
A Bioinformatics-Based Study on Methylation Alterations of the FBLN1 Gene in Hippocampal Tissue of Alzheimer’s Disease Model DKO and DTG Mice
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genotype Identification of Offspring in AD Model DKO and DTG Mice
2.2. Methylation Screening Results of RRBS Sequencing in Hippocampal Tissue of DKO Mice
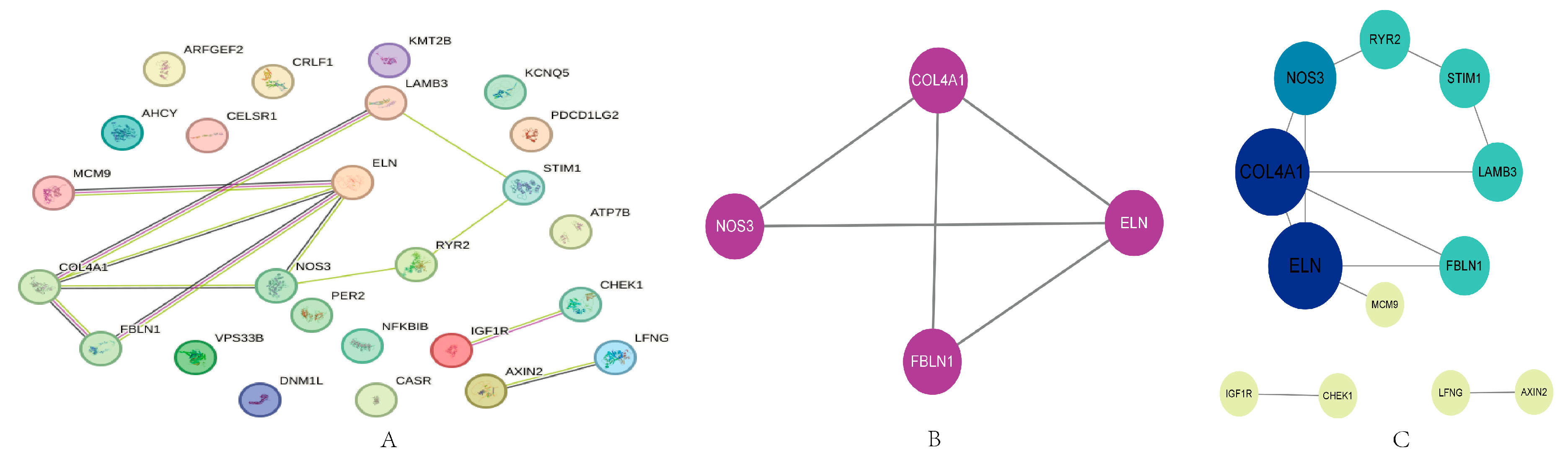
2.3. PPI Analysis
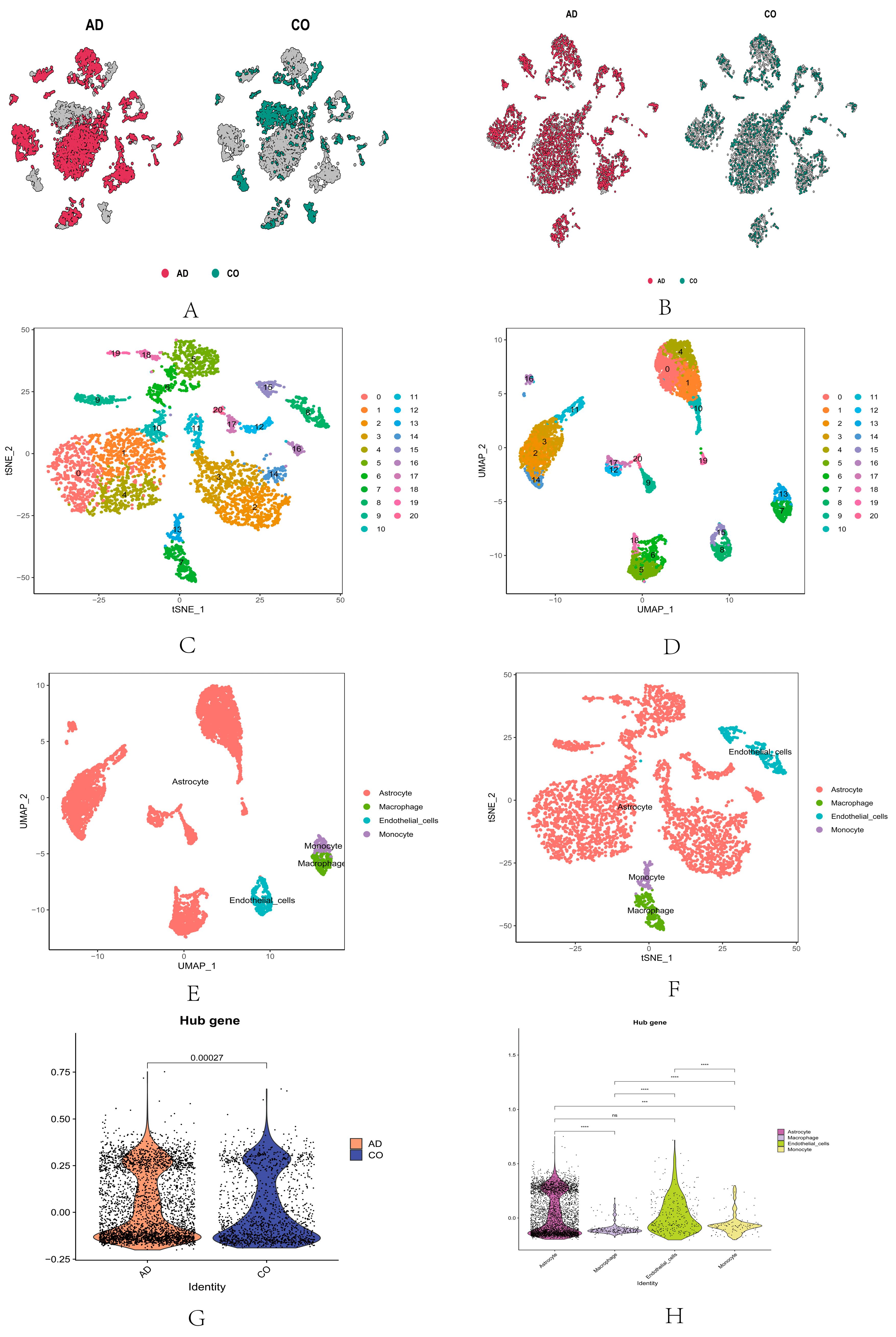
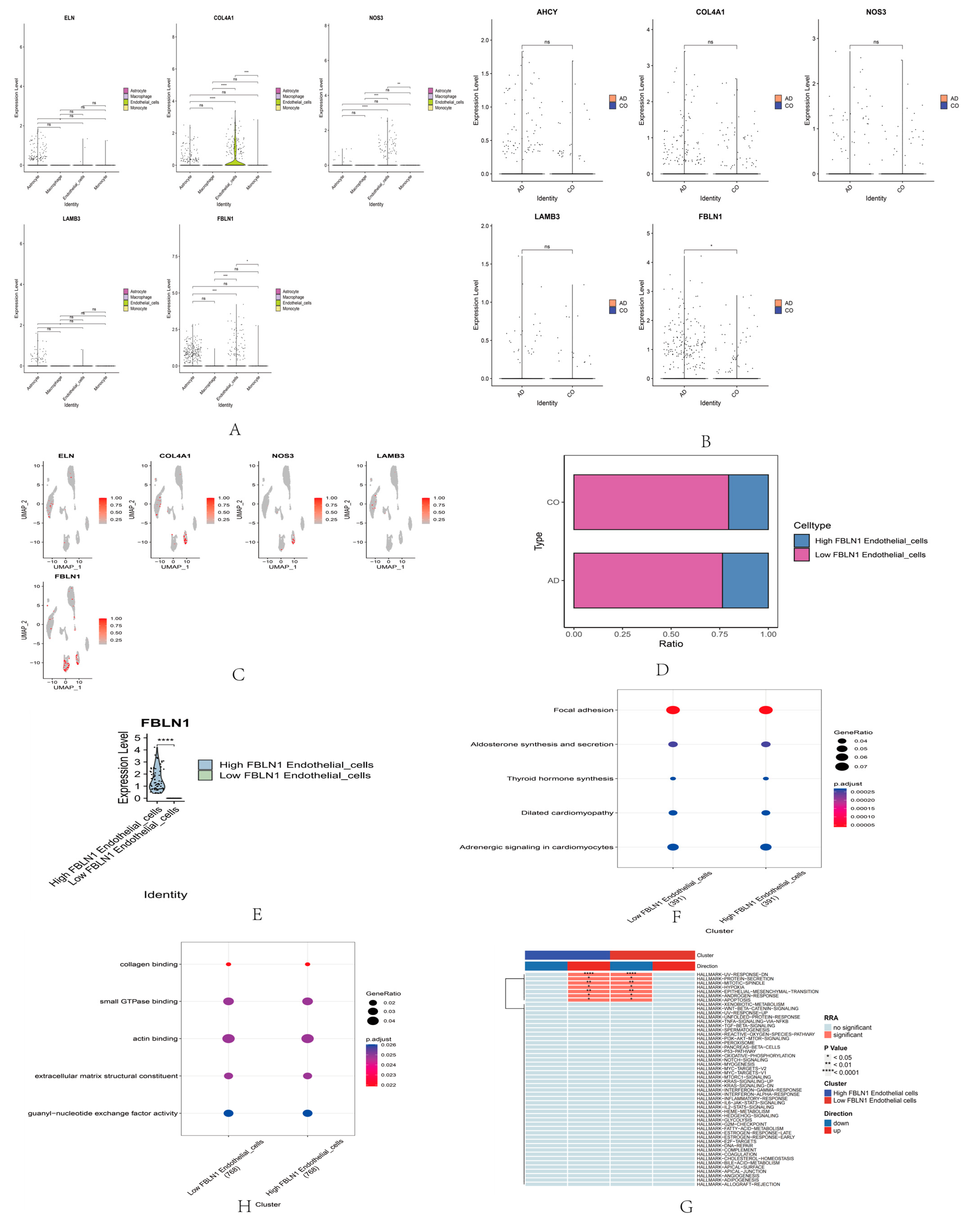
2.4. Single-Cell RNA Sequencing Analysis of Core Genes
2.5. Methylation Screening Results of FBLN1 Gene in Hippocampal Tissue of DKO Mice Based on RRBS Sequencing Data
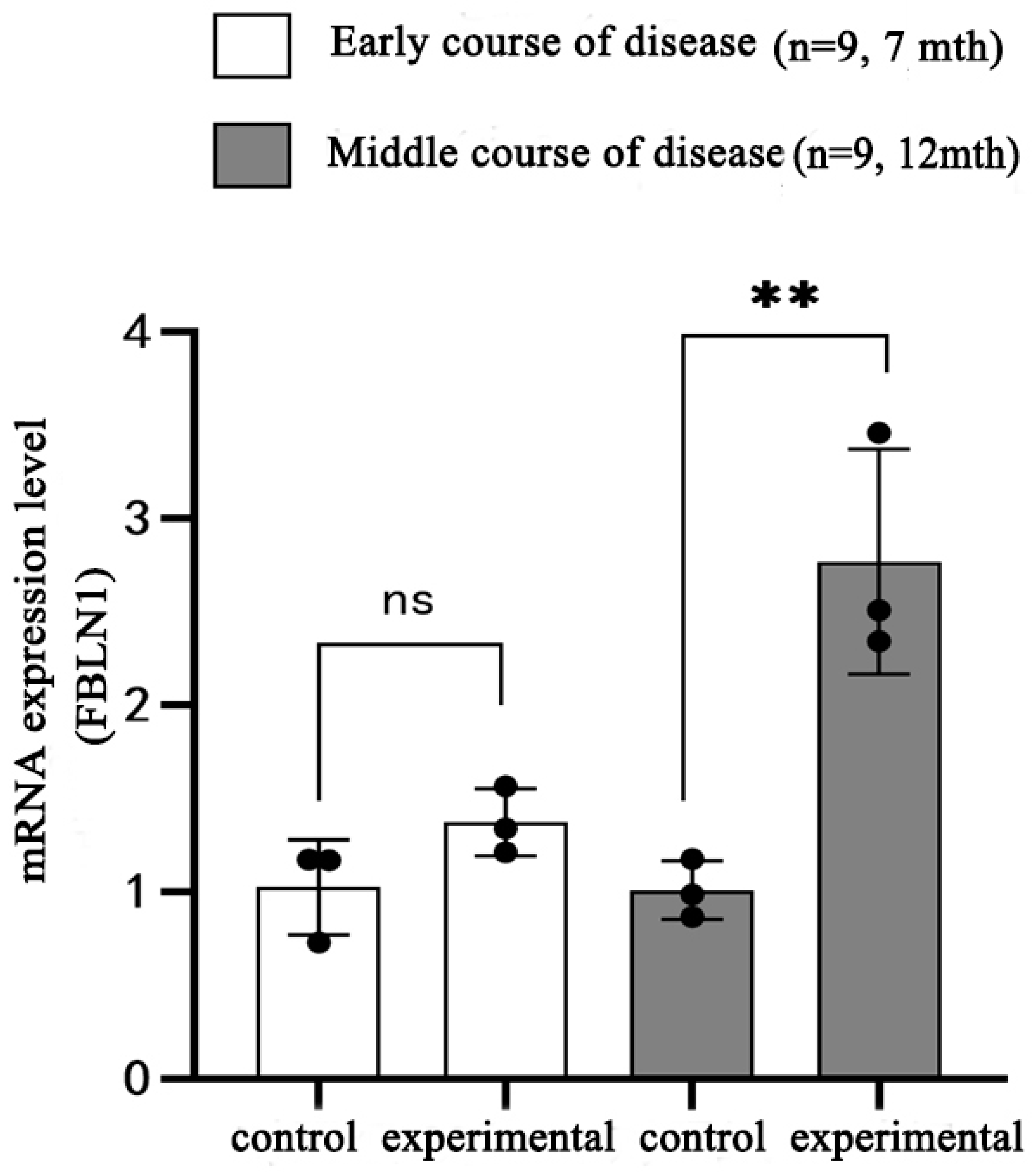
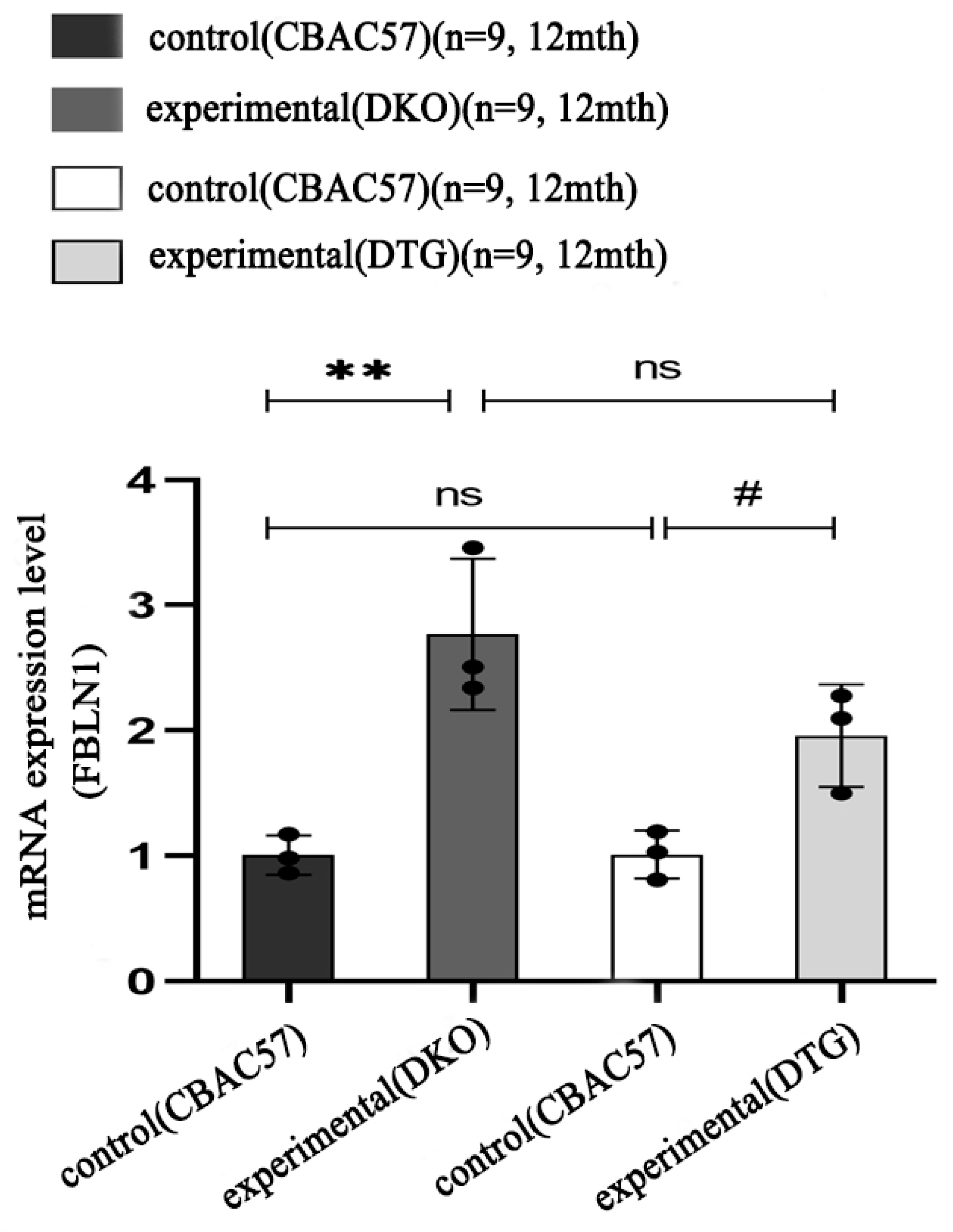
2.6. Transcriptional Expression of FBLN1 Gene in Hippocampal Tissue of DKO and DTG Mice
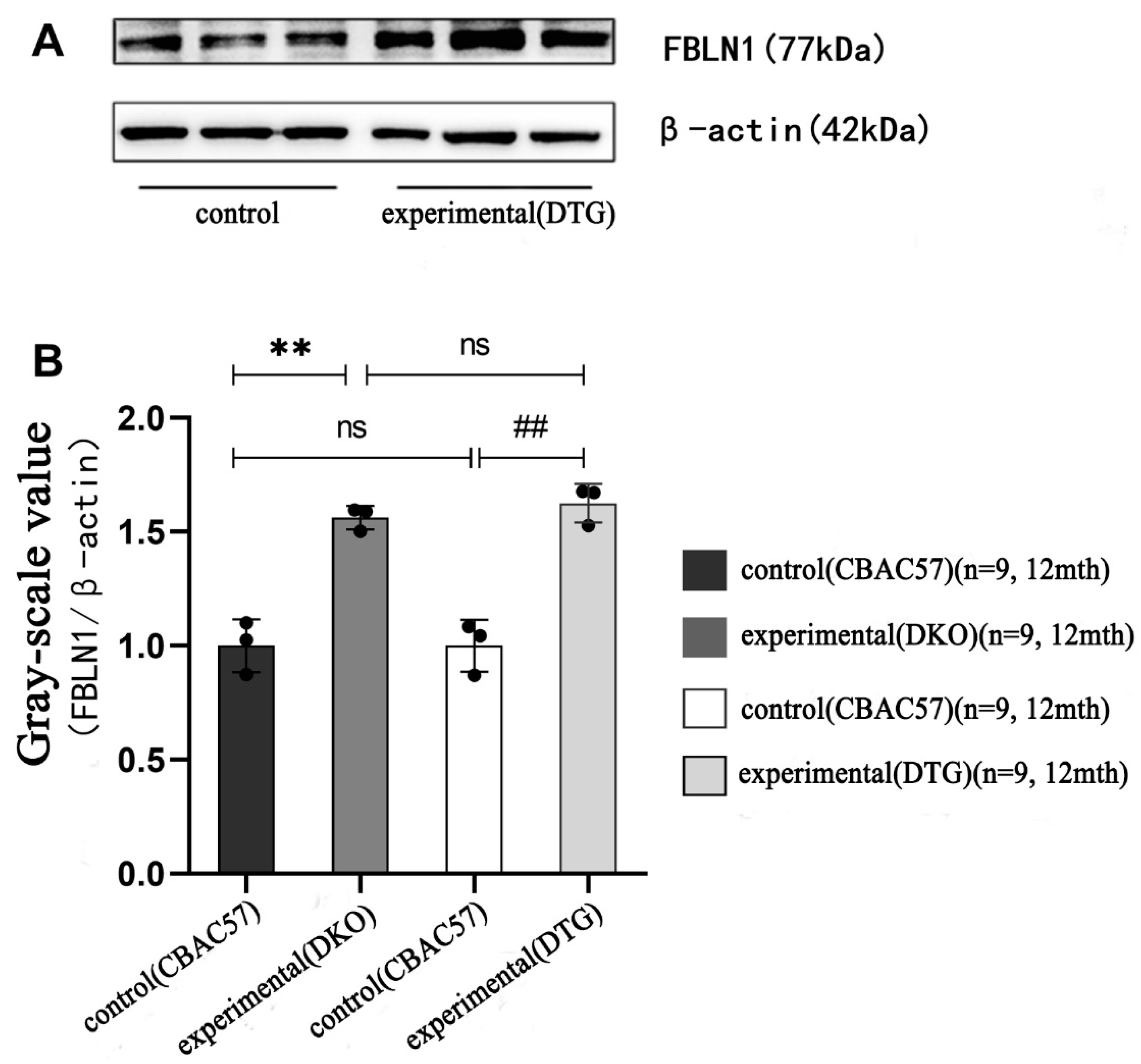
2.7. Translational Expression of FBLN1 Gene in Hippocampal Tissue of DKO and DTG Mice
3. Discussion
4. Materials and Methods
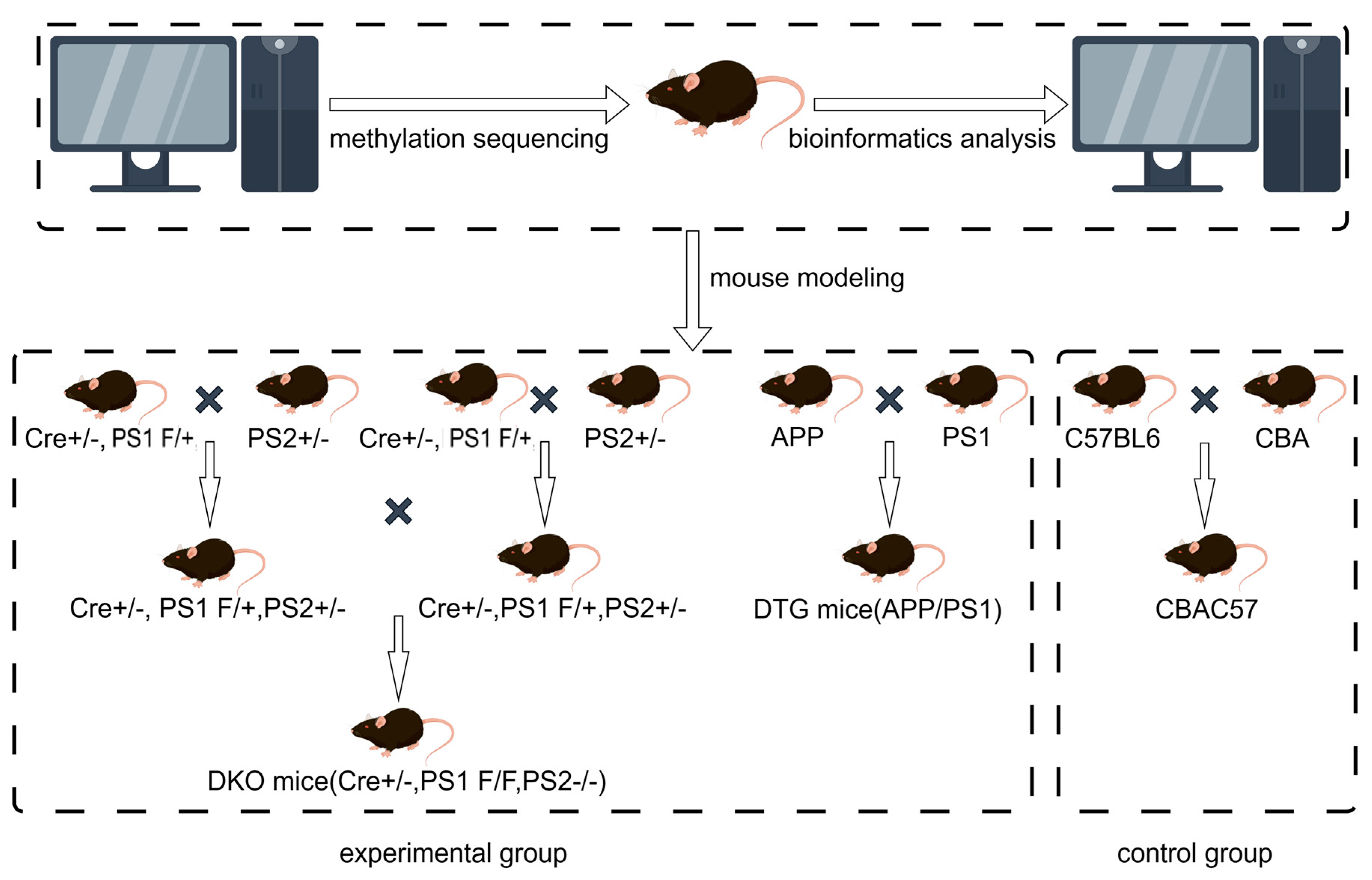
4.1. Experimental Animals
4.2. Experimental Grouping
4.3. Collection of Tail Tissue from Offspring Mice
4.4. Collection of Hippocampal Tissue
4.5. Methods
4.5.1. DNA Extraction from Mice
4.5.2. Agarose Gel Electrophoresis Detection
4.5.3. Reduced Representation Bisulfite Sequencing (RRBS)
4.5.4. PPI (Protein–Protein Interaction)
4.5.5. Single-Cell Sequencing (ScRNA-Seq)
4.5.6. Extraction of DNA from Hippocampal Tissue
4.5.7. DNA Quality Identification and Integrity Assessment
4.5.8. Bisulfite Treatment
4.5.9. PCR Amplification and Product Recovery
4.5.10. Real-Time Quantitative Polymerase Chain Reaction (RT-PCR/QPCR)
- RNA Extraction
- RNA Concentration and Purity Detection
- cDNA Template Preparation (Reverse Transcription)
- RT-PCR
- Total Protein Extraction
- Protein Concentration Measurement
- Western Blot Detection
- Gel Preparation and Sample Loading
- Electrophoresis
- Electrotransfer
- Blocking and Antibody Incubation
- Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Chen, Q.; Nakajima, A.; Choi, S.H.; Xiong, X.; Tang, Y.P. Loss of presenilin function causes Alzheimer’s disease-like neurodegeneration in the mouse. J. Neurosci. Res. 2008, 86, 1615–1625. [Google Scholar] [CrossRef]
- Wang, J.; Yu, J.T.; Tan, M.S.; Jiang, T.; Tan, L. Epigenetic mechanisms in Alzheimer’s disease: Implications for pathogenesis and therapy. Ageing Res. Rev. 2013, 12, 1024–1041. [Google Scholar] [CrossRef]
- Nikolac Perkovic, M.; Videtic Paska, A.; Konjevod, M.; Kouter, K.; Svob Strac, D.; Nedic Erjavec, G.; Pivac, N. Epigenetics of Alzheimer’s Disease. Biomolecules 2021, 11, 195. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.L.; Wang, T.; Meng, B.; Lin, L.N.; Mei, B. Presenilin-1/2 conditional double knockout mice: An ideal model for Alzheimer’s disease. Chin. J. New Drugs 2013, 22, 1530–1537. [Google Scholar]
- Luo, S.Y.; Tang, M.X.; Tang, X.Q.; Li, X.; Gao, Z.Q.; Tang, Q.Y. Methylation of hippocampal Ptprd gene in presenilin 1/presenilin 2 double-gene knockout mice with Alzheimer’s disease. China Med. Pharmacy 2018, 8, 4. [Google Scholar]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet. Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Esang, M.; Gupta, M. Aducanumab as a Novel Treatment for Alzheimer’s Disease: A Decade of Hope, Controversies, and the Future. Cureus 2021, 13, e17591. [Google Scholar] [CrossRef]
- H Ferreira-Vieira, T.; M Guimaraes, I.; R Silva, F.; M Ribeiro, F. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Li, R.; Sterling, K.; Song, W. Amyloid β-based therapy for Alzheimer’s disease: Challenges, successes and future. Signal Transduct. Target. Ther. 2023, 8, 248. [Google Scholar] [CrossRef]
- Levin, J.; Vöglein, J.; Quiroz, Y.T.; Bateman, R.J.; Ghisays, V.; Lopera, F.; McDade, E.; Reiman, E.; Tariot, P.N.; Morris, J.C. Testing the amyloid cascade hypothesis: Prevention trials in autosomal dominant Alzheimer disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2022, 18, 2687–2698. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The amyloid cascade hypothesis: An updated critical review. Brain A J. Neurol. 2023, 146, 3969–3990. [Google Scholar] [CrossRef]
- Bakota, L.; Brandt, R. Tau Biology and Tau-Directed Therapies for Alzheimer’s Disease. Drugs 2016, 76, 301–313. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain A J. Neurol. 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Berry, A.S.; Harrison, T.M. New perspectives on the basal forebrain cholinergic system in Alzheimer’s disease. Neurosci. Biobehav. Rev. 2023, 150, 105192. [Google Scholar] [CrossRef] [PubMed]
- Montavon, B.; Winter, L.E.; Gan, Q.; Arasteh, A.; Montaño, A.M. Mucopolysaccharidosis Type IVA: Extracellular Matrix Biomarkers in Cardiovascular Disease. Front. Cardiovasc. Med. 2022, 9, 829111. [Google Scholar] [CrossRef]
- Tanis, T.; Cincin, Z.B.; Gokcen-Rohlig, B.; Bireller, E.S.; Ulusan, M.; Tanyel, C.R.; Cakmakoglu, B. The role of components of the extracellular matrix and inflammation on oral squamous cell carcinoma metastasis. Arch. Oral Biol. 2014, 59, 1155–1163. [Google Scholar] [CrossRef]
- Harikrishnan, K.; Joshi, O.; Madangirikar, S.; Balasubramanian, N. Cell Derived Matrix Fibulin-1 Associates With Epidermal Growth Factor Receptor to Inhibit Its Activation, Localization and Function in Lung Cancer Calu-1 Cells. Front. Cell Dev. Biol. 2020, 8, 522. [Google Scholar] [CrossRef]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef]
- Lepelletier, F.X.; Mann, D.M.; Robinson, A.C.; Pinteaux, E.; Boutin, H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.M.; Özkan, E.; Gürsoy-Özdemir, Y. The role of extracellular matrix alterations in mediating astrocyte damage and pericyte dysfunction in Alzheimer’s disease: A comprehensive review. Eur. J. Neurosci. 2022, 56, 5453–5475. [Google Scholar] [CrossRef]
- Crapser, J.D.; Spangenberg, E.E.; Barahona, R.A.; Arreola, M.A.; Hohsfield, L.A.; Green, K.N. Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 2020, 58, 102919. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Hung, W.W.; Chang, W.A.; Wu, P.H.; Wu, L.Y.; Lee, S.C.; Kuo, M.C.; Hsu, Y.L. Autocrine Exosomal Fibulin-1 as a Target of MiR-1269b Induces Epithelial-Mesenchymal Transition in Proximal Tubule in Diabetic Nephropathy. Front. Cell Dev. Biol. 2021, 9, 789716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ji, P.; Xia, P.; Song, H.; Guo, Z.; Hu, X.; Guo, Y.; Yuan, X.; Song, Y.; Shen, R.; et al. Identification and targeting of cancer-associated fibroblast signature genes for prognosis and therapy in Cutaneous melanoma. Comput. Biol. Med. 2023, 167, 107597. [Google Scholar] [CrossRef]
- Ito, S.; Yokoyama, U.; Nakakoji, T.; Cooley, M.A.; Sasaki, T.; Hatano, S.; Kato, Y.; Saito, J.; Nicho, N.; Iwasaki, S.; et al. Fibulin-1 Integrates Subendothelial Extracellular Matrices and Contributes to Anatomical Closure of the Ductus Arteriosus. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2212–2226. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, A.; Dave, K.M.; Stetler, R.A.; Manickam, D.S. Targeting the blood-brain barrier for the delivery of stroke therapies. Adv. Drug Deliv. Rev. 2021, 171, 332–351. [Google Scholar] [CrossRef]
- Wong, B.W.; Marsch, E.; Treps, L.; Baes, M.; Carmeliet, P. Endothelial cell metabolism in health and disease: Impact of hypoxia. EMBO J. 2017, 36, 2187–2203. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chen, C.; Kang, S.S.; Liu, X.; Gu, X.; Yu, S.P.; Keene, C.D.; Cheng, L.; Ye, K. Neurotrophic signaling deficiency exacerbates environmental risks for Alzheimer’s disease pathogenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2100986118. [Google Scholar] [CrossRef] [PubMed]
- El Ouaamari, Y.; Van den Bos, J.; Willekens, B.; Cools, N.; Wens, I. Neurotrophic Factors as Regenerative Therapy for Neurodegenerative Diseases: Current Status, Challenges and Future Perspectives. Int. J. Mol. Sci. 2023, 24, 3866. [Google Scholar] [CrossRef]
- Kuznetsova, E.; Schliebs, R. β-Amyloid, cholinergic transmission, and cerebrovascular system—A developmental study in a mouse model of Alzheimer’s disease. Curr. Pharm. Des. 2013, 19, 6749–6765. [Google Scholar] [CrossRef]
- Ohsawa, I.; Takamura, C.; Kohsaka, S. Fibulin-1 binds the amino-terminal head of beta-amyloid precursor protein and modulates its physiological function. J. Neurochem. 2001, 76, 1411–1420. [Google Scholar] [CrossRef]
- Liu, X.T.; Liu, T.T.; Wu, M.Y.; Chen, Q.X.; Zhuang, J.X.; Wang, Q. Identifying FBLN1 (Gene ID: 2192) as a Potential Melanoma Biomarker for Melanoma based on an Analysis of microRNA Expression Profiles in the GEO and TCGA Databases. Genet. Test. Mol. Biomark. 2021, 25, 68–78. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, L.; Zhou, J.; Chen, Z.; Yang, G.; Liao, Y.; Zhu, M. Epidermal Growth Factor-Containing Fibulin-Like Extracellular Matrix Protein 1 (EFEMP1) Acts as a Potential Diagnostic Biomarker for Prostate Cancer. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2017, 23, 216–222. [Google Scholar] [CrossRef]
- Bardin, A.; Moll, F.; Margueron, R.; Delfour, C.; Chu, M.L.; Maudelonde, T.; Cavailles, V.; Pujol, P. Transcriptional and posttranscriptional regulation of fibulin-1 by estrogens leads to differential induction of messenger ribonucleic acid variants in ovarian and breast cancer cells. Endocrinology 2005, 146, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Nomoto, S.; Okamura, Y.; Hayashi, M.; Hishida, M.; Fujii, T.; Nishikawa, Y.; Sugimoto, H.; Takeda, S.; Nakao, A. Promoter hypermethylation of fibulin 1 gene is associated with tumor progression in hepatocellular carcinoma. Mol. Carcinog. 2011, 50, 571–579. [Google Scholar] [CrossRef]
- Yang, F.; Shi, J.; Zhang, L.; Wang, H.; Li, Y. Role of fibulin-1 gene promoter methylation in the carcinogenesis and development of tongue squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2022, 133, 432–440. [Google Scholar] [CrossRef]
- Wu, B.J.; Zhou, Z.P.; Li, W.P.; Ding, W.; Wu, Y.Z.; Zhou, Z.W.; Zhang, R.Q.; Liu, Q.F.; Jiang, H. Abnormal hypermethylation and clinicopathological significance of FBLN1 gene in cutaneous melanoma. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2014, 35, 123–127. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Qazi, T.J.; Quan, Z.; Mir, A.; Qing, H. Epigenetics in Alzheimer’s Disease: Perspective of DNA Methylation. Mol. Neurobiol. 2018, 55, 1026–1044. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Tong, Z.; Han, C.; Qiang, M.; Wang, W.; Lv, J.; Zhang, S.; Luo, W.; Li, H.; Luo, H.; Zhou, J.; et al. Age-related formaldehyde interferes with DNA methyltransferase function, causing memory loss in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Li, X.; Cheng, J.; Hou, L. Drug Development for Alzheimer’s Disease: Microglia Induced Neuroinflammation as a Target? Int. J. Mol. Sci. 2019, 20, 558. [Google Scholar] [CrossRef]
- Khan, S.; Shukla, S.; Sinha, S.; Meeran, S.M. Epigenetic targets in cancer and aging: Dietary and therapeutic interventions. Expert Opin. Ther. Targets 2016, 20, 689–703. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement. 2018, 4, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Brant, J.O.; Riva, A.; Resnick, J.L.; Yang, T.P. Influence of the Prader-Willi syndrome imprinting center on the DNA methylation landscape in the mouse brain. Epigenetics 2014, 9, 1540–1556. [Google Scholar] [CrossRef] [PubMed]
- Zhai, T.; Krass, E.; Zhang, F.; Huang, Z. A computational framework for identifying chemical compounds to bind Apolipoprotein E4 for Alzheimer’s disease intervention. Front. Syst. Biol. 2023, 3, 1188430. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, R.; Jian, Y.; Hu, Z.; Li, L.; Wang, H.; Miao, P.; Yang, Z.; Tang, M. A Bioinformatics-Based Study on Methylation Alterations of the FBLN1 Gene in Hippocampal Tissue of Alzheimer’s Disease Model DKO and DTG Mice. Int. J. Mol. Sci. 2024, 25, 9036. https://doi.org/10.3390/ijms25169036
Tang R, Jian Y, Hu Z, Li L, Wang H, Miao P, Yang Z, Tang M. A Bioinformatics-Based Study on Methylation Alterations of the FBLN1 Gene in Hippocampal Tissue of Alzheimer’s Disease Model DKO and DTG Mice. International Journal of Molecular Sciences. 2024; 25(16):9036. https://doi.org/10.3390/ijms25169036
Chicago/Turabian StyleTang, Rui, Yue Jian, Zhimin Hu, Li Li, Haitao Wang, Pengyu Miao, Zhihui Yang, and Mingxi Tang. 2024. "A Bioinformatics-Based Study on Methylation Alterations of the FBLN1 Gene in Hippocampal Tissue of Alzheimer’s Disease Model DKO and DTG Mice" International Journal of Molecular Sciences 25, no. 16: 9036. https://doi.org/10.3390/ijms25169036