
Identification and Characterization of Novel Founder Mutations in NDRG1: Refining the Genetic Landscape of Charcot–Marie–Tooth Disease Type 4D in Bulgaria

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
2.1. Identification of Novel Founder Variants in the NDRG1 Gene
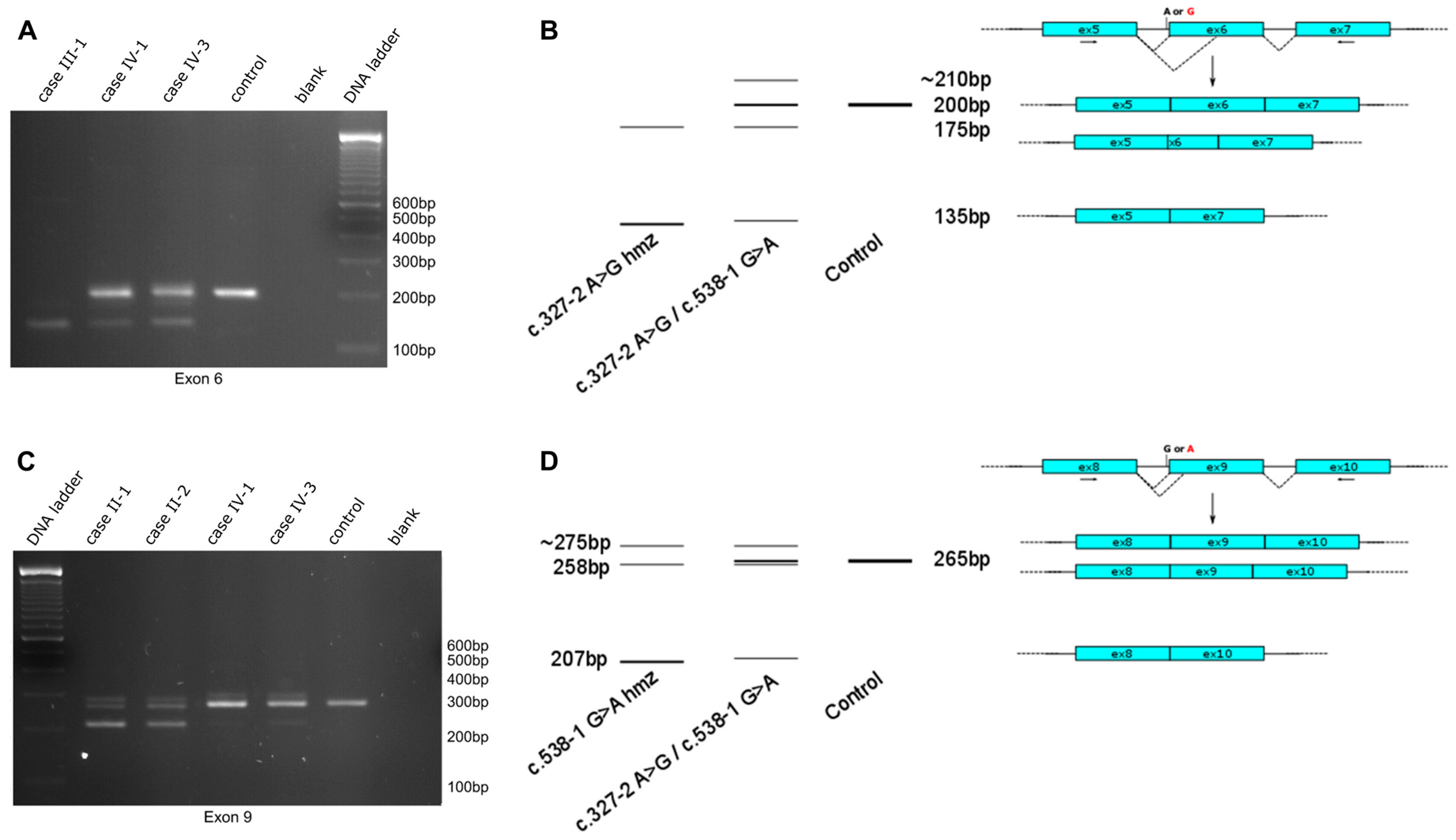
2.2. Functional Impact of Novel NDRG1 Variants
2.3. Clinical Findings
3. Discussion
4. Materials and Methods
4.1. Patient Cohort
4.2. Genetic Studies
4.3. Functional Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalaydjieva, L.; Nikolova, A.; Turnev, I.; Petrova, J.; Hristova, A.; Ishpekova, B.; Petkova, I.; Shmarov, A.; Stancheva, S.; Middleton, L.; et al. Hereditary Motor and Sensory Neuropathy—Lom, a Novel Demyelinating Neuropathy Associated with Deafness in Gypsies. Clinical, Electrophysiological and Nerve Biopsy Findings. Brain 1998, 121, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Kalaydjieva, L.; Gresham, D.; Gooding, R.; Heather, L.; Baas, F.; De Jonge, R.; Blechschmidt, K.; Angelicheva, D.; Chandler, D.; Worsley, P.; et al. N-Myc Downstream-Regulated Gene 1 Is Mutated in Hereditary Motor and Sensory Neuropathy-Lom. Am. J. Hum. Genet. 2000, 67, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Lachat, P.; Shaw, P.; Gebhard, S.; Van Belzen, N.; Chaubert, P.; Bosman, F.T. Expression of NDRG1, a Differentiation-Related Gene, in Human Tissues. Histochem. Cell Biol. 2002, 118, 399–408. [Google Scholar] [CrossRef] [PubMed]
- King, R.H.M.; Tournev, I.; Colomer, J.; Merlini, L.; Kalaydjieva, L.; Thomas, P.K. Ultrastructural Changes in Peripheral Nerve in Hereditary Motor and Sensory Neuropathy-Lom. Neuropathol. Appl. Neurobiol. 1999, 25, 306–312. [Google Scholar] [CrossRef] [PubMed]
- King, R.H.M.; Chandler, D.; Lopaticki, S.; Huang, D.; Blake, J.; Muddle, J.R.; Kilpatrick, T.; Nourallah, M.; Miyata, T.; Okuda, T.; et al. Ndrg1 in Development and Maintenance of the Myelin Sheath. Neurobiol. Dis. 2011, 42, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.T.; Ongusaha, P.P.; Hong, Y.K.; Kurdisiani, S.K.; Nakamura, M.; Lu, K.P.; Lee, S.W. Function of Drg1/Rit42 in P53-Dependent Mitotic Spindle Checkpoint. J. Biol. Chem. 2004, 279, 38597–38602. [Google Scholar] [CrossRef] [PubMed]
- Kokame, K.; Kato, H.; Miyata, T. Homocysteine-Respondent Genes in Vascular Endothelial Cells Identified by Differential Display Analysis: GRP78/BiP and Novel Genes. J. Biol. Chem. 1996, 271, 29659–29665. [Google Scholar] [CrossRef] [PubMed]
- Taketomi, Y.; Sugiki, T.; Saito, T.; Ishii, S.I.; Hisada, M.; Suzuki-Nishimura, T.; Uchida, M.K.; Moon, T.C.; Chang, H.W.; Natori, Y.; et al. Identification of NDRG1 as an Early Inducible Gene during in Vitro Maturation of Cultured Mast Cells. Biochem. Biophys. Res. Commun. 2003, 306, 339–346. [Google Scholar] [CrossRef]
- Parman, Y.; Battaloǧlu, E.; Bariş, I.; Bilir, B.; Poyraz, M.; Bissar-Tadmouri, N.; Williams, A.; Ammar, N.; Nelis, E.; Timmerman, V.; et al. Clinicopathological and Genetic Study of Early-Onset Demyelinating Neuropathy. Brain 2004, 127, 2540–2550. [Google Scholar] [CrossRef]
- Claramunt, R.; Sevilla, T.; Lupo, V.; Cuesta, A.; Millán, J.M.; Vílchez, J.J.; Palau, F.; Espinós, C. The p.R1109X Mutation in SH3TC2 Gene Is Predominant in Spanish Gypsies with Charcot-Marie-Tooth Disease Type 4. Clin. Genet. 2007, 71, 343–349. [Google Scholar] [CrossRef]
- Dačković, J.; Keckarević-Marković, M.; Komazec, Z.; Rakočević-Stojanović, V.; Lavrnić, D.; Stević, Z.; Ribarić, K.; Romac, S.; Apostolski, S. Hereditary Motor and Sensory Neuropathy Lom Type in a Serbian Family. Acta Myol. 2008, 27, 59–62. [Google Scholar]
- Ricard, E.; Mathis, S.; Magdelaine, C.; Delisle, M.; Magy, L.; Funalot, B.; Vallat, J. CMT4D (NDRG1 Mutation): Genotype-Phenotype Correlations. J. Peripher. Nerv. Syst. 2013, 18, 261–265. [Google Scholar] [CrossRef]
- Gabrikova, D.; Mistrik, M.; Bernasovska, J.; Bozikova, A.; Behulova, R.; Tothova, I.; Macekova, S. Founder Mutations in NDRG1 and HK1 Genes Are Common Causes of Inherited Neuropathies among Roma/Gypsies in Slovakia. J. Appl. Genet. 2013, 54, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Taroni, F.; Milani, M.; Del Grande, A.; Romano, A.; Bisogni, G.; Conte, A.; Contaldo, I.; Mercuri, E.; Sabatelli, M. Clinical, Electrophysiological and Pathological Findings in a Patient with Charcot-Marie-Tooth Disease 4D Caused by the NDRG1 Lom Mutation. J. Neurol. Sci. 2014, 345, 271–273. [Google Scholar] [CrossRef]
- Okamoto, Y.; Goksungur, M.T.; Pehlivan, D.; Beck, C.R.; Gonzaga-Jauregui, C.; Muzny, D.M.; Atik, M.M.; Carvalho, C.M.B.; Matur, Z.; Bayraktar, S.; et al. Exonic Duplication CNV of NDRG1 Associated with Autosomal-Recessive HMSN-Lom/CMT4D. Genet. Med. 2014, 16, 386–394. [Google Scholar] [CrossRef]
- Piscosquito, G.; Magri, S.; Saveri, P.; Milani, M.; Ciano, C.; Farina, L.; Taroni, F.; Pareyson, D. A Novel NDRG1 Mutation in a Non-Romani Patient with CMT4D/HMSN-Lom. J. Peripher. Nerv. Syst. 2017, 22, 47–50. [Google Scholar] [CrossRef]
- Dohrn, M.F.; Glöckle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hörtnagel, K.; et al. Frequent Genes in Rare Diseases: Panel-Based next Generation Sequencing to Disclose Causal Mutations in Hereditary Neuropathies. J. Neurochem. 2017, 143, 507–522. [Google Scholar] [CrossRef]
- Candayan, A.; Çakar, A.; Yunisova, G.; Acarlı, A.N.Ö.; Atkinson, D.; Topaloglu, P.; Durmuş, H.; Yapıcı, Z.; Jordanova, A.; Parman, Y.; et al. Genetic Survey of Autosomal Recessive Peripheral Neuropathy Cases Unravels High Genetic Heterogeneity in a Turkish Cohort. Neurol. Genet. 2021, 7, e621. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Niu, S.; Chen, N.; Pan, H.; Wang, X.; Zhang, Z. A Novel Homozygous NDRG1 Mutation in a Chinese Patient with Charcot-Marie-Tooth Disease 4D. J. Clin. Neurosci. 2018, 53, 231–234. [Google Scholar] [CrossRef]
- Pravinbabu, P.; Holla, V.V.; Phulpagar, P.; Kamble, N.; Netravathi, M.; Yadav, R.; Pal, P.K.; Muthusamy, B. A Splice Altering Variant in NDRG1 Gene Causes Charcot-Marie-Tooth Disease, Type 4D. Neurol. Sci. 2022, 43, 4463–4472. [Google Scholar] [CrossRef]
- Hunter, M.; Bernard, R.; Freitas, E.; Boyer, A.; Morar, B.; Martins, I.J.; Tournev, I.; Jordanova, A.; Guergelcheva, V.; Ishpekova, B.; et al. Mutation Screening of the N-Myc Downstream. Regulated Gene 1 (NDRG1) in Patients with Charcot-Marie-Tooth Disease. Hum. Mutat. 2003, 22, 129–135. [Google Scholar] [CrossRef]
- Chen, C.X.; Dong, H.L.; Wei, Q.; Li, L.X.; Yu, H.; Li, J.Q.; Liu, G.L.; Li, H.F.; Bai, G.; Ma, H.; et al. Genetic Spectrum and Clinical Profiles in a Southeast Chinese Cohort of Charcot-Marie-Tooth Disease. Clin. Genet. 2019, 96, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Alfares, A.; Alfadhel, M.; Wani, T.; Alsahli, S.; Alluhaydan, I.; Al Mutairi, F.; Alothaim, A.; Albalwi, M.; Al Subaie, L.; Alturki, S.; et al. A Multicenter Clinical Exome Study in Unselected Cohorts from a Consanguineous Population of Saudi Arabia Demonstrated a High Diagnostic Yield. Mol. Genet. Metab. 2017, 121, 91–95. [Google Scholar] [CrossRef]
- Li, L.X.; Liu, G.L.; Liu, Z.J.; Lu, C.; Wu, Z.Y. Identification and Functional Characterization of Two Missense Mutations in NDRG1 Associated with Charcot-Marie-Tooth Disease Type 4D. Hum. Mutat. 2017, 38, 1569–1578. [Google Scholar] [CrossRef]
- Dubourg, O.; Azzedine, H.; Verny, C.; Durosier, G.; Birouk, N.; Gouider, R.; Salih, M.; Bouhouche, A.; Thiam, A.; Grid, D.; et al. Autosomal-Recessive Forms of Demyelinating Charcot-Marie-Tooth Disease. NeuroMolecular Med. 2006, 8, 75–85. [Google Scholar] [CrossRef]
- Ishpekova, B.A.; Christova, L.G.; Alexandrov, A.S.; Thomas, P.K. The Electrophysiological Profile of Hereditary Motor and Sensory Neuropathy-Lom. J. Neurol. Neurosurg. Psychiatry 2005, 76, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Guergueltcheva, V.; Tournev, I.; Bojinova, V.; Hantke, J.; Litvinenko, I.; Ishpekova, B.; Shmarov, A.; Petrova, J.; Jordanova, A.; Kalaydjieva, L. Early Clinical and Electrophysiologic Features of the Two Most Common Autosomal Recessive Forms of Charcot-Marie-Tooth Disease in the Roma (Gypsies). J. Child. Neurol. 2006, 21, 20–25. [Google Scholar] [CrossRef]
- Merlini, L.; Villanova, M.; Sabatelli, P.; Trogu, A.; Malandrini, A.; Yanakiev, P.; Maraldi, N.; Kalaydjieva, L. Hereditary Motor and Sensory Neuropathy Lom Type in an Italian Gypsy Family. Neuromuscul. Disord. 1998, 8, 182–185. [Google Scholar] [CrossRef]
- Colomer, J.; Iturriaga, C.; Kalaydjieva, L.; Angelicheva, D.; King, R.H.M.; Thomas, P.K. Hereditary Motor and Sensory Neuropathy-Lom (HMSNL) in a Spanish Family: Clinical, Electrophysiological, Pathological and Genetic Studies. Neuromuscul. Disord. 2000, 10, 578–583. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Murphy, S.M.; Herrmann, D.N.; McDermott, M.P.; Scherer, S.S.; Shy, M.E.; Reilly, M.M.; Pareyson, D. Reliability of the CMT Neuropathy Score (Second Version) in Charcot-Marie-Tooth Disease. J. Peripher. Nerv. Syst. 2011, 16, 191–198. [Google Scholar] [CrossRef]
- Mustonen, V.; Muruganandam, G.; Loris, R.; Kursula, P.; Ruskamo, S. Crystal and Solution Structure of NDRG1, a Membrane-Binding Protein Linked to Myelination and Tumour Suppression. FEBS J. 2021, 288, 3507–3529. [Google Scholar] [CrossRef] [PubMed]
- World Directory of Minorities and Indigenous Peoples—Bulgaria: Bulgarian-Speaking Muslims (Pomaks)|Refworld. Available online: https://www.refworld.org/reference/countryrep/mrgi/2018/en/65202 (accessed on 5 July 2024).
- Chamova, T.; Bichev, S.; Todorov, T.; Gospodinova, M.; Taneva, A.; Kastreva, K.; Zlatareva, D.; Krupev, M.; Hadjiivanov, R.; Guergueltcheva, V.; et al. Limb Girdle Muscular Dystrophy 2G in a Religious Minority of Bulgarian Muslims Homozygous for the c.75G>A, p.Trp25X Mutation. Neuromuscul. Disord. 2018, 28, 625–632. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Degos, B.; Bonnet, C.; Latour, P.; Hamadouche, T.; Lévy, N.; Leheup, B. NDRG1-Linked Charcot-Marie-Tooth Disease (CMT4D) with Central Nervous System Involvement. Neuromuscul. Disord. 2007, 17, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Baethmann, M.; Göhlich-Ratmann, G.; Schröder, J.M.; Kalaydjieva, L.; Voit, T. HMSNL in a 13-Year-Old Bulgarian Girl. Neuromuscul. Disord. 1998, 8, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Butinar, D.; Zidar, J.; Leonardis, L.; Popovic, M.; Kalaydjieva, L.; Angelicheva, D.; Sininger, Y.; Keats, B.; Starr, A. Hereditary Auditory, Vestibular, Motor, and Sensory Neuropathy in a Slovenian Roma (Gypsy) Kindred. Ann. Neurol. 1999, 46, 36–44. [Google Scholar] [CrossRef]
- Chandler, D.; Angelicheva, D.; Heather, L.; Gooding, R.; Gresham, D.; Yanakiev, P.; De Jonge, R.; Baas, F.; Dye, D.; Karagyozov, L.; et al. Hereditary Motor and Sensory Neuropathy—Lom (HMSNL): Refined Genetic Mapping in Romani (Gypsy) Families from Several European Countries. Neuromuscul. Disord. 2000, 10, 584–591. [Google Scholar] [CrossRef]
- Medical Research Council; Riddoch, G.; Rowley-Bristow, W.; Cairns, H.; Carmichael, E.; Critchley, M.; Greenfield, J.; Learmonth, J.; Seddon, H.; Seymonds, C.; et al. Aids to the Examination of the Peripheral Nervous System; HMSO: London, UK, 1943; Volume 45. [Google Scholar]
- Zimoń, M.; Baets, J.; Almeida-Souza, L.; De Vriendt, E.; Nikodinovic, J.; Parman, Y.; Battaloǧlu, E.; Matur, Z.; Guergueltcheva, V.; Tournev, I.; et al. Loss-of-Function Mutations in HINT1 Cause Axonal Neuropathy with Neuromyotonia. Nat. Genet. 2012, 44, 1080–1083. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Family | Individual | Ethnicity | Variant in NDRG1 | Zygosity | ACMG Criteria | ACMG Classification | Interpretation |
|---|---|---|---|---|---|---|---|
| I * | I-1 | Bulgarian Muslims | c.538-1G>A | Homozygous | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant |
| II | II-1 | Bulgarian Muslims | c.538-1G>A | Homozygous | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant |
| II-2 | c.538-1G>A | Homozygous | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant | ||
| III | III-1 | Bulgarian Muslims | c.327-2A>G | Homozygous | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant |
| IV | IV-1 | Bulgarian Muslims | c.538-1G>A | Compound heterozygous | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant |
| c.327-2A>G | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant | ||||
| IV-2 | c.538-1G>A | Compound heterozygous | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant | ||
| c.327-2A>G | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant | ||||
| IV-3 | c.538-1G>A | Compound heterozygous | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant | ||
| c.327-2A>G | PVS1, PS3, PM2 | Pathogenic | Novel splice-site variant | ||||
| V | V-1 | Roma | c.508dup, p.E170Gfs*35 | Compound heterozygous | PVS1, PM2, PM3 | Pathogenic | Novel frameshift variant |
| c.442C>T, p.R148X | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant | ||||
| VI | VI-1 | Roma | c.442C>T, p.R148X | Homozygous | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant |
| VII | VII-1 | Roma | c.442C>T, p.R148X | Homozygous | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant |
| VIII | VIII-1 | Roma | c.442C>T, p.R148X | Homozygous | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant |
| IX | IX-1 | Roma | c.442C>T, p.R148X | Homozygous | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant |
| IX-2 | c.442C>T, p.R148X | Homozygous | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant | ||
| X | X-1 | Roma | c.442C>T, p.R148X | Homozygous | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant |
| XI | XI-1 | Roma | c.442C>T, p.R148X | Homozygous | PVS1, PS3, PM2 | Pathogenic | Recurrent disease-causing variant |
| Case ID | Age at Starting to Walk | Age at Onset | Initial Symptoms | Age of Involvement of Upper Limbs | Age at Assessment | Tendon Reflexes | Superficial Sensation | Deep Sensation | Limb Deformities | Hearing Loss | CMTNS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Biceps | Triceps | Brachioradial | Knee | Achille | Upper Limbs | Lower Limbs | Upper Limbs | Lower Limbs | Upper Limbs * | Lower Limbs ** | ||||||||
| I-1 | NA | 7 y | Impaired gait, distal weakness and impaired sensation in LL | 10 y | 43 y | - | - | - | - | - | D | D | D | D | + | + Pes equinovarus, scoliosis | + | 38 |
| II-1 | 1 y 3 mo | 7 y | Weakness in distal parts of lower limbs | 12 y | 30 y | + | + | - | - | - | D | D | N | D | + R>L | + | - | 23 |
| II-2 | 1 y 4 mo | 8 y | Weakness in distal parts of lower limbs | 13 y | 31 y | + | + | - | - | - | D | D | N | D | + R>L | + The right limb is 7 cm shorter due to congenital luxation of right art. coxae | - | 28 |
| III-1 | 1 y | 9 y | Weakness in the lower limbs, impaired gait | 16 y | 20 y | + | + | - | - | - | D | D | N | D | + | + Pes equinovarus | + | 24 |
| IV-1 | 1 y | 7 y | Frequent falls, impaired gait | 11 y | 15 y | + | + | - | - | - | D | D | N | D | + | + | + | 34 |
| IV-2 | 1 y 2 mo | 6 y | Frequent falls, impaired gait | 10 y | 13 y | + | + | - | - | - | D | D | N | D | + | + | + | 29 |
| IV-3 | 1 y 2 mo | 12 y | Weakness in distal parts of lower limbs | 16 y | 36 y | - | - | - | - | - | D | D | D | D | + | + Pes equinovarus, more severe on the right | + | 34 |
| Case | N. medianus CV/DL/A | N. ulnaris CV/DL/A | N. peroneus CV/DL/A | N. tibialis CV/DL/A | Audiometry | Auditory Evoked Potentials | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Latencies ms | Interpeak Latencies ms | ||||||||||
| Wave I R/L | Wave III R/L | Wave V R/L | I-III R/L | III-V R/L | I-V R/L | ||||||
| Case I-1 at 9 years | NA | 10.3/NA/NA | NM | NA | Severe sensorineural hearing loss | NA | |||||
| Case I-1 at 43 years | NM | NM | NM | NM | Severe sensorineural hearing loss | Severely affected. Not possible to identify the different waves. | |||||
| Case II-1 at 30 years | 9.7/8.6/1.2 | 11.5/9.2/0.3 | NM | NM | Normal | 1.9/2.0 | 4.8/5.0 | 7.5/7.8 | 2.9/3.0 | 2.7/2.8 | 5.6/5.8 |
| Case II-2 at 31 years | 16.7/11.9/0.27 | 12.4/9.51/0.34 | NM | NM | Normal | 1.9/1.6 | 4.4/4.4 | 7.2/7.1 | 2.5/2.8 | 2.8/2.7 | 5.3/5.5 |
| Case III-1 at 20 years | 10.8/23.8/1.8 | 12.5/21.3/1.9 | NM | NM | Severe sensorineural hearing loss | Severely affected. Not possible to identify the different waves. | |||||
| Case IV-1 at 9 years | 13.7/11.7/2.8 | NA | NM | NM | ND | ND | ND | ND | ND | ND | ND |
| Case IV-1 at 15 years | NM | NM | NM | NM | Sensorineural hearing loss, especially for high frequencies | 2.96/2.87 | 6.04/6.00 | 8.84/8.46 | 3.08/3.13 | 2.8/2.46 | 5.88/5.59 |
| Case IV-2 at 2.5 years | 15.6/10.4/2.2 | NA | 12.8/12.5/0.2 | NA | ND | ND | ND | ND | ND | ND | ND |
| Case IV-2 at 12 years | 5.8/13.8/0.1 | NM | NM | NM | Sensorineural hearing loss, especially for high frequencies | 3.05/3.07 | 5.87/5.98 | 7.95/7.99 | 2.82/3.91 | 2.08/2.01 | 4.9/4.92 |
| Case IV-3 at 36 years | NM | NM | NM | NM | Sensorineural hearing loss, especially for high frequencies | 2.1/2.2 | 4.8/5.0 | 7.7/8.0 | 2.7/2.8 | 2.9/3.0 | 5.6/5.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atkinson, D.; Chamova, T.; Candayan, A.; Kastreva, K.; Asenov, O.; Litvinenko, I.; Estrada-Cuzcano, A.; De Vriendt, E.; Kukushev, G.; Tournev, I.; et al. Identification and Characterization of Novel Founder Mutations in NDRG1: Refining the Genetic Landscape of Charcot–Marie–Tooth Disease Type 4D in Bulgaria. Int. J. Mol. Sci. 2024, 25, 9047. https://doi.org/10.3390/ijms25169047
Atkinson D, Chamova T, Candayan A, Kastreva K, Asenov O, Litvinenko I, Estrada-Cuzcano A, De Vriendt E, Kukushev G, Tournev I, et al. Identification and Characterization of Novel Founder Mutations in NDRG1: Refining the Genetic Landscape of Charcot–Marie–Tooth Disease Type 4D in Bulgaria. International Journal of Molecular Sciences. 2024; 25(16):9047. https://doi.org/10.3390/ijms25169047
Chicago/Turabian StyleAtkinson, Derek, Teodora Chamova, Ayse Candayan, Kristina Kastreva, Ognian Asenov, Ivan Litvinenko, Alejandro Estrada-Cuzcano, Els De Vriendt, Georgi Kukushev, Ivailo Tournev, and et al. 2024. "Identification and Characterization of Novel Founder Mutations in NDRG1: Refining the Genetic Landscape of Charcot–Marie–Tooth Disease Type 4D in Bulgaria" International Journal of Molecular Sciences 25, no. 16: 9047. https://doi.org/10.3390/ijms25169047