
Pyroptosis in Skeleton Diseases: A Potential Therapeutic Target Based on Inflammatory Cell Death


Abstract
1. Introduction
1.1. Pyroptosis: From Discovery to Development
1.2. A Quick Look at Pyroptosis

{kind=link}
{kind=link}
| Cell Death Types | Morphological Characteristics | Key Components | Refs. | ||
|---|---|---|---|---|---|
| Cell Membrane | Cytoplasm and Organelles | Nucleus | |||
| Pyroptosis | Formation of membrane pores; membrane blebbing; formation of pyroptotic bodies; membrane rupture | Cytoplasmic swelling; swollen mitochondria with reduced matrix density | Intact nucleus; chromatin condensation | Caspase-1/3/4/5/8/11; gasdermins; inflammasomes | [24,25,27,32,34,37,38] |
| Apoptosis | Intact cell membrane; membrane blebbing; formation of apoptotic bodies | Reduced cellular volume; swollen mitochondria; MOMP | Nuclear fragmentation; chromatin condensation | Caspase-2/3/6/10; Bcl-2 family; death receptors; TNF receptor superfamily members | [28,33,35,38,47] |
| Necroptosis | Formation of membrane pores; membrane permeabilization and rupture | Cytoplasmic and organelle swelling | Dilated perinuclear space; sickle nucleus | MLKL; RIPK1; RIPK3 | [30,31,36,48,49] |
2. Signaling Pathways of Pyroptosis and Their Significance in Skeleton Diseases
2.1. Pro-Inflammatory Pathway
2.1.1. Canonical Inflammasome Pathway
A Quick Look at the Canonical Inflammasome Pathway
Canonical Inflammasome Pathway in Skeleton Disease
2.1.2. Non-Canonical Inflammasome Pathway

2.2. Alternative Pathways
2.2.1. Apoptotic Caspases Pathway
2.2.2. Granzyme Pathway
2.2.3. GSDMA-Mediated Pathway
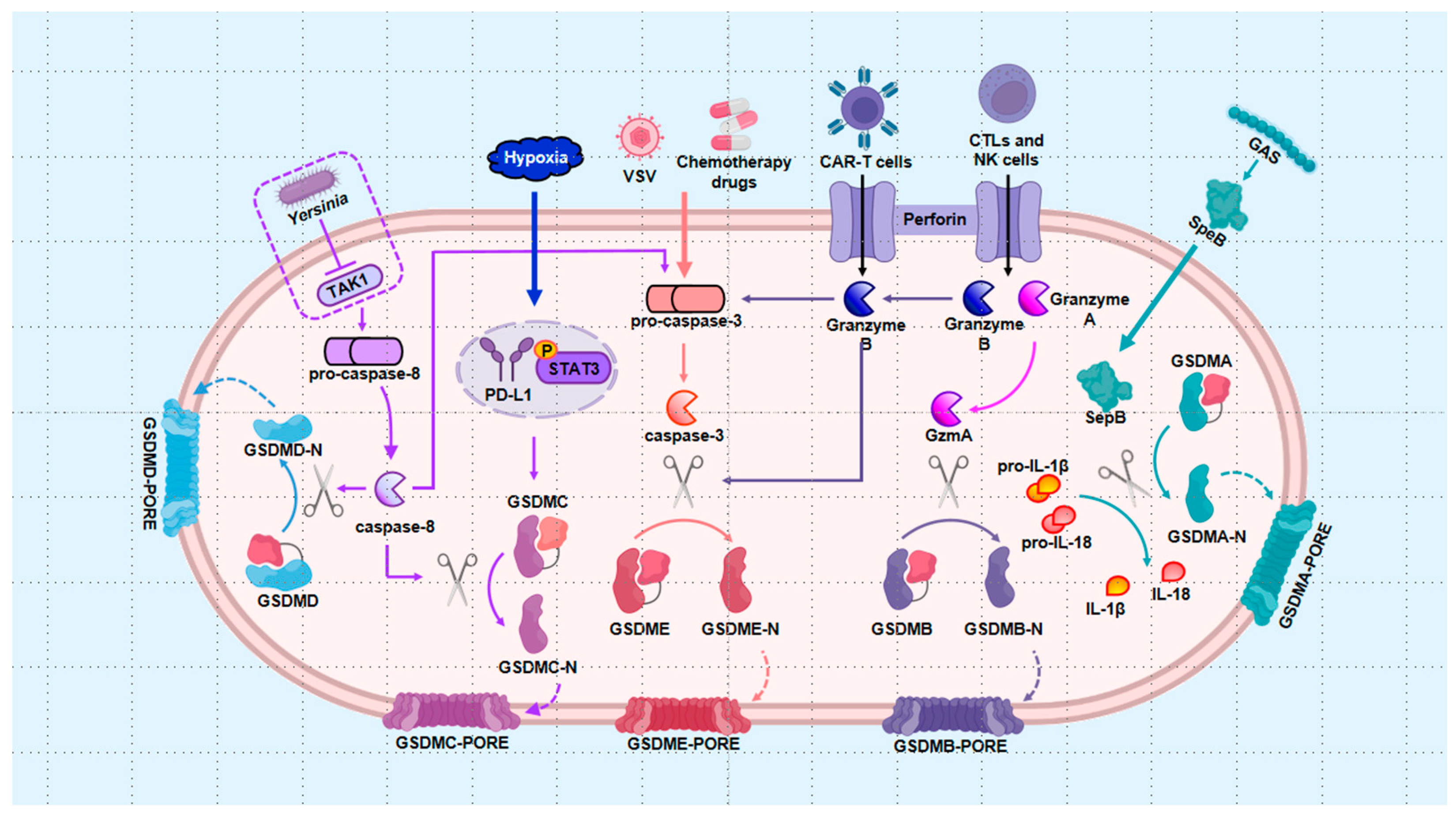
3. The Regulation of Pyroptosis in Cells with Bone Diseases
3.1. Macrophages
3.2. Chondrocytes
3.3. Synovial Cells
3.4. Nucleus Pulposus Cell
4. Pyroptosis as an Intervention Target to Treat Skeleton Diseases
4.1. Exosomes and miRNAs
4.2. Non-Drug Therapy
4.3. Drug Therapy
4.3.1. Melatonin
4.3.2. Common Drugs
5. Conclusions and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Buttner, S.; Kroemer, G.; Madeo, F. The mitochondrial pathway in yeast apoptosis. Apoptosis 2007, 12, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [PubMed]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Hersh, D.; Monack, D.M.; Smith, M.R.; Ghori, N.; Falkow, S.; Zychlinsky, A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA 1999, 96, 2396–2401. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Boise, L.H.; Collins, C.M. Salmonella-induced cell death: Apoptosis, necrosis or programmed cell death? Trends Microbiol. 2001, 9, 64–67. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Flores-Romero, H.; Ros, U.; Garcia-Saez, A.J. Pore formation in regulated cell death. EMBO J. 2020, 39, e105753. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Chan, A.H.; von Pein, J.B.; Maddugoda, M.P.; Boucher, D.; Schroder, K. Inflammatory Caspases: Toward a Unified Model for Caspase Activation by Inflammasomes. Annu. Rev. Immunol. 2022, 40, 249–269. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Wang, R.; Tan, H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int. J. Biol. Sci. 2019, 15, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, B.A.; Dixit, V.M.; Power, C. Fiery Cell Death: Pyroptosis in the Central Nervous System. Trends Neurosci. 2020, 43, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, S.; Wan, B.; Velani, B.; Zhu, Y. Pyroptosis in Liver Disease: New Insights into Disease Mechanisms. Aging Dis. 2019, 10, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Loveless, R.; Bloomquist, R.; Teng, Y. Pyroptosis at the forefront of anticancer immunity. J. Exp. Clin. Cancer Res. 2021, 40, 264. [Google Scholar] [CrossRef]
- Allen, K.D.; Thoma, L.M.; Golightly, Y.M. Epidemiology of osteoarthritis. Osteoarthr. Cartil. 2022, 30, 184–195. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.A.; Hoy, D.; Smith, E.; Bettampadi, D.; Mansournia, M.A.; Almasi-Hashiani, A.; Ashrafi-Asgarabad, A.; Moradi-Lakeh, M.; Qorbani, M.; et al. Global, regional and national burden of rheumatoid arthritis 1990–2017: A systematic analysis of the Global Burden of Disease study 2017. Ann. Rheum. Dis. 2019, 78, 1463–1471. [Google Scholar] [CrossRef]
- Wang, L.; Yu, W.; Yin, X.; Cui, L.; Tang, S.; Jiang, N.; Cui, L.; Zhao, N.; Lin, Q.; Chen, L.; et al. Prevalence of Osteoporosis and Fracture in China: The China Osteoporosis Prevalence Study. JAMA Netw. Open 2021, 4, e2121106. [Google Scholar] [CrossRef]
- Storheim, K.; Zwart, J.A. Musculoskeletal disorders and the Global Burden of Disease study. Ann. Rheum. Dis. 2014, 73, 949–950. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xing, R.; Huang, Z.; Zhang, N.; Zhang, L.; Li, X.; Wang, P. Inhibition of Synovial Macrophage Pyroptosis Alleviates Synovitis and Fibrosis in Knee Osteoarthritis. Mediat. Inflamm. 2019, 2019, 2165918. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yuan, P.; Yu, W.; Lin, J.; Xu, A.; Xu, X.; Lou, J.; Yu, T.; Qian, C.; Liu, B.; et al. Mitochondria-Targeting Polymer Micelle of Dichloroacetate Induced Pyroptosis to Enhance Osteosarcoma Immunotherapy. ACS Nano 2022, 16, 10327–10340. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Sborgi, L.; Ruhl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Muller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, W.T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef] [PubMed]
- de Vasconcelos, N.M.; Van Opdenbosch, N.; Van Gorp, H.; Parthoens, E.; Lamkanfi, M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019, 26, 146–161. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 2010, 8, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prevost, M.C.; et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000, 192, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in Cell Death, Inflammation, and Pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Ramirez, M.L.G.; Salvesen, G.S. A primer on caspase mechanisms. Semin Cell Dev. Biol. 2018, 82, 79–85. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Declercq, W.; Kalai, M.; Saelens, X.; Vandenabeele, P. Alice in caspase land. A phylogenetic analysis of caspases from worm to man. Cell Death Differ. 2002, 9, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Orning, P.; Weng, D.; Starheim, K.; Ratner, D.; Best, Z.; Lee, B.; Brooks, A.; Xia, S.; Wu, H.; Kelliher, M.A.; et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 2018, 362, 1064–1069. [Google Scholar] [CrossRef]
- Chen, K.W.; Gross, C.J.; Sotomayor, F.V.; Stacey, K.J.; Tschopp, J.; Sweet, M.J.; Schroder, K. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep. 2014, 8, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.Z.; Crawford, N.; Longley, D.B. The role of Ubiquitination in Apoptosis and Necroptosis. Cell Death Differ. 2022, 29, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Murai, S.; Kakuta, S.; Uchiyama, Y.; Nakano, H. Identification of the hallmarks of necroptosis and ferroptosis by transmission electron microscopy. Biochem. Biophys. Res. Commun. 2020, 527, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kuang, S.; Zheng, J.; Yang, H.; Li, S.; Duan, S.; Shen, Y.; Ji, C.; Gan, J.; Xu, X.W.; Li, J. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, 10642–10647. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B.; Deisenhofer, J. A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature 1995, 374, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Ratsimandresy, R.A.; Dorfleutner, A.; Stehlik, C. An Update on PYRIN Domain-Containing Pattern Recognition Receptors: From Immunity to Pathology. Front. Immunol. 2013, 4, 440. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [PubMed]
- Vance, R.E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 2015, 32, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745. [Google Scholar] [CrossRef]
- Kumari, P.; Russo, A.J.; Shivcharan, S.; Rathinam, V.A. AIM2 in health and disease: Inflammasome and beyond. Immunol. Rev. 2020, 297, 83–95. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Wood, G.; Masters, S.L.; Richard, K.; Park, G.; Smith, B.J.; Kastner, D.L. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc. Natl. Acad. Sci. USA 2006, 103, 9982–9987. [Google Scholar] [CrossRef] [PubMed]
- Ewald, S.E.; Chavarria-Smith, J.; Boothroyd, J.C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 2014, 82, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Boyden, E.D.; Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Nunez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Yang, X.D.; Li, W.; Zhang, S.; Wu, D.; Jiang, X.; Tan, R.; Niu, X.; Wang, Q.; Wu, X.; Liu, Z.; et al. PLK4 deubiquitination by Spata2-CYLD suppresses NEK7-mediated NLRP3 inflammasome activation at the centrosome. EMBO J. 2020, 39, e102201. [Google Scholar] [CrossRef]
- Reyes Ruiz, V.M.; Ramirez, J.; Naseer, N.; Palacio, N.M.; Siddarthan, I.J.; Yan, B.M.; Boyer, M.A.; Pensinger, D.A.; Sauer, J.D.; Shin, S. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2017, 114, 13242–13247. [Google Scholar] [CrossRef]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Lima Leite, E.; Gautron, A.; Deplanche, M.; Nicolas, A.; Ossemond, J.; Nguyen, M.T.; do Carmo, F.L.R.; Gilot, D.; Azevedo, V.; Gotz, F.; et al. Involvement of caspase-1 in inflammasomes activation and bacterial clearance in S. aureus-infected osteoblast-like MG-63 cells. Cell. Microbiol. 2020, 22, e13204. [Google Scholar] [CrossRef] [PubMed]
- McCall, S.H.; Sahraei, M.; Young, A.B.; Worley, C.S.; Duncan, J.A.; Ting, J.P.; Marriott, I. Osteoblasts express NLRP3, a nucleotide-binding domain and leucine-rich repeat region containing receptor implicated in bacterially induced cell death. J. Bone Miner. Res. 2008, 23, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhang, K.; Lu, K.; Shi, T.; Shen, S.; Chen, X.; Dong, J.; Gong, W.; Bao, Z.; Shi, Y.; et al. Inhibition of pyroptosis attenuates Staphylococcus aureus-induced bone injury in traumatic osteomyelitis. Ann. Transl. Med. 2019, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Compston, J.E.; McClung, M.R.; Leslie, W.D. Osteoporosis. Lancet 2019, 393, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, K.; Wan, X.; Wang, F.; Guo, Z.; Mo, Z. NLRP3 inflammasome activation in mesenchymal stem cells inhibits osteogenic differentiation and enhances adipogenic differentiation. Biochem. Biophys. Res. Commun. 2017, 484, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Du, J.; Li, D.; Yang, P.; Kou, Y.; Li, C.; Zhou, Q.; Lu, Y.; Hasegawa, T.; Li, M. Oxidative stress induced pyroptosis leads to osteogenic dysfunction of MG63 cells. J. Mol. Histol. 2020, 51, 221–232. [Google Scholar] [CrossRef]
- Tao, Z.; Wang, J.; Wen, K.; Yao, R.; Da, W.; Zhou, S.; Meng, Y.; Qiu, S.; Yang, K.; Zhu, Y.; et al. Pyroptosis in Osteoblasts: A Novel Hypothesis Underlying the Pathogenesis of Osteoporosis. Front. Endocrinol. 2020, 11, 548812. [Google Scholar] [CrossRef]
- Dai, S.M.; Nishioka, K.; Yudoh, K. Interleukin (IL) 18 stimulates osteoclast formation through synovial T cells in rheumatoid arthritis: Comparison with IL1 beta and tumour necrosis factor alpha. Ann. Rheum. Dis. 2004, 63, 1379–1386. [Google Scholar] [CrossRef]
- Sartoretto, S.; Gemini-Piperni, S.; da Silva, R.A.; Calasans, M.D.; Rucci, N.; Pires Dos Santos, T.M.; Lima, I.B.C.; Rossi, A.M.; Alves, G.; Granjeiro, J.M.; et al. Apoptosis-associated speck-like protein containing a caspase-1 recruitment domain (ASC) contributes to osteoblast differentiation and osteogenesis. J. Cell. Physiol. 2019, 234, 4140–4153. [Google Scholar] [CrossRef]
- Detzen, L.; Cheat, B.; Besbes, A.; Hassan, B.; Marchi, V.; Baroukh, B.; Lesieur, J.; Sadoine, J.; Torrens, C.; Rochefort, G.; et al. NLRP3 is involved in long bone edification and the maturation of osteogenic cells. J. Cell. Physiol. 2021, 236, 4455–4469. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, D.; Cao, Y.; Zhang, H.; Li, J.; Xu, J.; Yu, L.; Ye, S.; Yang, L. Screening of crosstalk and pyroptosis-related genes linking periodontitis and osteoporosis based on bioinformatics and machine learning. Front. Immunol. 2022, 13, 955441. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.R.G.; Delitto, A.E.; de Souza, J.A.C.; Gonzalez-Maldonado, L.A.; Wallet, S.M.; Rossa Junior, C. Relevance of Caspase-1 and Nlrp3 Inflammasome on Inflammatory Bone Resorption in A Murine Model of Periodontitis. Sci. Rep. 2020, 10, 7823. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Kurita-Ochiai, T.; Kobayashi, R.; Suzuki, T.; Ando, T. Regulation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated periodontal disease. Inflamm. Res. 2017, 66, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, Q.; Lv, C.; Chen, Y.; Zhao, W.; Li, W.; Chen, H.; Wang, H.; Sun, W.; Yuan, H. NLRP3 regulates alveolar bone loss in ligature-induced periodontitis by promoting osteoclastic differentiation. Cell Prolif. 2021, 54, e12973. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Song, J.H.; Oh, S.H.; Kim, J.W.; Lee, M.N.; Piao, X.; Yang, J.W.; Kim, O.S.; Kim, T.S.; Kim, S.H.; et al. Targeting NLRP3 Inflammasome Reduces Age-Related Experimental Alveolar Bone Loss. J. Dent. Res. 2020, 99, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.I.; Mae, M.; Farhana, F.; Oohira, M.; Yamashita, Y.; Ozaki, Y.; Sakai, E.; Yoshimura, A. NLRP3 Inflammasome Negatively Regulates RANKL-Induced Osteoclastogenesis of Mouse Bone Marrow Macrophages but Positively Regulates It in the Presence of Lipopolysaccharides. Int. J. Mol. Sci. 2022, 23, 6096. [Google Scholar] [CrossRef]
- Mateen, S.; Zafar, A.; Moin, S.; Khan, A.Q.; Zubair, S. Understanding the role of cytokines in the pathogenesis of rheumatoid arthritis. Clin. Chim. Acta 2016, 455, 161–171. [Google Scholar]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef]
- Jing, Y.; Han, D.; Xi, C.; Yan, J.; Zhuang, J. Identification of Cross-Talk and Pyroptosis-Related Genes Linking Periodontitis and Rheumatoid Arthritis Revealed by Transcriptomic Analysis. Dis. Markers 2021, 2021, 5074305. [Google Scholar] [CrossRef] [PubMed]
- Vande Walle, L.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.D.; van Loo, G.; et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Li, K.T.; Yang, H.X.; Yang, B.; Lu, X.; Zhao, L.D.; Fei, Y.Y.; Chen, H.; Wang, L.; Li, J.; et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J. Autoimmun. 2020, 106, 102336. [Google Scholar] [CrossRef]
- Dayer, J.M. The pivotal role of interleukin-1 in the clinical manifestations of rheumatoid arthritis. Rheumatology 2003, 42 (Suppl. S2), ii3–ii10. [Google Scholar] [CrossRef]
- Zhao, L.R.; Xing, R.L.; Wang, P.M.; Zhang, N.S.; Yin, S.J.; Li, X.C.; Zhang, L. NLRP1 and NLRP3 inflammasomes mediate LPS/ATPinduced pyroptosis in knee osteoarthritis. Mol. Med. Rep. 2018, 17, 5463–5469. [Google Scholar] [PubMed]
- Wang, S.; Mobasheri, A.; Zhang, Y.; Wang, Y.; Dai, T.; Zhang, Z. Exogenous stromal cell-derived factor-1 (SDF-1) suppresses the NLRP3 inflammasome and inhibits pyroptosis in synoviocytes from osteoarthritic joints via activation of the AMPK signaling pathway. Inflammopharmacology 2021, 29, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, L.; Huang, Z.; Xing, R.; Li, X.; Yin, S.; Mao, J.; Zhang, N.; Mei, W.; Ding, L.; et al. Increased HIF-1alpha in Knee Osteoarthritis Aggravate Synovial Fibrosis via Fibroblast-Like Synoviocyte Pyroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 6326517. [Google Scholar] [PubMed]
- An, S.; Hu, H.; Li, Y.; Hu, Y. Pyroptosis Plays a Role in Osteoarthritis. Aging Dis. 2020, 11, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Chang, X.; Kang, Y.; Yang, Y.; Chen, Y.; Shen, Y.; Jiang, C.; Shen, Y. Pyroptosis: A Novel Intervention Target in the Progression of Osteoarthritis. J. Inflamm. Res. 2022, 15, 3859–3871. [Google Scholar] [CrossRef]
- Wang, C.; Xu, C.X.; Alippe, Y.; Qu, C.; Xiao, J.; Schipani, E.; Civitelli, R.; Abu-Amer, Y.; Mbalaviele, G. Chronic inflammation triggered by the NLRP3 inflammasome in myeloid cells promotes growth plate dysplasia by mesenchymal cells. Sci. Rep. 2017, 7, 4880. [Google Scholar] [CrossRef]
- Goodman, S.B.; Gallo, J. Periprosthetic Osteolysis: Mechanisms, Prevention and Treatment. J. Clin. Med. 2019, 8, 2091. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, M.; Yu, A.; Mao, H.; Zhang, J. Inhibitory effects of beta-tricalciumphosphate wear particles on osteocytes via apoptotic response and Akt inactivation. Toxicology 2012, 297, 57–67. [Google Scholar] [CrossRef]
- Kennedy, O.D.; Herman, B.C.; Laudier, D.M.; Majeska, R.J.; Sun, H.B.; Schaffler, M.B. Activation of resorption in fatigue-loaded bone involves both apoptosis and active pro-osteoclastogenic signaling by distinct osteocyte populations. Bone 2012, 50, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, O.D.; Laudier, D.M.; Majeska, R.J.; Sun, H.B.; Schaffler, M.B. Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo. Bone 2014, 64, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, M.; Niu, W.; Mao, H.; Yang, P.; Xu, B.; Sun, Y. Tricalcium phosphate particles promote pyroptotic death of calvaria osteocytes through the ROS/NLRP3/Caspase-1 signaling axis in amouse osteolysis model. Int. Immunopharmacol. 2022, 107, 108699. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Jin, S.H.; Wang, M.Y.; Jin, X.L.; Lv, C.; Deng, Y.F.; Wang, J.L. Enhanced NLRP3, caspase-1, and IL-1beta levels in degenerate human intervertebral disc and their association with the grades of disc degeneration. Anat. Rec. 2015, 298, 720–726. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, P.; Zhou, Z.; Yuan, Y.; Jiao, Y.; Zheng, Y. Overview: The role of Propionibacterium acnes in nonpyogenic intervertebral discs. Int. Orthop. 2016, 40, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Zhou, M.; Bai, Z.; Wen, Y.; Shen, J.; Hu, Z. Propionibacterium acnes induces intervertebral disc degeneration by promoting nucleus pulposus cell pyroptosis via NLRP3-dependent pathway. Biochem. Biophys. Res. Commun. 2020, 526, 772–779. [Google Scholar] [CrossRef]
- Tang, G.; Han, X.; Lin, Z.; Qian, H.; Chen, B.; Zhou, C.; Chen, Y.; Jiang, W. Propionibacterium acnes Accelerates Intervertebral Disc Degeneration by Inducing Pyroptosis of Nucleus Pulposus Cells via the ROS-NLRP3 Pathway. Oxid. Med. Cell. Longev. 2021, 2021, 4657014. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Zhu, R.; Ji, W.P.; Wang, J.Y.; Chen, S.; Fan, S.W.; Hu, Z.J. The NLRP3/Caspase-1/Interleukin-1beta Axis Is Active in Human Lumbar Cartilaginous Endplate Degeneration. Clin. Orthop. Relat. Res. 2016, 474, 1818–1826. [Google Scholar] [CrossRef]
- Anjum, A.; Yazid, M.D.; Fauzi Daud, M.; Idris, J.; Ng, A.M.H.; Selvi Naicker, A.; Ismail, O.H.R.; Athi Kumar, R.K.; Lokanathan, Y. Spinal Cord Injury: Pathophysiology, Multimolecular Interactions, and Underlying Recovery Mechanisms. Int. J. Mol. Sci. 2020, 21, 7533. [Google Scholar] [CrossRef]
- Dai, W.; Wang, X.; Teng, H.; Li, C.; Wang, B.; Wang, J. Celastrol inhibits microglial pyroptosis and attenuates inflammatory reaction in acute spinal cord injury rats. Int. Immunopharmacol. 2019, 66, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, C.; Yu, D.D.; Su, H.; Chen, Y.; Ni, W. Piperine attenuates the inflammation, oxidative stress, and pyroptosis to facilitate recovery from spinal cord injury via autophagy enhancement. Phytother. Res. 2023, 37, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Yu, Q.; Fang, B.; Zhang, Z.L.; Ma, H. Knockdown of the AIM2 molecule attenuates ischemia-reperfusion-induced spinal neuronal pyroptosis by inhibiting AIM2 inflammasome activation and subsequent release of cleaved caspase-1 and IL-1beta. Neuropharmacology 2019, 160, 107661. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Aglietti, R.A.; Estevez, A.; Gupta, A.; Ramirez, M.G.; Liu, P.S.; Kayagaki, N.; Ciferri, C.; Dixit, V.M.; Dueber, E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 2016, 113, 7858–7863. [Google Scholar] [CrossRef] [PubMed]
- Lacey, C.A.; Mitchell, W.J.; Dadelahi, A.S.; Skyberg, J.A. Caspase-1 and Caspase-11 Mediate Pyroptosis, Inflammation, and Control of Brucella Joint Infection. Infect. Immun. 2018, 86, 10–1128. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Ruhl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, X.; Wang, D.; Zheng, J.; Chen, L.; Xie, Q.; Liu, X.; Niu, S.; Qu, G.; Lan, J.; et al. Periodontal Inflammation-Triggered by Periodontal Ligament Stem Cell Pyroptosis Exacerbates Periodontitis. Front. Cell Dev. Biol. 2021, 9, 663037. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, Z.; Zhang, H.; Lu, J.; Tian, Y.; Wei, Y.; Yang, Y.; Bai, L. P2X7 Receptor Induces Pyroptotic Inflammation and Cartilage Degradation in Osteoarthritis via NF-kappaB/NLRP3 Crosstalk. Oxid. Med. Cell. Longev. 2021, 2021, 8868361. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, H.; Zhang, B.; Hua, T.; Wang, H.; Wang, L.; Han, L.; Li, Q.; Wu, W.; Sun, Y.; et al. Inhibition of IL1R1 or CASP4 attenuates spinal cord injury through ameliorating NLRP3 inflammasome-induced pyroptosis. Front. Immunol. 2022, 13, 963582. [Google Scholar] [CrossRef]
- Op de Beeck, K.; Van Camp, G.; Thys, S.; Cools, N.; Callebaut, I.; Vrijens, K.; Van Nassauw, L.; Van Tendeloo, V.F.; Timmermans, J.P.; Van Laer, L. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur. J. Hum. Genet. 2011, 19, 965–973. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef]
- Zhou, B.; Abbott, D.W. Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases. Cell Rep. 2021, 35, 108998. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, T.; Xiao, J.; Xu, C.; Alippe, Y.; Sun, K.; Kanneganti, T.D.; Monahan, J.B.; Abu-Amer, Y.; Lieberman, J.; et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci. Immunol. 2021, 6, eabj3859. [Google Scholar] [CrossRef]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, P.; An, L.; Sun, N.; Peng, L.; Tang, W.; Ma, D.; Chen, J. Miltirone induces cell death in hepatocellular carcinoma cell through GSDME-dependent pyroptosis. Acta Pharm. Sin. B 2020, 10, 1397–1413. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, W.; Cheng, C.; Mo, F.; Chen, L.; Peng, G.; Cai, X.; Wang, J.; Yang, S.; Liu, X. Dioscin inhibits the growth of human osteosarcoma by inducing G2/M-phase arrest, apoptosis, and GSDME-dependent cell death in vitro and in vivo. J. Cell. Physiol. 2020, 235, 2911–2924. [Google Scholar] [CrossRef]
- Zhai, Z.; Yang, F.; Xu, W.; Han, J.; Luo, G.; Li, Y.; Zhuang, J.; Jie, H.; Li, X.; Shi, X.; et al. Attenuation of Rheumatoid Arthritis Through the Inhibition of Tumor Necrosis Factor-Induced Caspase 3/Gasdermin E-Mediated Pyroptosis. Arthritis Rheumatol. 2022, 74, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, X.P.; Zhang, Q.; Zou, Y.Y.; Ma, J.D.; Chen, L.F.; Zou, Y.W.; Xue, J.M.; Ma, R.F.; Chen, Z.; et al. Gasdermin-E Mediated Pyroptosis-A Novel Mechanism Regulating Migration, Invasion and Release of Inflammatory Cytokines in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Front. Cell Dev. Biol. 2021, 9, 810635. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef]
- Chen, K.W.; Demarco, B.; Heilig, R.; Shkarina, K.; Boettcher, A.; Farady, C.J.; Pelczar, P.; Broz, P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019, 38, e101638. [Google Scholar] [CrossRef]
- Jiang, H.; Moro, A.; Liu, Y.; Wang, J.; Meng, D.; Zhan, X.; Wei, Q. Two GWAS-identified variants are associated with lumbar spinal stenosis and Gasdermin-C expression in Chinese population. Sci. Rep. 2020, 10, 21069. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, Y.; Chen, X.; Wang, Z.; Liang, X.; Zhang, T.; Liu, M.; Zhou, N.; Lv, J.; Tang, K.; et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci. Immunol. 2020, 5, eaax7969. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- LaRock, C.N.; Todd, J.; LaRock, D.L.; Olson, J.; O’Donoghue, A.J.; Robertson, A.A.; Cooper, M.A.; Hoffman, H.M.; Nizet, V. IL-1beta is an innate immune sensor of microbial proteolysis. Sci. Immunol. 2016, 1, eaah3539. [Google Scholar] [CrossRef]
- Deng, W.; Bai, Y.; Deng, F.; Pan, Y.; Mei, S.; Zheng, Z.; Min, R.; Wu, Z.; Li, W.; Miao, R.; et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 2022, 602, 496–502. [Google Scholar] [CrossRef]
- LaRock, D.L.; Johnson, A.F.; Wilde, S.; Sands, J.S.; Monteiro, M.P.; LaRock, C.N. Group A Streptococcus induces GSDMA-dependent pyroptosis in keratinocytes. Nature 2022, 605, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Chen, S.; Wu, X.; Zhu, J.; Li, J. Resveratrol Relieves Gouty Arthritis by Promoting Mitophagy to Inhibit Activation of NLRP3 Inflammasomes. J. Inflamm. Res. 2021, 14, 3523–3536. [Google Scholar] [CrossRef]
- Hsieh, C.Y.; Li, L.H.; Lam, Y.; Fang, Z.; Gan, C.H.; Rao, Y.K.; Chiu, H.W.; Wong, W.T.; Ju, T.C.; Chen, F.H.; et al. Synthetic 4-Hydroxy Auxarconjugatin B, a Novel Autophagy Inducer, Attenuates Gouty Inflammation by Inhibiting the NLRP3 Inflammasome. Cells 2020, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, W.; Chen, Y.; Chen, Y.; Shi, J.; Qin, R.; Wang, H.; Wang, R.; Yuan, H.; Sun, W. RelA/MicroRNA-30a/NLRP3 signal axis is involved in rheumatoid arthritis via regulating NLRP3 inflammasome in macrophages. Cell Death Dis. 2021, 12, 1060. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-kappaB/GSDMD signal in mice adipose tissue. J. Pineal Res. 2017, 63, e12414. [Google Scholar] [CrossRef]
- Yan, Z.; Qi, W.; Zhan, J.; Lin, Z.; Lin, J.; Xue, X.; Pan, X.; Zhou, Y. Activating Nrf2 signalling alleviates osteoarthritis development by inhibiting inflammasome activation. J. Cell. Mol. Med. 2020, 24, 13046–13057. [Google Scholar] [CrossRef]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef]
- Yu, H.; Yao, S.; Zhou, C.; Fu, F.; Luo, H.; Du, W.; Jin, H.; Tong, P.; Chen, D.; Wu, C.; et al. Morroniside attenuates apoptosis and pyroptosis of chondrocytes and ameliorates osteoarthritic development by inhibiting NF-kappaB signaling. J. Ethnopharmacol. 2021, 266, 113447. [Google Scholar] [CrossRef]
- Jia, S.; Yang, Y.; Bai, Y.; Wei, Y.; Zhang, H.; Tian, Y.; Liu, J.; Bai, L. Mechanical Stimulation Protects Against Chondrocyte Pyroptosis Through Irisin-Induced Suppression of PI3K/Akt/NF-kappaB Signal Pathway in Osteoarthritis. Front. Cell Dev. Biol. 2022, 10, 797855. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 Receptor in Infection and Inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef]
- Li, W.; Wang, K.; Liu, Y.; Wu, H.; He, Y.; Li, C.; Wang, Q.; Su, X.; Yan, S.; Su, W.; et al. A Novel Drug Combination of Mangiferin and Cinnamic Acid Alleviates Rheumatoid Arthritis by Inhibiting TLR4/NFkappaB/NLRP3 Activation-Induced Pyroptosis. Front. Immunol. 2022, 13, 912933. [Google Scholar]
- Gu, L.; Sun, Y.; Wu, T.; Chen, G.; Tang, X.; Zhao, L.; He, L.; Hu, Z.; Sun, L.; Pan, F.; et al. A novel mechanism for macrophage pyroptosis in rheumatoid arthritis induced by Pol beta deficiency. Cell Death Dis. 2022, 13, 583. [Google Scholar] [CrossRef] [PubMed]
- Jhun, J.; Woo, J.S.; Kwon, J.Y.; Na, H.S.; Cho, K.H.; Kim, S.A.; Kim, S.J.; Moon, S.J.; Park, S.H.; Cho, M.L. Vitamin D Attenuates Pain and Cartilage Destruction in OA Animals via Enhancing Autophagic Flux and Attenuating Inflammatory Cell Death. Immune Netw. 2022, 22, e34. [Google Scholar] [CrossRef]
- Yan, J.; Feng, G.; Yang, Y.; Ding, D.; Ma, L.; Zhao, X.; Chen, X.; Wang, H.; Chen, Z.; Jin, Q. Autophagy attenuates osteoarthritis in mice by inhibiting chondrocyte pyroptosis and improving subchondral bone remodeling. Bosn. J. Basic Med. Sci. 2023, 23, 77–88. [Google Scholar] [CrossRef]
- Wu, X.; Ren, G.; Zhou, R.; Ge, J.; Chen, F.H. The role of Ca2+ in acid-sensing ion channel 1a-mediated chondrocyte pyroptosis in rat adjuvant arthritis. Lab. Investig. 2019, 99, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, F. Acid-Sensing Ion Channel-1a in Articular Chondrocytes and Synovial Fibroblasts: A Novel Therapeutic Target for Rheumatoid Arthritis. Front. Immunol. 2020, 11, 580936. [Google Scholar] [CrossRef]
- Kim, M.J.; Jo, D.G.; Hong, G.S.; Kim, B.J.; Lai, M.; Cho, D.H.; Kim, K.W.; Bandyopadhyay, A.; Hong, Y.M.; Kim, D.H.; et al. Calpain-dependent cleavage of cain/cabin1 activates calcineurin to mediate calcium-triggered cell death. Proc. Natl. Acad. Sci. USA 2002, 99, 9870–9875. [Google Scholar] [CrossRef] [PubMed]
- Zu, S.Q.; Feng, Y.B.; Zhu, C.J.; Wu, X.S.; Zhou, R.P.; Li, G.; Dai, B.B.; Wang, Z.S.; Xie, Y.Y.; Li, Y.; et al. Acid-sensing ion channel 1a mediates acid-induced pyroptosis through calpain-2/calcineurin pathway in rat articular chondrocytes. Cell Biol. Int. 2020, 44, 2140–2152. [Google Scholar] [CrossRef]
- Gong, W.; Kolker, S.J.; Usachev, Y.; Walder, R.Y.; Boyle, D.L.; Firestein, G.S.; Sluka, K.A. Acid-sensing ion channel 3 decreases phosphorylation of extracellular signal-regulated kinases and induces synoviocyte cell death by increasing intracellular calcium. Arthritis Res. Ther. 2014, 16, R121. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, X.; Yang, X.; Niu, R.; Song, S.; Zhu, F.; Zhu, C.; Peng, X.; Chen, F. ASIC1a induces synovial inflammation via the Ca(2+)/NFATc3/ RANTES pathway. Theranostics 2020, 10, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Liu, W.; He, D.; Wang, Y.; Yi, W.; Luo, C.; Shen, J.; Hu, Z. Protective effects of autophagy and NFE2L2 on reactive oxygen species-induced pyroptosis of human nucleus pulposus cells. Aging 2020, 12, 7534–7548. [Google Scholar] [CrossRef]
- Hong, J.; Li, S.; Markova, D.Z.; Liang, A.; Kepler, C.K.; Huang, Y.; Zhou, J.; Yan, J.; Chen, W.; Huang, D.; et al. Bromodomain-containing protein 4 inhibition alleviates matrix degradation by enhancing autophagy and suppressing NLRP3 inflammasome activity in NP cells. J. Cell. Physiol. 2020, 235, 5736–5749. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Li, S.; Liu, R.; Feng, X.; Shi, Y.; Wang, K.; Li, S.; Zhang, Y.; Wu, X.; Yang, C. Autophagic Degradation of Gasdermin D Protects against Nucleus Pulposus Cell Pyroptosis and Retards Intervertebral Disc Degeneration In Vivo. Oxid. Med. Cell. Longev. 2021, 2021, 5584447. [Google Scholar] [CrossRef]
- Tschoeke, S.K.; Hellmuth, M.; Hostmann, A.; Robinson, Y.; Ertel, W.; Oberholzer, A.; Heyde, C.E. Apoptosis of human intervertebral discs after trauma compares to degenerated discs involving both receptor-mediated and mitochondrial-dependent pathways. J. Orthop. Res. 2008, 26, 999–1006. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Zeng, Z.; Fang, B.; Tao, M.; Gu, C.; Zheng, L.; Wang, Y.; Shi, Y.; Fang, C.; Mei, S.; et al. Mesenchymal stem cell-derived exosomes ameliorate intervertebral disc degeneration via anti-oxidant and anti-inflammatory effects. Free Radic. Biol. Med. 2019, 143, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qiu, C.; Wang, W.; Peng, J.; Cheng, X.; Shangguan, Y.; Xu, M.; Li, J.; Qu, R.; Chen, X.; et al. Cortistatin protects against intervertebral disc degeneration through targeting mitochondrial ROS-dependent NLRP3 inflammasome activation. Theranostics 2020, 10, 7015–7033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, G.; Luo, R.; Lei, J.; Song, Y.; Wang, B.; Ma, L.; Liao, Z.; Ke, W.; Liu, H.; et al. Cytosolic escape of mitochondrial DNA triggers cGAS-STING-NLRP3 axis-dependent nucleus pulposus cell pyroptosis. Exp. Mol. Med. 2022, 54, 129–142. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, P.; Zhao, Y.; Yu, F.; Wang, S.; Liu, K.; Cheng, X.; Shi, J.; He, Q.; Xia, Y.; et al. Scutellarin Protects Against Mitochondrial Reactive Oxygen Species-Dependent NLRP3 Inflammasome Activation to Attenuate Intervertebral Disc Degeneration. Front. Bioeng. Biotechnol. 2022, 10, 883118. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Ma, Z.; Tang, P.; Dong, W.; Lu, Y.; Tan, B.; Zhou, N.; Hao, J.; Shen, J.; Hu, Z. SIRT1 alleviates IL-1beta induced nucleus pulposus cells pyroptosis via mitophagy in intervertebral disc degeneration. Int. Immunopharmacol. 2022, 107, 108671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Mao, F.; Wang, S.; Wu, H.; Wang, S.; Liao, Y. Role of Transcription Factor Nrf2 in Pyroptosis in Spinal Cord Injury by Regulating GSDMD. Neurochem. Res. 2023, 48, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Xie, C.; Chen, Z.; He, G.; Dai, Z.; Cai, H.; Zhang, H.; Lu, H.; Wu, H.; Hu, X.; et al. MFG-E8 alleviates intervertebral disc degeneration by suppressing pyroptosis and extracellular matrix degradation in nucleus pulposus cells via Nrf2/TXNIP/NLRP3 axis. Cell Death Discov. 2022, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; Zhang, Y.; Liu, W.; Ni, W.; Huang, X.; Yuan, J.; Zhao, B.; Xiao, H.; Xue, F. Mesenchymal stem cells-derived exosomes ameliorate intervertebral disc degeneration through inhibiting pyroptosis. J. Cell. Mol. Med. 2020, 24, 11742–11754. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Li, T.; Shi, L.; Miao, J.; Guo, Y.; Chen, Y. Human umbilical cord mesenchymal stem cells deliver exogenous miR-26a-5p via exosomes to inhibit nucleus pulposus cell pyroptosis through METTL14/NLRP3. Mol. Med. 2021, 27, 91. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Zhang, Z.; Mao, Q.; Wang, C.; Zhou, Y.; Zhou, X.; Ying, L.; Xu, H.; Hu, S.; Zhang, N. Injectable exosome-functionalized extracellular matrix hydrogel for metabolism balance and pyroptosis regulation in intervertebral disc degeneration. J. Nanobiotechnol. 2021, 19, 264. [Google Scholar] [CrossRef]
- Wittmann, J.; Jack, H.M. microRNAs in rheumatoid arthritis: Midget RNAs with a giant impact. Ann. Rheum. Dis. 2011, 70 (Suppl. S1), i92–i96. [Google Scholar] [CrossRef]
- Tian, J.; Zhou, D.; Xiang, L.; Liu, X.; Zhang, H.; Wang, B.; Xie, B. MiR-223-3p inhibits inflammation and pyroptosis in monosodium urate-induced rats and fibroblast-like synoviocytes by targeting NLRP3. Clin. Exp. Immunol. 2021, 204, 396–410. [Google Scholar] [CrossRef]
- Zhang, L.; Qiu, J.; Shi, J.; Liu, S.; Zou, H. MicroRNA-140-5p represses chondrocyte pyroptosis and relieves cartilage injury in osteoarthritis by inhibiting cathepsin B/Nod-like receptor protein 3. Bioengineered 2021, 12, 9949–9964. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, P.Y.; Wu, J.G.; Zhang, T.Q.; Li, L.F.; Huang, L.D.; Yu, Y.M.; Wang, M.H.; He, J. miR-219a-5p inhibits the pyroptosis in knee osteoarthritis by inactivating the NLRP3 signaling via targeting FBXO3. Environ. Toxicol. 2022, 37, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Xu, B. BMSC-Derived Exosomes Ameliorate Osteoarthritis by Inhibiting Pyroptosis of Cartilage via Delivering miR-326 Targeting HDAC3 and STAT1//NF-kappaB p65 to Chondrocytes. Mediat. Inflamm. 2021, 2021, 9972805. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Fu, P.; Li, S.; Li, X.; Chen, Y.; Lin, Z. miR-107 affects cartilage matrix degradation in the pathogenesis of knee osteoarthritis by regulating caspase-1. J. Orthop. Surg. Res. 2021, 16, 40. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Dong, X.; Sun, Y.; Liu, Z.; Liu, L.; Gu, H. Dietary Fatty Acid Regulation of the NLRP3 Inflammasome via the TLR4/NF-kappaB Signaling Pathway Affects Chondrocyte Pyroptosis. Oxid. Med. Cell. Longev. 2022, 2022, 3711371. [Google Scholar] [CrossRef]
- Wang, Y.H.; Li, Y.; Wang, J.N.; Zhao, Q.X.; Jin, J.; Wen, S.; Wang, S.C.; Sun, T. Maresin 1 Attenuates Radicular Pain Through the Inhibition of NLRP3 Inflammasome-Induced Pyroptosis via NF-kappaB Signaling. Front. Neurosci. 2020, 14, 831. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Huang, Z.; Zhang, H.; Lu, J.; Tian, Y.; Piao, S.; Lin, Z.; Bai, L. Moderate-intensity exercise alleviates pyroptosis by promoting autophagy in osteoarthritis via the P2X7/AMPK/mTOR axis. Cell Death Discov. 2021, 7, 346. [Google Scholar] [CrossRef] [PubMed]
- Vadala, G.; Di Giacomo, G.; Ambrosio, L.; Cannata, F.; Cicione, C.; Papalia, R.; Denaro, V. Irisin Recovers Osteoarthritic Chondrocytes In Vitro. Cells 2020, 9, 1478. [Google Scholar] [CrossRef]
- Shen, P.; Jia, S.; Wang, Y.; Zhou, X.; Zhang, D.; Jin, Z.; Wang, Z.; Liu, D.; Bai, L.; Yang, Y. Mechanical stress protects against chondrocyte pyroptosis through lipoxin A4 via synovial macrophage M2 subtype polarization in an osteoarthritis model. Biomed. Pharmacother. 2022, 153, 113361. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef]
- Arioz, B.I.; Tastan, B.; Tarakcioglu, E.; Tufekci, K.U.; Olcum, M.; Ersoy, N.; Bagriyanik, A.; Genc, K.; Genc, S. Melatonin Attenuates LPS-Induced Acute Depressive-Like Behaviors and Microglial NLRP3 Inflammasome Activation Through the SIRT1/Nrf2 Pathway. Front. Immunol. 2019, 10, 1511. [Google Scholar] [CrossRef]
- Chen, F.; Jiang, G.; Liu, H.; Li, Z.; Pei, Y.; Wang, H.; Pan, H.; Cui, H.; Long, J.; Wang, J.; et al. Melatonin alleviates intervertebral disc degeneration by disrupting the IL-1beta/NF-kappaB-NLRP3 inflammasome positive feedback loop. Bone Res. 2020, 8, 10. [Google Scholar] [CrossRef]
- Huang, Y.; Peng, Y.; Sun, J.; Li, S.; Hong, J.; Zhou, J.; Chen, J.; Yan, J.; Huang, Z.; Wang, X.; et al. Nicotinamide Phosphoribosyl Transferase Controls NLRP3 Inflammasome Activity Through MAPK and NF-kappaB Signaling in Nucleus Pulposus Cells, as Suppressed by Melatonin. Inflammation 2020, 43, 796–809. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, Z.; Hu, D.; Zhang, L.; Wang, L.; Liu, G. Low dose of indomethacin and Hedgehog signaling inhibitor administration synergistically attenuates cartilage damage in osteoarthritis by controlling chondrocytes pyroptosis. Gene 2019, 712, 143959. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, J.; Shi, J.; Ning, D.; Feng, J.; Lin, W.; He, F.; Xie, Z. Ipriflavone suppresses NLRP3 inflammasome activation in host response to biomaterials and promotes early bone healing. J. Clin. Periodontol. 2022, 49, 814–827. [Google Scholar] [CrossRef]
- Zhang, Q.; Yu, W.; Lee, S.; Xu, Q.; Naji, A.; Le, A.D. Bisphosphonate Induces Osteonecrosis of the Jaw in Diabetic Mice via NLRP3/Caspase-1-Dependent IL-1beta Mechanism. J. Bone Miner. Res. 2015, 30, 2300–2312. [Google Scholar] [CrossRef]
- Cortet, B.; Lucas, S.; Legroux-Gerot, I.; Penel, G.; Chauveau, C.; Paccou, J. Bone disorders associated with diabetes mellitus and its treatments. Jt. Bone Spine 2019, 86, 315–320. [Google Scholar] [CrossRef]
- Yang, X.; Qu, C.; Jia, J.; Zhan, Y. NLRP3 inflammasome inhibitor glyburide expedites diabetic-induced impaired fracture healing. Immunobiology 2019, 224, 786–791. [Google Scholar] [CrossRef]
- Nie, L.; Zhao, P.; Yue, Z.; Zhang, P.; Ji, N.; Chen, Q.; Wang, Q. Diabetes induces macrophage dysfunction through cytoplasmic dsDNA/AIM2 associated pyroptosis. J. Leukoc. Biol. 2021, 110, 497–510. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, L.; Shi, X.; Yang, L.; Hua, F.; Ma, J.; Zhu, W.; Liu, X.; Xuan, R.; Shen, Y.; et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 2020, 12, 24270–24287. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Ding, D.; Feng, G.; Yang, Y.; Zhou, Y.; Ma, L.; Guo, H.; Lu, Z.; Jin, Q. Metformin reduces chondrocyte pyroptosis in an osteoarthritis mouse model by inhibiting NLRP3 inflammasome activation. Exp. Ther. Med. 2022, 23, 222. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Fan, X.; Guo, Z.; Zhou, Z.; Gao, W. Metformin Protects against Spinal Cord Injury and Cell Pyroptosis via AMPK/NLRP3 Inflammasome Pathway. Anal. Cell. Pathol. 2022, 2022, 3634908. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Q.; Du, J.; Bae, E.J.; Choi, Y. Pyroptosis in Skeleton Diseases: A Potential Therapeutic Target Based on Inflammatory Cell Death. Int. J. Mol. Sci. 2024, 25, 9068. https://doi.org/10.3390/ijms25169068
Wu Q, Du J, Bae EJ, Choi Y. Pyroptosis in Skeleton Diseases: A Potential Therapeutic Target Based on Inflammatory Cell Death. International Journal of Molecular Sciences. 2024; 25(16):9068. https://doi.org/10.3390/ijms25169068
Chicago/Turabian StyleWu, Qian, Jiacheng Du, Eun Ju Bae, and Yunjung Choi. 2024. "Pyroptosis in Skeleton Diseases: A Potential Therapeutic Target Based on Inflammatory Cell Death" International Journal of Molecular Sciences 25, no. 16: 9068. https://doi.org/10.3390/ijms25169068
APA StyleWu, Q., Du, J., Bae, E. J., & Choi, Y. (2024). Pyroptosis in Skeleton Diseases: A Potential Therapeutic Target Based on Inflammatory Cell Death. International Journal of Molecular Sciences, 25(16), 9068. https://doi.org/10.3390/ijms25169068