[3,2-d]pyrimidine Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Design, Synthesis, and Biological Evaluation. International Journal of Molecular Sciences. 2024; 25(16):9028. https://doi.org/10.3390/ijms25169028
Chicago/Turabian StyleZhuo, Zongji, Zhao Wang, Lanlan Jing, Tao Zhang, Anchao Ge, Zhenzhen Zhou, Ying Liu, Xin Li, Erik De Clercq, Christophe Pannecouque, and et al. 2024. "Discovery of Novel Amino Acids (Analogues)-Substituted Thiophene[3,2-d]pyrimidine Derivatives as Potent HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Design, Synthesis, and Biological Evaluation" International Journal of Molecular Sciences 25, no. 16: 9028. https://doi.org/10.3390/ijms25169028