
Disturbance of Immune Microenvironment in Androgenetic Alopecia through Spatial Transcriptomics
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Participants’ Demographic Data
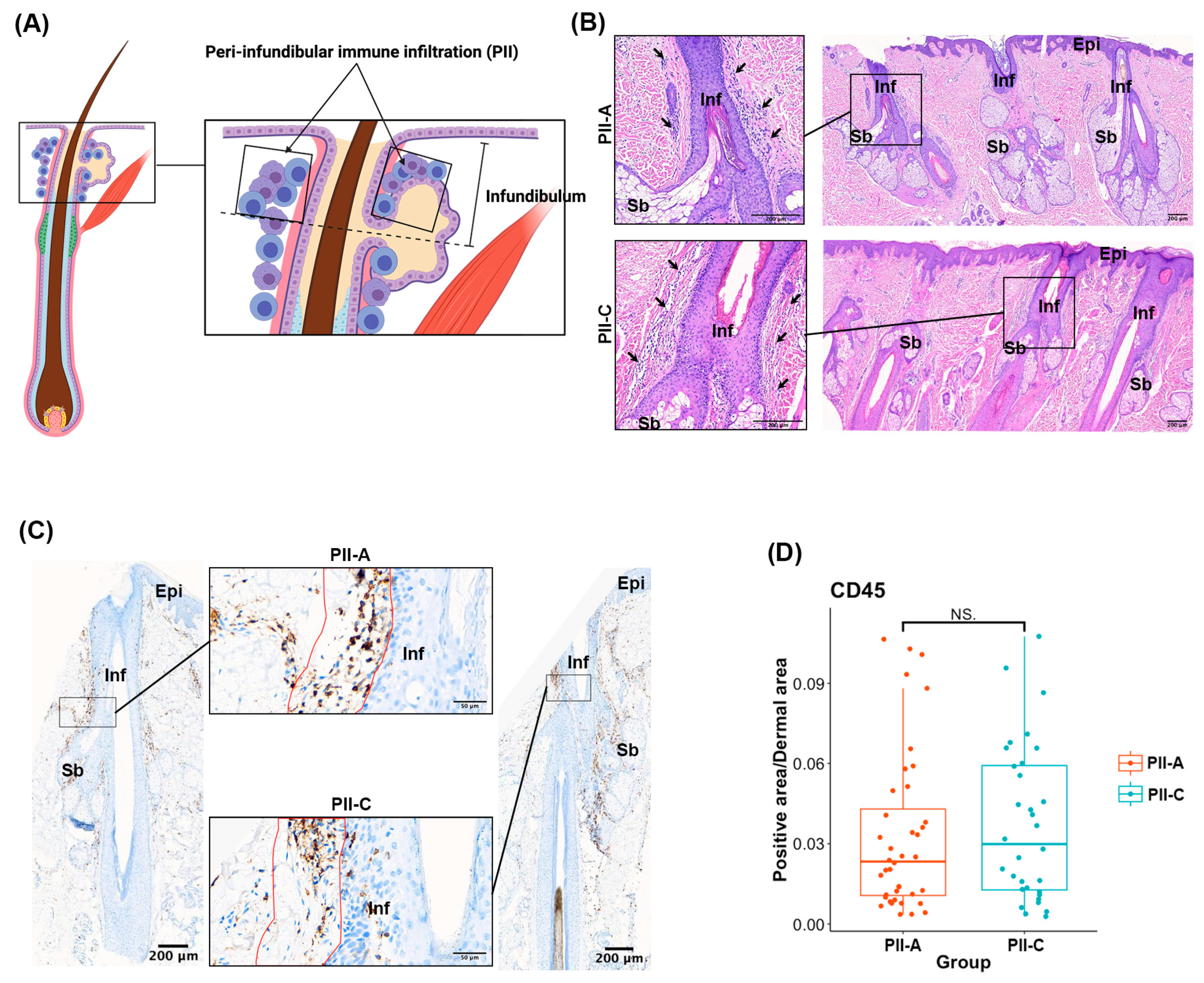
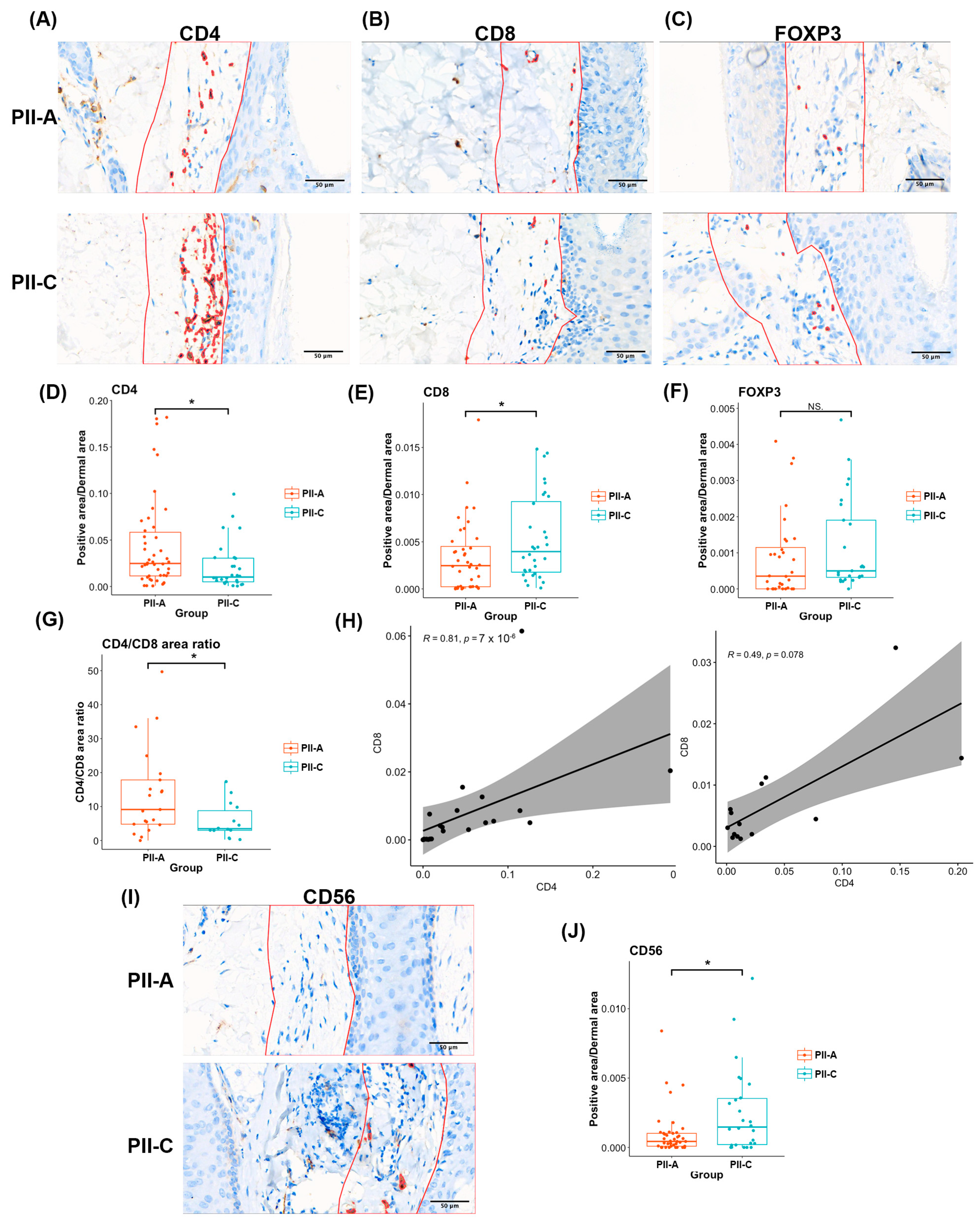
2.2. Assessing Overall Immune Cell Presence and Density in PII Regions with Immunostaining
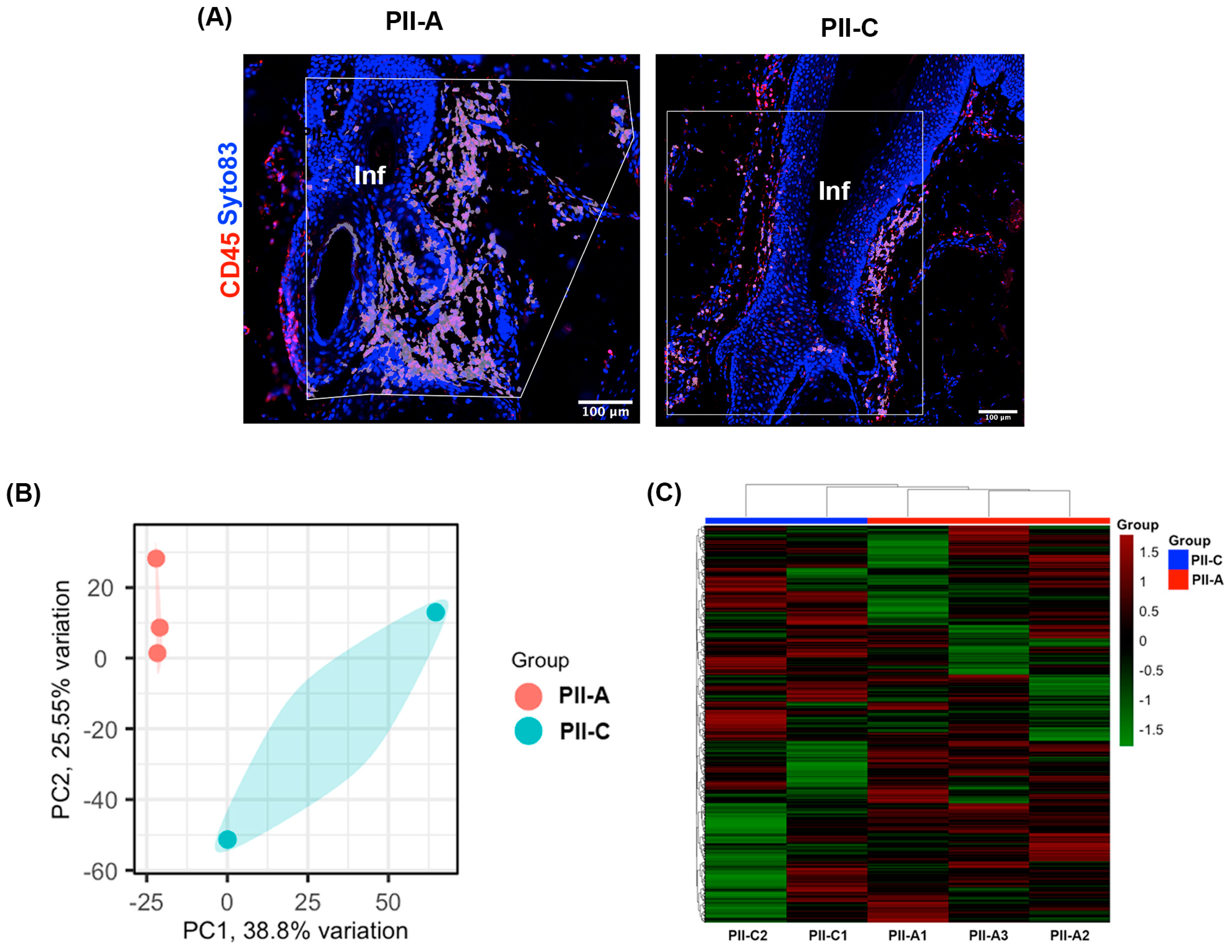
2.3. Selecting Regions of Interest (ROIs) and Validating Immune Cell Enrichment in PII Regions Using Spatial Profiling Techniques
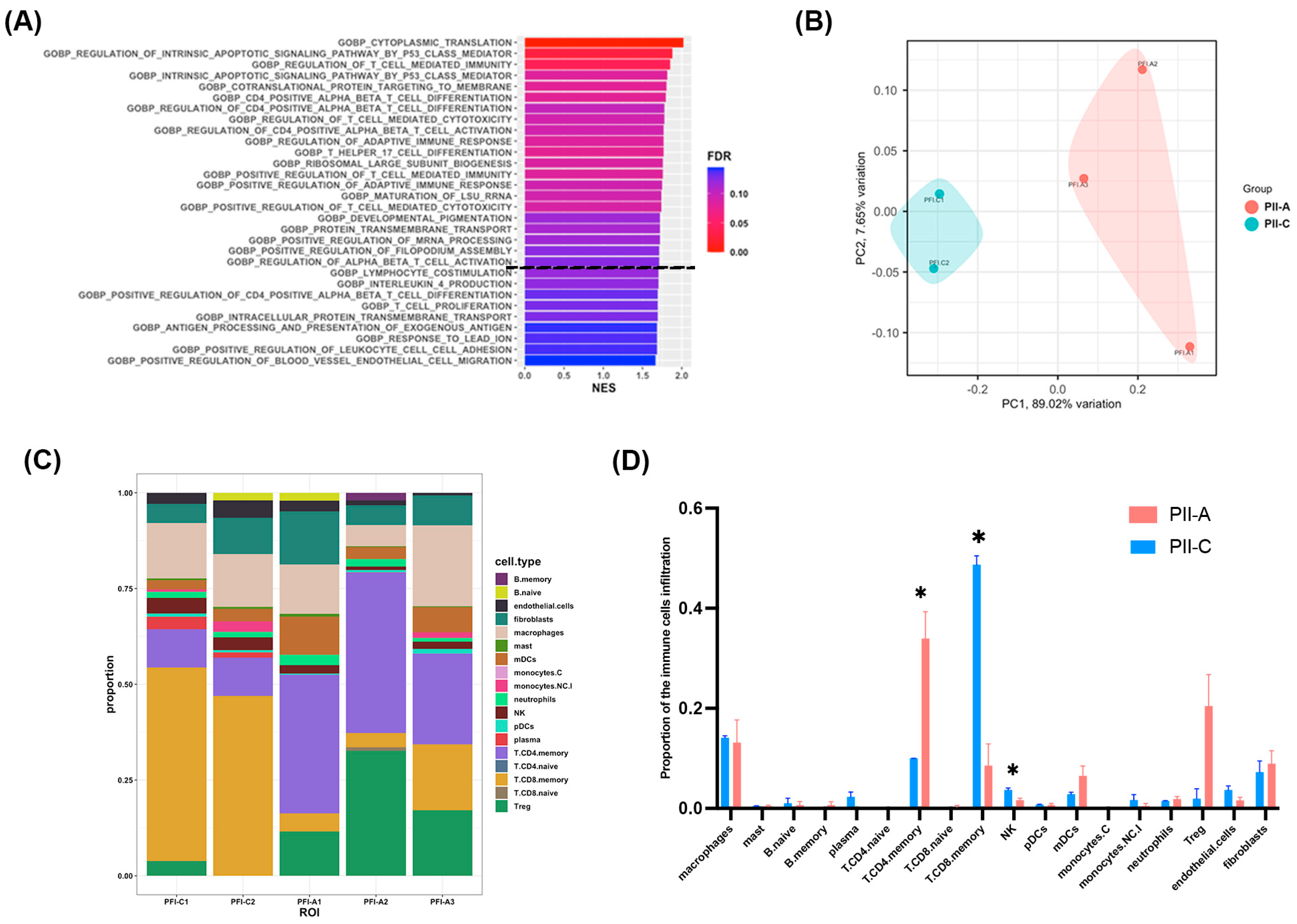
2.4. Enhanced CD4+ T Cell-Mediated Immunity in PII-A Regions
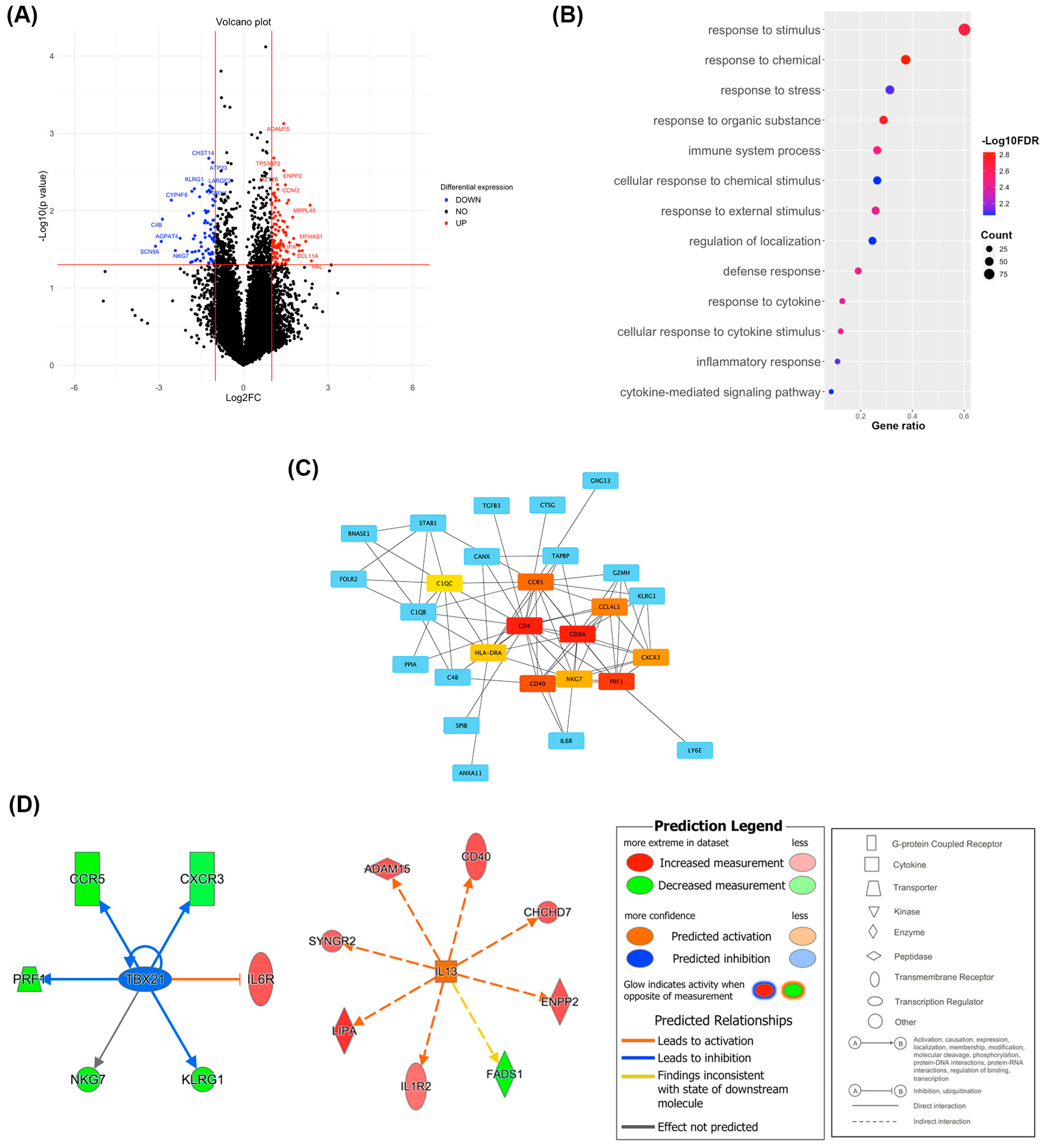
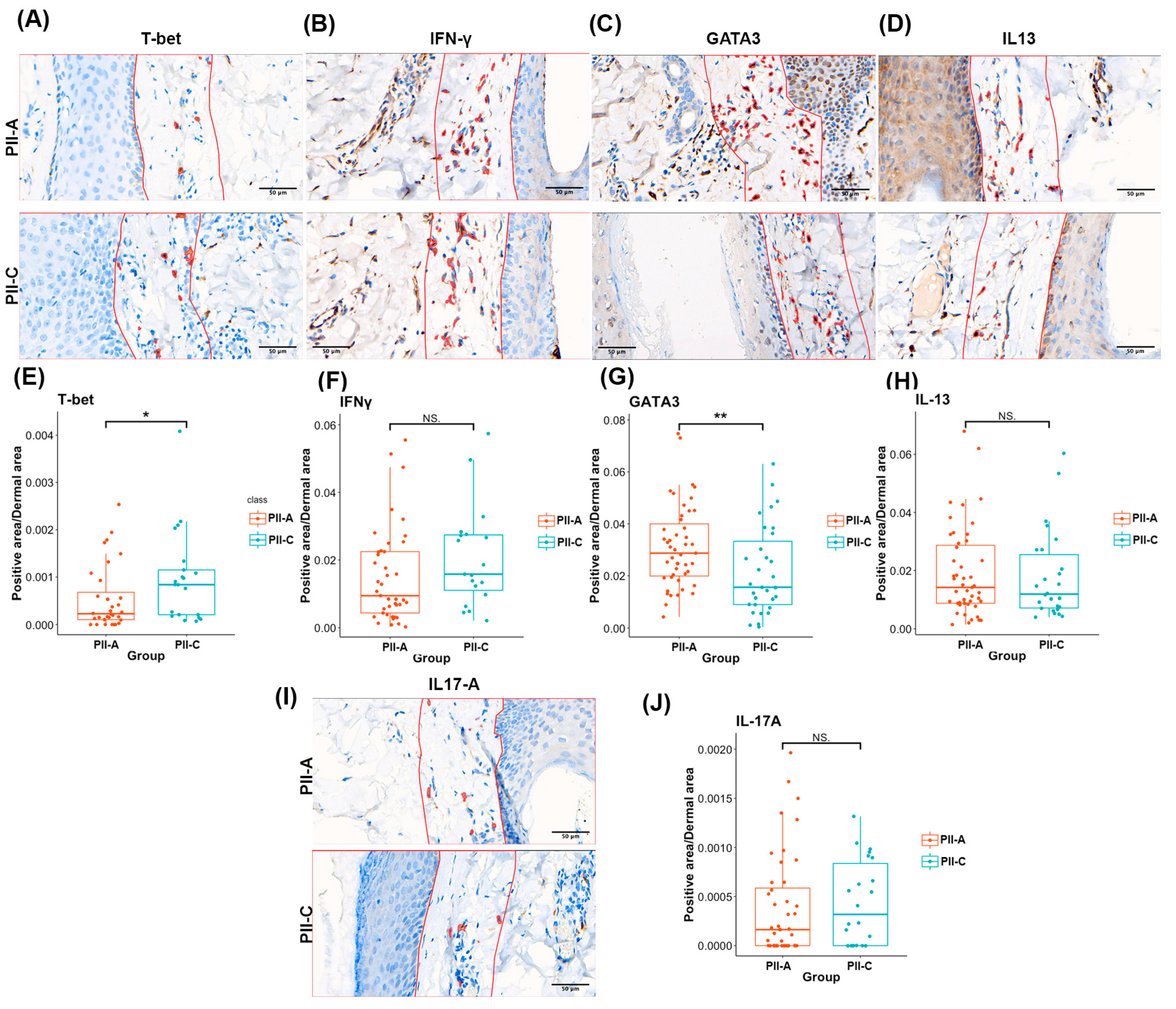
2.5. PII-A Shows Enhanced Antigen-Presenting Cell-Related Molecules and a Potential Th2 Immune Bias
2.6. Density of CD4+ T Cells and Th2 Markers Predominates in PII-A
3. Discussion
4. Materials and Methods
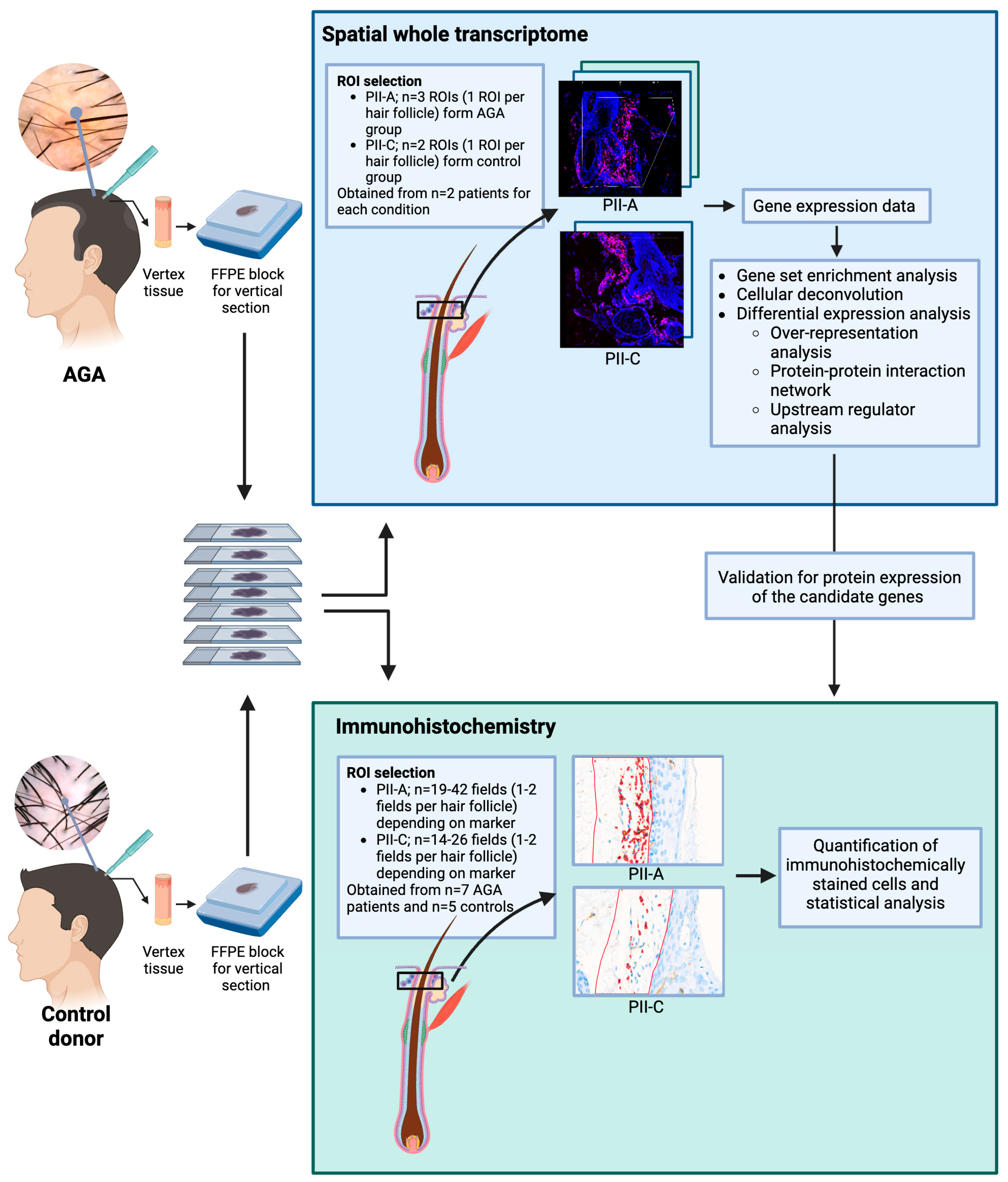
4.1. Patient Selection and Tissue Biopsy
4.2. Spatial Transcriptome Profiling
4.3. Gene Set Enrichment Analysis
4.4. Cellular Deconvolution
4.5. Differential Expression and Functional Analysis
4.6. Immunohistochemistry Assay and Digital Image Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lolli, F.; Pallotti, F.; Rossi, A.; Fortuna, M.C.; Caro, G.; Lenzi, A.; Sansone, A.; Lombardo, F. Androgenetic alopecia: A review. Endocrine 2017, 57, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Stough, D.; Stenn, K.; Haber, R.; Parsley, W.M.; Vogel, J.E.; Whiting, D.A.; Washenik, K. Psychological effect, pathophysiology, and management of androgenetic alopecia in men. Mayo Clin. Proc. 2005, 80, 1316–1322. [Google Scholar] [CrossRef]
- Varothai, S.; Bergfeld, W.F. Androgenetic alopecia: An evidence-based treatment update. Am. J. Clin. Dermatol. 2014, 15, 217–230. [Google Scholar] [CrossRef]
- Coskuner, E.R.; Ozkan, B.; Culha, M.G. Sexual Problems of Men With Androgenic Alopecia Treated with 5-Alpha Reductase Inhibitors. Sex. Med. Rev. 2019, 7, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Cantisani, C.; Melis, L.; Iorio, A.; Scali, E.; Calvieri, S. Minoxidil use in dermatology, side effects and recent patents. Recent. Pat. Inflamm. Allergy Drug Discov. 2012, 6, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Guarrera, M.; Rebora, A. The Higher Number and Longer Duration of Kenogen Hairs Are the Main Cause of the Hair Rarefaction in Androgenetic Alopecia. Skin. Appendage Disord. 2019, 5, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Courtois, M.; Loussouarn, G.; Hourseau, C.; Grollier, J.F. Hair cycle and alopecia. Skin. Pharmacol. 1994, 7, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Hagenaars, S.P.; Hill, W.D.; Harris, S.E.; Ritchie, S.J.; Davies, G.; Liewald, D.C.; Gale, C.R.; Porteous, D.J.; Deary, I.J.; Marioni, R.E. Genetic prediction of male pattern baldness. PLoS Genet. 2017, 13, e1006594. [Google Scholar] [CrossRef] [PubMed]
- Hibberts, N.A.; Howell, A.E.; Randall, V.A. Balding hair follicle dermal papilla cells contain higher levels of androgen receptors than those from non-balding scalp. J. Endocrinol. 1998, 156, 59–65. [Google Scholar] [CrossRef]
- Gatherwright, J.; Liu, M.T.; Amirlak, B.; Gliniak, C.; Totonchi, A.; Guyuron, B. The contribution of endogenous and exogenous factors to male alopecia: A study of identical twins. Plast. Reconstr. Surg. 2013, 131, 794e–801e. [Google Scholar] [CrossRef]
- Premanand, A.; Reena Rajkumari, B. Androgen modulation of Wnt/β-catenin signaling in androgenetic alopecia. Arch. Dermatol. Res. 2018, 310, 391–399. [Google Scholar] [CrossRef]
- Muneeb, F.; Hardman, J.A.; Paus, R. Hair growth control by innate immunocytes: Perifollicular macrophages revisited. Exp. Dermatol. 2019, 28, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Hardman, J.A.; Muneeb, F.; Pople, J.; Bhogal, R.; Shahmalak, A.; Paus, R. Human Perifollicular Macrophages Undergo Apoptosis, Express Wnt Ligands, and Switch their Polarization during Catagen. J. Investig. Dermatol. 2019, 139, 2543–2546.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.C.E.; Higgins, C.A. Immune cell regulation of the hair cycle. Exp. Dermatol. 2020, 29, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Schmidt-Ullrich, R.; Paus, R. The hair follicle as a dynamic miniorgan. Curr. Biol. 2009, 19, R132–R142. [Google Scholar] [CrossRef]
- Paus, R.; Cotsarelis, G. The biology of hair follicles. N. Engl. J. Med. 1999, 341, 491–497. [Google Scholar] [CrossRef]
- Deloche, C.; de Lacharrière, O.; Misciali, C.; Piraccini, B.M.; Vincenzi, C.; Bastien, P.; Tardy, I.; Bernard, B.A.; Tosti, A. Histological features of peripilar signs associated with androgenetic alopecia. Arch. Dermatol. Res. 2004, 295, 422–428. [Google Scholar] [CrossRef]
- Bhat, Y.J.; Saqib, N.U.; Latif, I.; Hassan, I. Female Pattern Hair Loss-An Update. Indian. Dermatol. Online J. 2020, 11, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Stefanato, C.M. Histopathology of alopecia: A clinicopathological approach to diagnosis. Histopathology 2010, 56, 24–38. [Google Scholar] [CrossRef]
- Christoph, T.; Müller-Röver, S.; Audring, H.; Tobin, D.J.; Hermes, B.; Cotsarelis, G.; Rückert, R.; Paus, R. The human hair follicle immune system: Cellular composition and immune privilege. Br. J. Dermatol. 2000, 142, 862–873. [Google Scholar] [CrossRef]
- Chen, C.L.; Huang, W.Y.; Wang, E.H.C.; Tai, K.Y.; Lin, S.J. Functional complexity of hair follicle stem cell niche and therapeutic targeting of niche dysfunction for hair regeneration. J. Biomed. Sci. 2020, 27, 43. [Google Scholar] [CrossRef]
- Heymann, W.R. The inflammatory component of androgenetic alopecia. J. Am. Acad. Dermatol. 2022, 86, 301–302. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Qu, Q.; Jiang, W.; Liu, X.M.; Shi, P.L.; Fan, Z.X.; Du, L.J.; Wang, G.F.; Liu, X.N.; Guo, Z.H.; et al. Identification of Functional Patterns of Androgenetic Alopecia Using Transcriptome Profiling in Distinct Locations of Hair Follicles. J. Investig. Dermatol. 2018, 138, 972–975. [Google Scholar] [CrossRef] [PubMed]
- El-Domyati, M.; Attia, S.; Saleh, F.; Abdel-Wahab, H. Androgenetic alopecia in males: A histopathological and ultrastructural study. J. Cosmet. Dermatol. 2009, 8, 83–91. [Google Scholar] [CrossRef]
- Ho, B.S.; Vaz, C.; Ramasamy, S.; Chew, E.G.Y.; Mohamed, J.S.; Jaffar, H.; Hillmer, A.; Tanavde, V.; Bigliardi-Qi, M.; Bigliardi, P.L. Progressive expression of PPARGC1α is associated with hair miniaturization in androgenetic alopecia. Sci. Rep. 2019, 9, 8771. [Google Scholar] [CrossRef] [PubMed]
- Garza, L.A.; Liu, Y.; Yang, Z.; Alagesan, B.; Lawson, J.A.; Norberg, S.M.; Loy, D.E.; Zhao, T.; Blatt, H.B.; Stanton, D.C.; et al. Prostaglandin D2 inhibits hair growth and is elevated in bald scalp of men with androgenetic alopecia. Sci. Transl. Med. 2012, 4, 126ra134. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.; Reygagne, P.; Benech, P.; Jean-Louis, F.; Scalvino, S.; Ly Ka So, S.; Hamidou, Z.; Bianovici, S.; Pouch, J.; Ducos, B.; et al. Study of gene expression alteration in male androgenetic alopecia: Evidence of predominant molecular signalling pathways. Br. J. Dermatol. 2017, 177, 1322–1336. [Google Scholar] [CrossRef]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-omics: Novel approaches to probe cell heterogeneity and extracellular matrix biology. Matrix Biol. 2020, 91–92, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Labra, S.R.; Pitkin, R.M.; Hoxha, K.; Narasimhan, S.; Changolkar, L.; Rosenbloom, A.; Lee, V.M.; Trojanowski, J.Q. Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropathol. 2019, 138, 613–630. [Google Scholar] [CrossRef]
- Hoefsmit, E.P.; Rozeman, E.A.; Van, T.M.; Dimitriadis, P.; Krijgsman, O.; Conway, J.W.; Pires da Silva, I.; van der Wal, J.E.; Ketelaars, S.L.C.; Bresser, K.; et al. Comprehensive analysis of cutaneous and uveal melanoma liver metastases. J. Immunother. Cancer 2020, 8, e001501. [Google Scholar] [CrossRef]
- Woodford-Thomas, T.; Thomas, M.L. The leukocyte common antigen, CD45 and other protein tyrosine phosphatases in hematopoietic cells. Semin. Cell Biol. 1993, 4, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Qurat Ul, A.; Frei, N.F.; Khoshiwal, A.M.; Stougie, P.; Odze, R.; Camilleri-Broet, S.; Ferri, L.; Duits, L.C.; Bergman, J.; Stachler, M.D. Feasibility Study Utilizing NanoString’s Digital Spatial Profiling (DSP) Technology for Characterizing the Immune Microenvironment in Barrett’s Esophagus Formalin-Fixed Paraffin-Embedded Tissues. Cancers 2023, 15, 5895. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Xiong, A.; Wu, F.; Li, X.; Wang, J.; Jiang, T.; Chen, P.; Zhang, X.; Zhao, Z.; Liu, H.; et al. Spatial multi-omics revealed the impact of tumor ecosystem heterogeneity on immunotherapy efficacy in patients with advanced non-small cell lung cancer treated with bispecific antibody. J. Immunother. Cancer 2023, 11, e006234. [Google Scholar] [CrossRef]
- Kiuru, M.; Kriner, M.A.; Wong, S.; Zhu, G.; Terrell, J.R.; Li, Q.; Hoang, M.; Beechem, J.; McPherson, J.D. High-Plex Spatial RNA Profiling Reveals Cell Type-Specific Biomarker Expression during Melanoma Development. J. Investig. Dermatol. 2022, 142, 1401–1412.e20. [Google Scholar] [CrossRef] [PubMed]
- Danaher, P.; Kim, Y.; Nelson, B.; Griswold, M.; Yang, Z.; Piazza, E.; Beechem, J.M. Advances in mixed cell deconvolution enable quantification of cell types in spatial transcriptomic data. Nat. Commun. 2022, 13, 385. [Google Scholar] [CrossRef] [PubMed]
- St Paul, M.; Ohashi, P.S. The Roles of CD8(+) T Cell Subsets in Antitumor Immunity. Trends Cell Biol. 2020, 30, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Bolitho, P.; Voskoboinik, I.; Trapani, J.A.; Smyth, M.J. Apoptosis induced by the lymphocyte effector molecule perforin. Curr. Opin. Immunol. 2007, 19, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.S.; De Labastida Rivera, F.; Yan, J.; Corvino, D.; Das, I.; Zhang, P.; Kuns, R.; Chauhan, S.B.; Hou, J.; Li, X.Y.; et al. The NK cell granule protein NKG7 regulates cytotoxic granule exocytosis and inflammation. Nat. Immunol. 2020, 21, 1205–1218. [Google Scholar] [CrossRef]
- Qin, S.; Rottman, J.B.; Myers, P.; Kassam, N.; Weinblatt, M.; Loetscher, M.; Koch, A.E.; Moser, B.; Mackay, C.R. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Investig. 1998, 101, 746–754. [Google Scholar] [CrossRef]
- Szabo, S.J.; Kim, S.T.; Costa, G.L.; Zhang, X.; Fathman, C.G.; Glimcher, L.H. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 2000, 100, 655–669. [Google Scholar] [CrossRef]
- Zhu, J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine 2015, 75, 14–24. [Google Scholar] [CrossRef]
- Ono, M. Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes. Immunology 2020, 160, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef]
- Zhou, M.; Ouyang, W. The function role of GATA-3 in Th1 and Th2 differentiation. Immunol. Res. 2003, 28, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Murphy, T.L.; Ouyang, W.; Murphy, K.M. Induction of interferon-gamma production in Th1 CD4+ T cells: Evidence for two distinct pathways for promoter activation. Eur. J. Immunol. 1999, 29, 548–555. [Google Scholar] [CrossRef]
- Su, Y.; Huang, J.; Zhao, X.; Lu, H.; Wang, W.; Yang, X.O.; Shi, Y.; Wang, X.; Lai, Y.; Dong, C. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci. Immunol. 2019, 4, eaau9657. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Moresi, J.M.; Horn, T.D. Distribution of Langerhans cells in human hair follicle. J. Cutan. Pathol. 1997, 24, 636–640. [Google Scholar] [CrossRef]
- Nagao, K.; Kobayashi, T.; Moro, K.; Ohyama, M.; Adachi, T.; Kitashima, D.Y.; Ueha, S.; Horiuchi, K.; Tanizaki, H.; Kabashima, K.; et al. Stress-induced production of chemokines by hair follicles regulates the trafficking of dendritic cells in skin. Nat. Immunol. 2012, 13, 744–752. [Google Scholar] [CrossRef]
- Polak-Witka, K.; Rudnicka, L.; Blume-Peytavi, U.; Vogt, A. The role of the microbiome in scalp hair follicle biology and disease. Exp. Dermatol. 2020, 29, 286–294. [Google Scholar] [CrossRef]
- Lattanand, A.; Johnson, W.C. Male pattern alopecia a histopathologic and histochemical study. J. Cutan. Pathol. 1975, 2, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Jaworsky, C.; Kligman, A.M.; Murphy, G.F. Characterization of inflammatory infiltrates in male pattern alopecia: Implications for pathogenesis. Br. J. Dermatol. 1992, 127, 239–246. [Google Scholar] [CrossRef]
- Whiting, D.A. Chronic telogen effluvium: Increased scalp hair shedding in middle-aged women. J. Am. Acad. Dermatol. 1996, 35, 899–906. [Google Scholar] [CrossRef]
- Sueki, H.; Stoudemayer, T.; Kligman, A.M.; Murphy, G.F. Quantitative and ultrastructural analysis of inflammatory infiltrates in male pattern alopecia. Acta Derm. Venereol. 1999, 79, 347–350. [Google Scholar] [PubMed]
- Valdebran, M.; Mo, J.; Elston, D.M.; Doan, L. Pattern hair loss: Assessment of inflammation and fibrosis on histologic sections. J. Am. Acad. Dermatol. 2020, 82, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kobayashi, T.; Sugihara, E.; Yamada, T.; Ikuta, K.; Pittaluga, S.; Saya, H.; Amagai, M.; Nagao, K. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med. 2015, 21, 1272–1279. [Google Scholar] [CrossRef]
- Fabbrocini, G.; Cantelli, M.; Masarà, A.; Annunziata, M.C.; Marasca, C.; Cacciapuoti, S. Female pattern hair loss: A clinical, pathophysiologic, and therapeutic review. Int. J. Womens Dermatol. 2018, 4, 203–211. [Google Scholar] [CrossRef]
- Merlotto, M.R.; Ramos, P.M.; Miot, H.A. Pattern hair loss: Assessment of microinflammation in miniaturized and terminal hair follicles through horizontal histologic sections. J. Am. Acad. Dermatol. 2020, 83, e145–e146. [Google Scholar] [CrossRef]
- Mahé, Y.F.; Michelet, J.F.; Billoni, N.; Jarrousse, F.; Buan, B.; Commo, S.; Saint-Léger, D.; Bernard, B.A. Androgenetic alopecia and microinflammation. Int. J. Dermatol. 2000, 39, 576–584. [Google Scholar] [CrossRef]
- Neefjes, J.; Jongsma, M.L.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [CrossRef]
- Suzuki, K.; Inoue, M.; Cho, O.; Mizutani, R.; Shimizu, Y.; Nagahama, T.; Sugita, T. Scalp Microbiome and Sebum Composition in Japanese Male Individuals with and without Androgenetic Alopecia. Microorganisms 2021, 9, 2132. [Google Scholar] [CrossRef]
- Constant, S.L.; Bottomly, K. Induction of Th1 and Th2 CD4+ T cell responses: The alternative approaches. Annu. Rev. Immunol. 1997, 15, 297–322. [Google Scholar] [CrossRef] [PubMed]
- Bulfone-Paus, S.; Bahri, R. Mast Cells as Regulators of T Cell Responses. Front. Immunol. 2015, 6, 394. [Google Scholar] [CrossRef] [PubMed]
- Won, C.H.; Kwon, O.S.; Kim, Y.K.; Kang, Y.J.; Kim, B.J.; Choi, C.W.; Eun, H.C.; Cho, K.H. Dermal fibrosis in male pattern hair loss: A suggestive implication of mast cells. Arch. Dermatol. Res. 2008, 300, 147–152. [Google Scholar] [CrossRef]
- Nelson, A.M.; Loy, D.E.; Lawson, J.A.; Katseff, A.S.; Fitzgerald, G.A.; Garza, L.A. Prostaglandin D2 inhibits wound-induced hair follicle neogenesis through the receptor, Gpr44. J. Investig. Dermatol. 2013, 133, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Colombe, L.; Michelet, J.F.; Bernard, B.A. Prostanoid receptors in anagen human hair follicles. Exp. Dermatol. 2008, 17, 63–72. [Google Scholar] [CrossRef]
- Xue, L.; Gyles, S.L.; Wettey, F.R.; Gazi, L.; Townsend, E.; Hunter, M.G.; Pettipher, R. Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J. Immunol. 2005, 175, 6531–6536. [Google Scholar] [CrossRef]
- Gieseck, R.L., 3rd; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.K.; Austin, E.; Huang, A.; Mamalis, A.; Jagdeo, J. The IL-4/IL-13 axis in skin fibrosis and scarring: Mechanistic concepts and therapeutic targets. Arch. Dermatol. Res. 2020, 312, 81–92. [Google Scholar] [CrossRef]
- Elston, D.M. Vertical vs. transverse sections: Both are valuable in the evaluation of alopecia. Am. J. Dermatopathol. 2005, 27, 353–356. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef]
- Li, C.Y.; Cai, J.H.; Tsai, J.J.P.; Wang, C.C.N. Identification of Hub Genes Associated With Development of Head and Neck Squamous Cell Carcinoma by Integrated Bioinformatics Analysis. Front. Oncol. 2020, 10, 681. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Emmanuel, T.; Brent, M.B.; Iversen, L.; Johansen, C. Quantification of Immunohistochemically Stained Cells in Skin Biopsies. Dermatopathology 2022, 9, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Hiss, S.; Eckstein, M.; Segschneider, P.; Mantsopoulos, K.; Iro, H.; Hartmann, A.; Agaimy, A.; Haller, F.; Mueller, S.K. Tumour-Infiltrating Lymphocytes (TILs) and PD-L1 Expression Correlate with Lymph Node Metastasis, High-Grade Transformation and Shorter Metastasis-Free Survival in Patients with Acinic Cell Carcinoma (AciCC) of the Salivary Glands. Cancers 2021, 13, 965. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charoensuksira, S.; Tantiwong, S.; Pongklaokam, J.; Hanvivattanakul, S.; Surinlert, P.; Krajarng, A.; Thanasarnaksorn, W.; Hongeng, S.; Ponnikorn, S. Disturbance of Immune Microenvironment in Androgenetic Alopecia through Spatial Transcriptomics. Int. J. Mol. Sci. 2024, 25, 9031. https://doi.org/10.3390/ijms25169031
Charoensuksira S, Tantiwong S, Pongklaokam J, Hanvivattanakul S, Surinlert P, Krajarng A, Thanasarnaksorn W, Hongeng S, Ponnikorn S. Disturbance of Immune Microenvironment in Androgenetic Alopecia through Spatial Transcriptomics. International Journal of Molecular Sciences. 2024; 25(16):9031. https://doi.org/10.3390/ijms25169031
Chicago/Turabian StyleCharoensuksira, Sasin, Supasit Tantiwong, Juthapa Pongklaokam, Sirashat Hanvivattanakul, Piyaporn Surinlert, Aungkana Krajarng, Wilai Thanasarnaksorn, Suradej Hongeng, and Saranyoo Ponnikorn. 2024. "Disturbance of Immune Microenvironment in Androgenetic Alopecia through Spatial Transcriptomics" International Journal of Molecular Sciences 25, no. 16: 9031. https://doi.org/10.3390/ijms25169031