
Inhibition of Calcineurin with FK506 Reduces Tau Levels and Attenuates Synaptic Impairment Driven by Tau Oligomers in the Hippocampus of Male Mouse Models
 ,
,
Abstract
:1. Introduction
2. Results
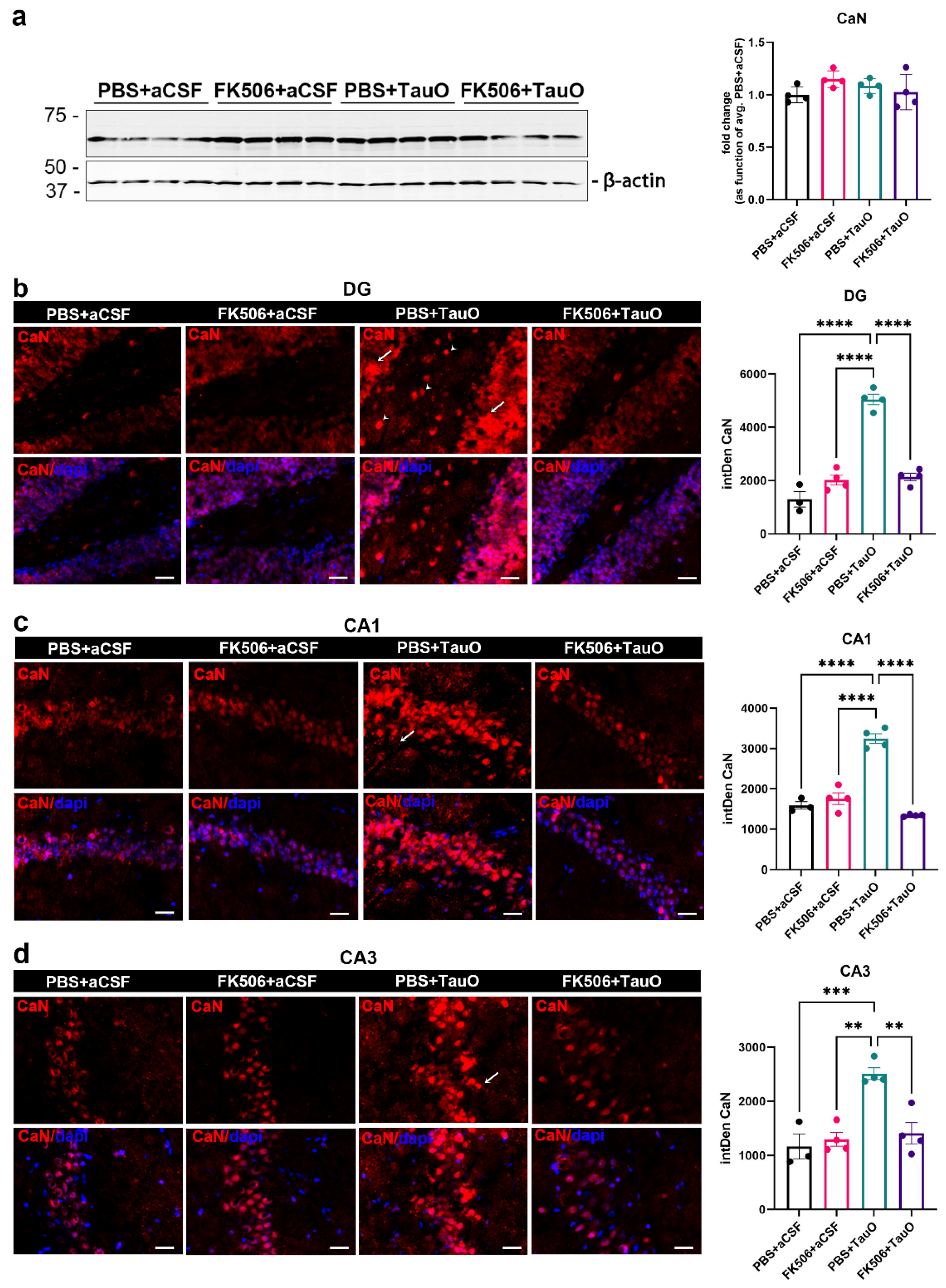
2.1. Modulation of TauO-Induced Changes in CaN Protein Levels by FK506 in Mouse Hippocampus
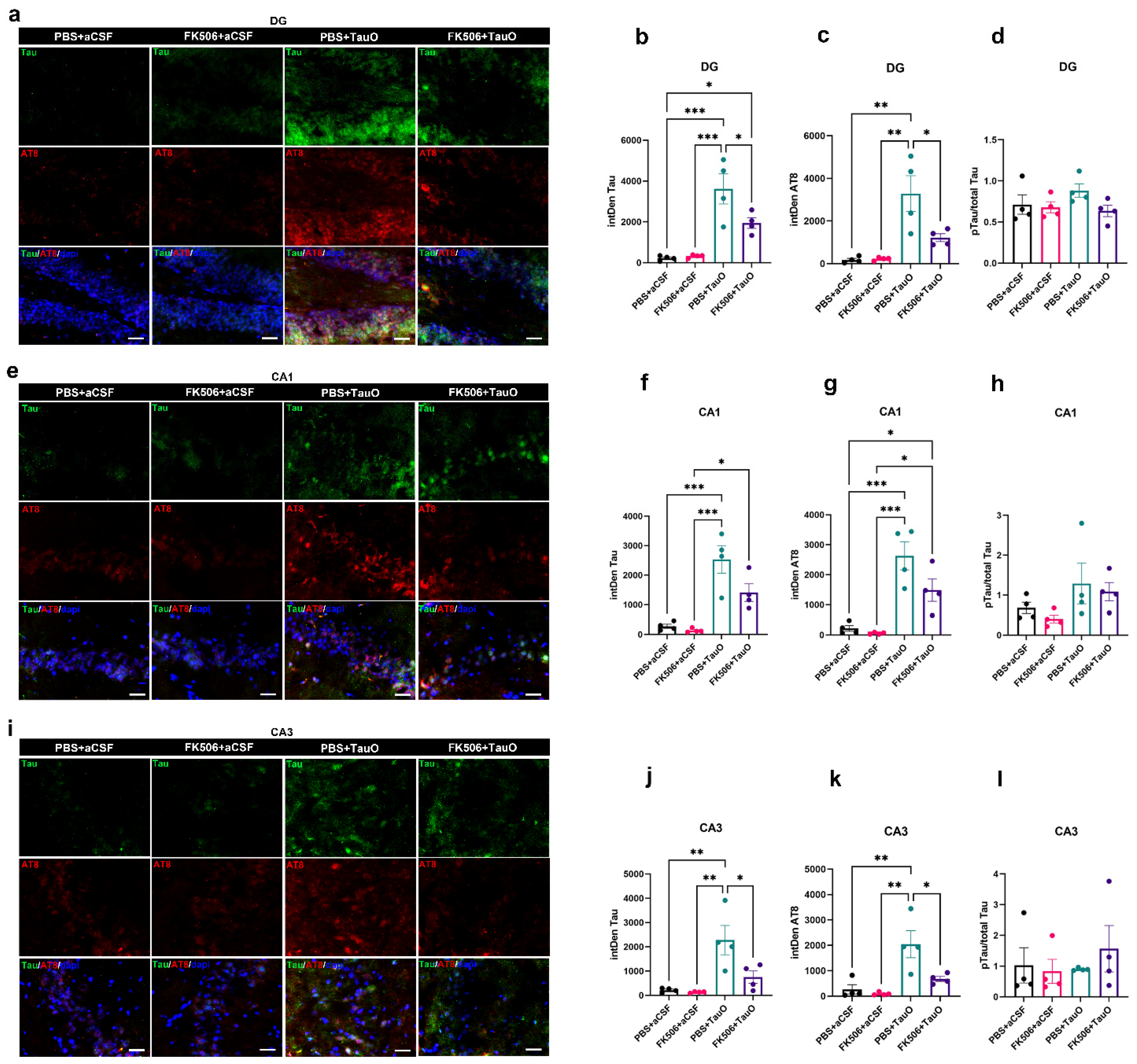
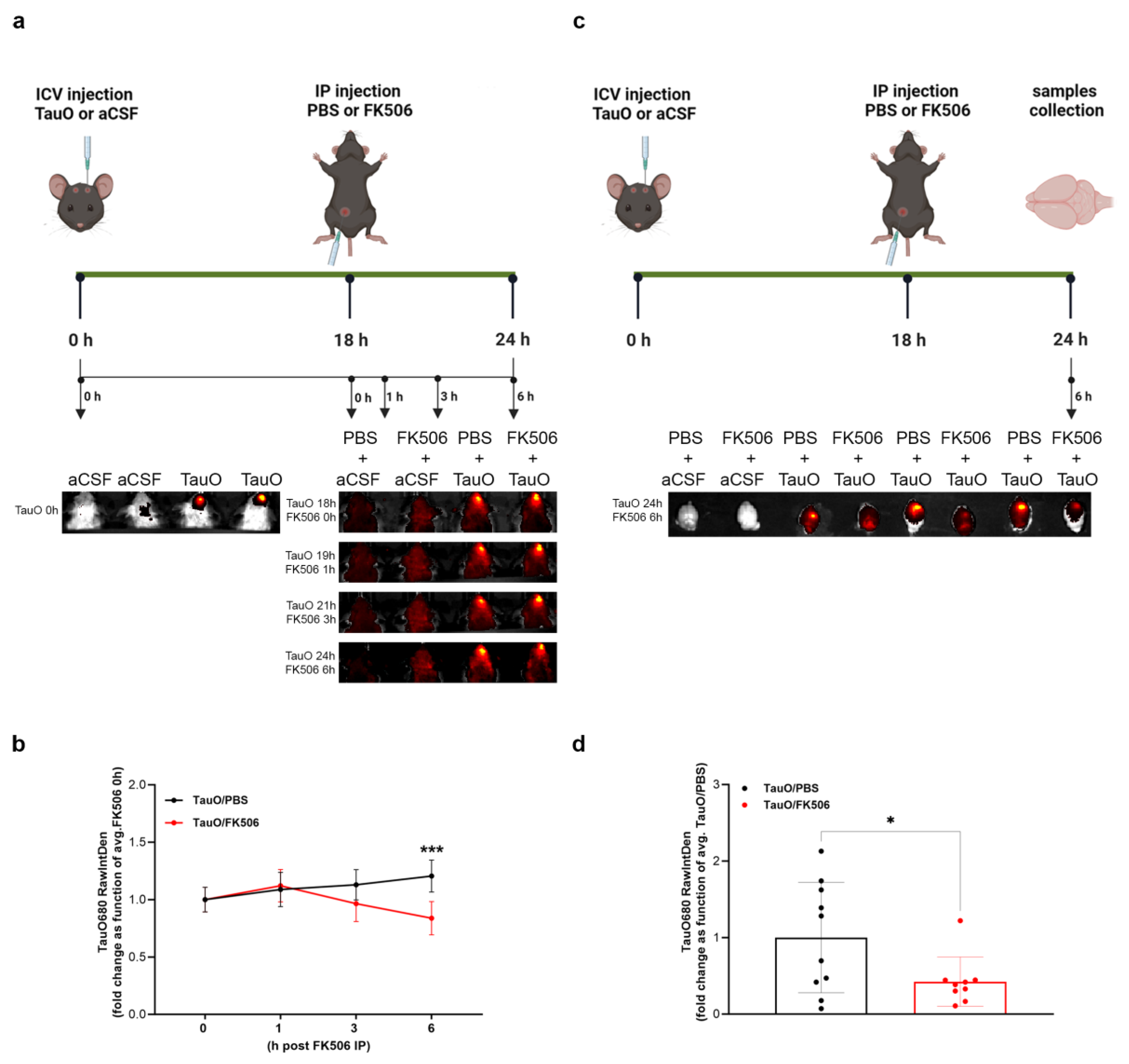
2.2. FK506 Treatment Decreases Levels and Phosphorylation of ICV-Injected TauO
2.3. FK506 Treatment Prevents the Formation of CaN-GSK3β Complex
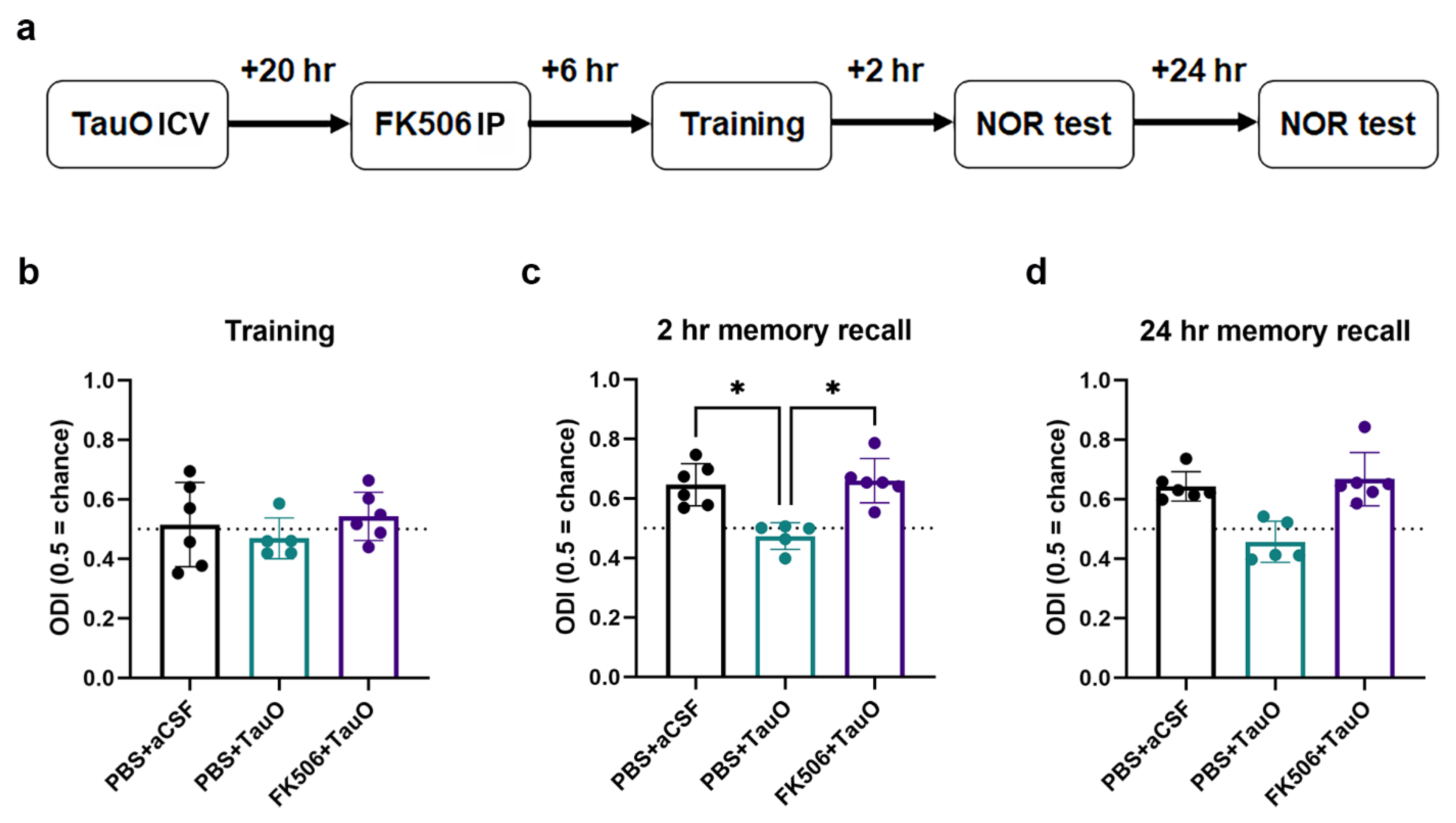
2.4. FK506 Restores TauO-Induced Memory Deficits and Synaptic Alterations
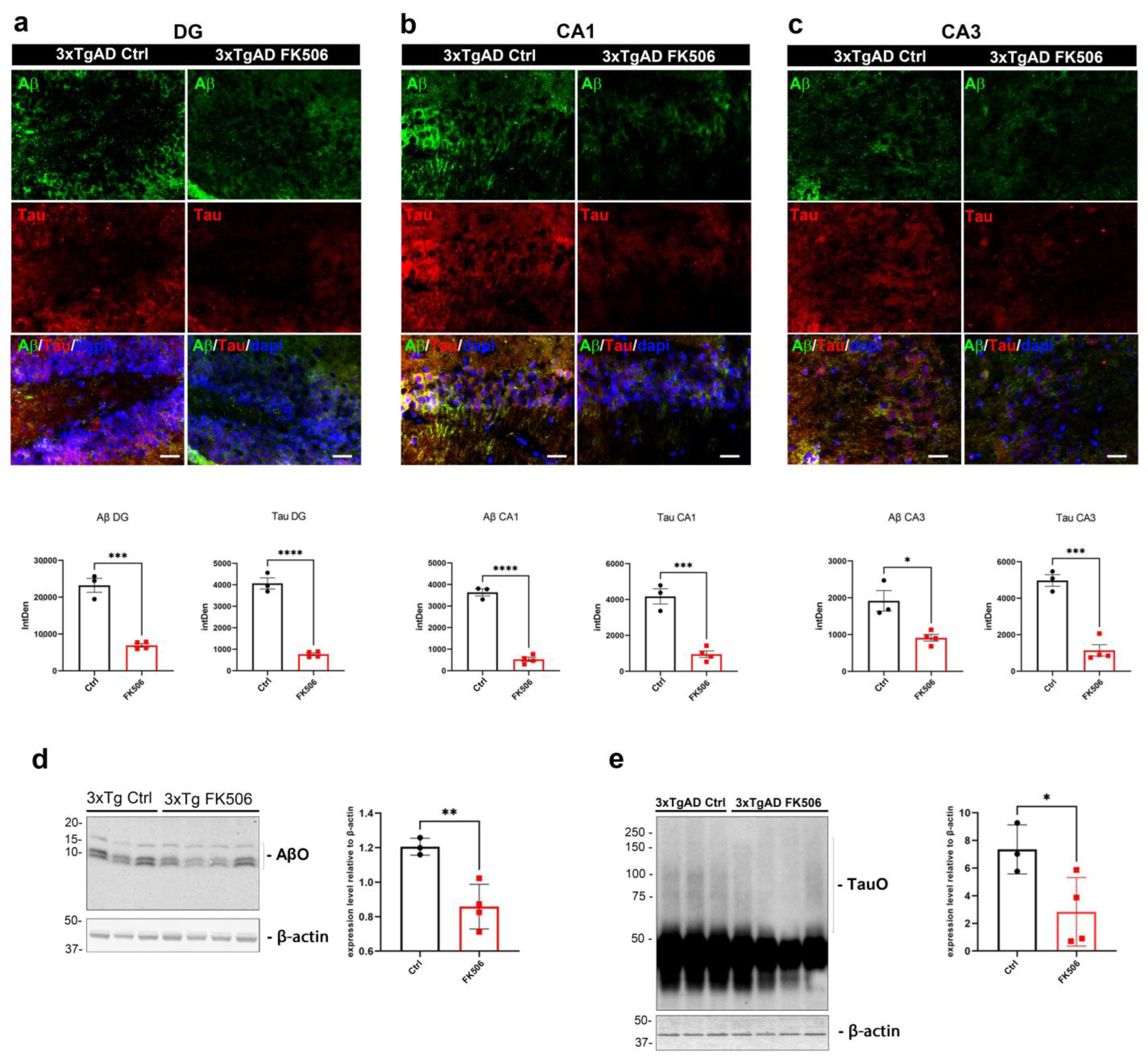
2.5. The Impact of Chronic FK506 on Neuropathology and Autophagy in 3xTgAD Mice
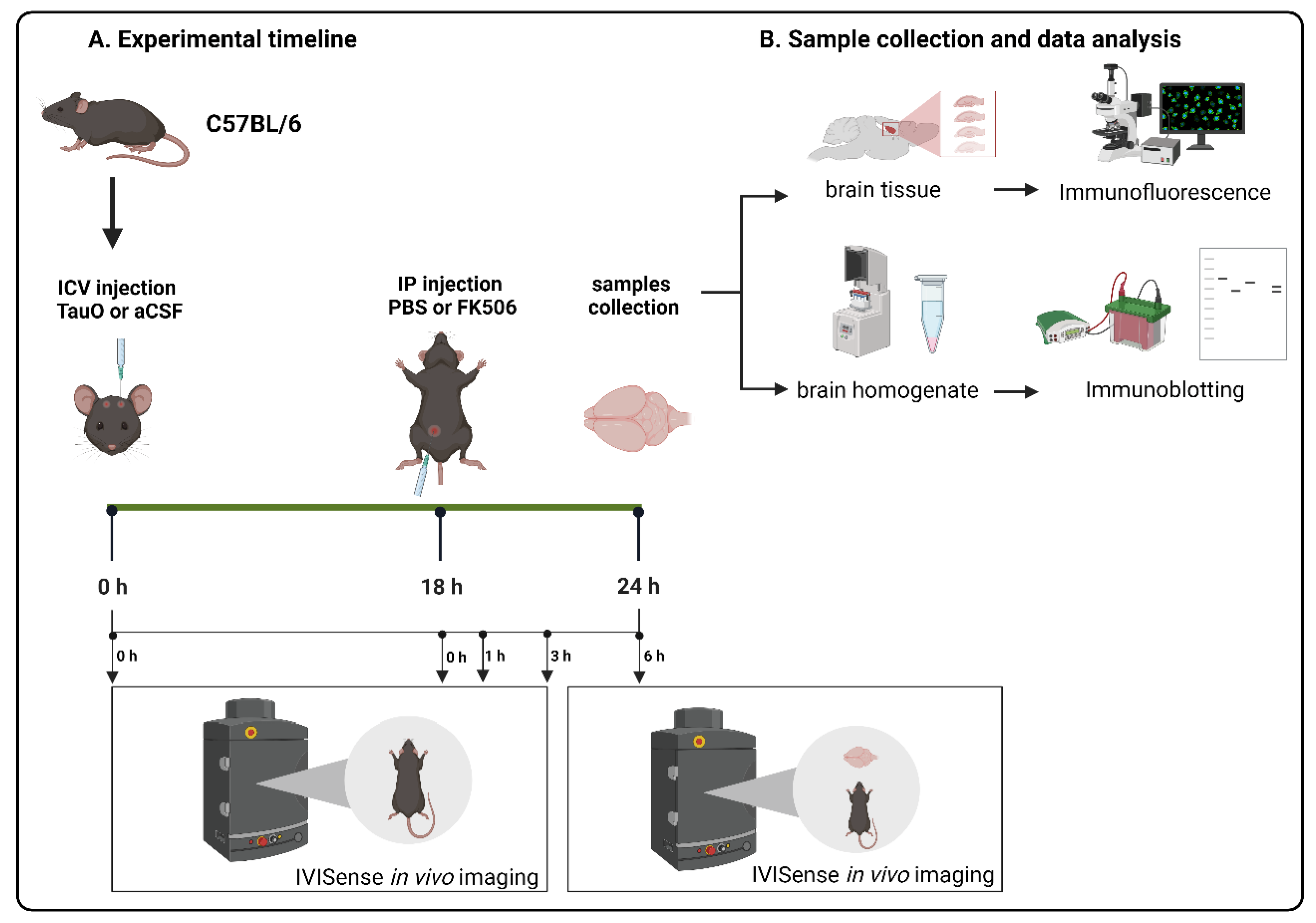
3. Materials and Methods
3.1. Animals
3.2. In Vivo Imaging of the Effect of FK506 Treatment on Mice ICV Injected with Labeled TauO
3.3. Chronic Treatment of 3xTgAD Mice with FK506
3.4. Immunofluorescence
3.5. Proximity Ligation Assay (PLA)
3.6. Western Blot Analyses
3.7. Field Excitatory Postsynaptic Potential Recordings
3.8. Behavioral Testing
3.9. Statistical Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- M Ashraf, G.; H Greig, N.; A Khan, T.; Hassan, I.; Tabrez, S.; Shakil, S.; A Sheikh, I.; K Zaidi, S.; Akram, M.; R Jabir, N.; et al. Protein Misfolding and Aggregation in Alzheimer’s Disease and Type 2 Diabetes Mellitus HHS Public Access. CNS Neurol. Disord. Drug. Targets 2014, 13, 1280–1293. [Google Scholar] [CrossRef]
- Marcatti, M.; Fracassi, A.; Montalbano, M.; Natarajan, C.; Krishnan, B.; Kayed, R.; Taglialatela, G. Aβ/tau oligomer interplay at human synapses supports shifting therapeutic targets for Alzheimer’s disease. Cell. Mol. Life Sci. 2022, 79, 222. [Google Scholar] [CrossRef]
- Tzioras, M.; McGeachan, R.I.; Durrant, C.S.; Spires-Jones, T.L. Synaptic degeneration in Alzheimer disease. Nat. Rev. Neurol. 2023, 19, 19–38. [Google Scholar] [CrossRef]
- Colom-Cadena, M.; Davies, C.; Sirisi, S.; Lee, J.-E.; Simzer, E.M.; Tzioras, M.; Querol-Vilaseca, M.; Sánchez-Aced, É.; Chang, Y.Y.; Holt, K.; et al. Synaptic oligomeric tau in Alzheimer’s disease—A potential culprit in the spread of tau pathology through the brain. Neuron 2023, 111, 2170–2183.e6. [Google Scholar] [CrossRef]
- Walsh, D.M.; Selkoe, D.J. Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 2004, 44, 181–193. [Google Scholar] [CrossRef]
- Fotuhi, S.N.; Khalaj-Kondori, M.; Feizi, M.A.H.; Talebi, M. Memory-related process in physiological status and alzheimer’s disease. Mol. Biol. Rep. 2020, 47, 4651–4657. [Google Scholar] [CrossRef]
- Fà, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Puma, D.D.L.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef]
- Lei, M.; Xu, H.; Li, Z.; Wang, Z.; O’Malley, T.T.; Zhang, D.; Walsh, D.M.; Xu, P.; Selkoe, D.J.; Li, S. Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol. Dis. 2016, 85, 111–121. [Google Scholar] [CrossRef]
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.-Y.; Hyman, B.T.; Bacskai, B.J. Aβ Plaques Lead to Aberrant Regulation of Calcium Homeostasis In Vivo Resulting in Structural and Functional Disruption of Neuronal Networks. Neuron 2008, 59, 214–225. [Google Scholar] [CrossRef]
- Glabe, C.G.; Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66, S74–S78. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric A β-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 48. [Google Scholar] [CrossRef]
- Dineley, K.T.; Kayed, R.; Neugebauer, V.; Fu, Y.; Zhang, W.; Reese, L.C.; Taglialatela, G. Amyloid-β oligomers impair fear conditioned memory in a calcineurin-dependent fashion in mice. J. Neurosci. Res. 2010, 88, 2923–2932. [Google Scholar] [CrossRef]
- Martin, Z.S.; Neugebauer, V.; Dineley, K.T.; Kayed, R.; Zhang, W.; Reese, L.C.; Taglialatela, G. α-Synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: Relevance to human synucleopathic diseases. J. Neurochem. 2012, 120, 440–452. [Google Scholar] [CrossRef]
- Reese, L.C.; Zhang, W.R.; Dineley, K.T.; Kayed, R.; Taglialatela, G. Selective induction of calcineurin activity and signaling by oligomeric amyloid beta. Aging Cell 2008, 7, 824–835. [Google Scholar] [CrossRef]
- Dineley, K.T.; Hogan, D.; Zhang, W.R.; Taglialatela, G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol. Learn Mem. 2007, 88, 217–224. [Google Scholar] [CrossRef]
- Cavallucci, V.; Berretta, N.; Nobili, A.; Nisticò, R.; Mercuri, N.B.; D’amelio, M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer’s disease. Neuromol. Med. 2013, 15, 541–548. [Google Scholar] [CrossRef]
- Rozkalne, A.; Hyman, B.T.; Spires-Jones, T.L. Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model mice. Neurobiol. Dis. 2011, 41, 650–654. [Google Scholar] [CrossRef]
- Taglialatela, G.; Rastellini, C.; Cicalese, L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J. Alzheimer’s Dis. 2015, 47, 329–333. [Google Scholar] [CrossRef]
- Silva, J.D.; Taglialatela, G.; Jupiter, D.C. Reduced Prevalence of Dementia in Patients Prescribed Tacrolimus, Sirolimus, or Cyclosporine. J. Alzheimer’s Dis. 2023, 95, 585–597. [Google Scholar] [CrossRef]
- Gerson, J.E.; Sengupta, U.; Kayed, R. Tau oligomers as pathogenic seeds: Preparation and propagation in vitro and in vivo. Methods Mol. Biol. 2017, 1523, 141–157. [Google Scholar]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, Y.I.; Seo, M.; Kim, S.Y.; Lee, J.E.; Youn, H.D.; Kim, Y.S.; Juhnn, Y.S. Calcineurin dephosphorylates glycogen synthase kinase-3 beta at serine-9 in neuroblast-derived cells. J. Neurochem. 2009, 111, 344–354. [Google Scholar] [CrossRef]
- Hill, E.; Karikari, T.K.; Moffat, K.G.; Richardson, M.J.E.; Wall, M.J. Introduction of Tau oligomers into cortical neurons alters action potential dynamics and disrupts synaptic transmission and plasticity. eNeuro 2019, 6, ENEURO.0166-19.2019. [Google Scholar] [CrossRef]
- Tumurbaatar, B.; Fracassi, A.; Scaduto, P.; Guptarak, J.; Woltjer, R.; Jupiter, D.; Taglialatela, G. Preserved autophagy in cognitively intact non-demented individuals with Alzheimer’s neuropathology. Alzheimer’s Dement. 2023, 19, 5355–5370. [Google Scholar] [CrossRef]
- Sengupta, U.; Carretero-Murillo, M.; Kayed, R. Preparation and Characterization of Tau Oligomer Strains. Methods Mol. Biol. 2018, 1779, 113–146. [Google Scholar]
- Butcher, S.P.; Henshall, D.C.; Teramura, Y.; Iwasaki, K.; Sharkey, J. Neuroprotective Actions of FK506 in Experimental Stroke: In Vivo Evidence against an Antiexcitotoxic Mechanism. J. Neurosci. 1997, 17, 6939–6946. [Google Scholar] [CrossRef]
- Homayoun, H.; Khavandgar, S.; Mehr, S.E.; Namiranian, K.; Dehpour, A.R. The effects of FK506 on the development and expression of morphine tolerance and dependence in mice. Behav. Pharmacol. 2003, 14, 121–127. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, F.; Li, J.; Chen, S.; Kong, Y.; Mo, C.; Leng, X.; Liu, Y.; Xu, Y.; Wang, Y. A quinazoline derivative suppresses B cell hyper-activation and ameliorates the severity of systemic lupus erythematosus in mice. Front. Pharmacol. 2023, 14, 1159075. [Google Scholar] [CrossRef]
- Li, Z.; Sun, F.; Zhang, Y.; Chen, H.; He, N.; Chen, H.; Song, P.; Wang, Y.; Yan, S.; Zheng, S. Tacrolimus induces insulin resistance and increases the glucose absorption in the jejunum: A potential mechanism of the diabetogenic effects. PLoS ONE 2015, 10, e0143405. [Google Scholar] [CrossRef]
- Bourne, K.Z.; Natarajan, C.; Perez, C.X.M.; Tumurbaatar, B.; Taglialatela, G.; Krishnan, B. Suppressing aberrant phospholipase D1 signaling in 3xTg Alzheimer’s disease mouse model promotes synaptic resilience. Sci. Rep. 2019, 9, 18342. [Google Scholar] [CrossRef]
- Comerota, M.M.; Tumurbaatar, B.; Krishnan, B.; Kayed, R.; Taglialatela, G. Near Infrared Light Treatment Reduces Synaptic Levels of Toxic Tau Oligomers in Two Transgenic Mouse Models of Human Tauopathies. Mol. Neurobiol. 2019, 56, 3341–3355. [Google Scholar] [CrossRef]
- Krishnan, B.; Kayed, R.; Taglialatela, G. Elevated phospholipase D isoform 1 in Alzheimer’s disease patients’ hippocampus: Relevance to synaptic dysfunction and memory deficits. Alzheimers Dement 2018, 4, 89–102. [Google Scholar] [CrossRef]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef]
- Mateos-Aparicio, P.; Rodríguez-Moreno, A. Calcium Dynamics and Synaptic Plasticity. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2020; Volume 1131, pp. 965–984. [Google Scholar]
- Groth, R.D.; Dunbar, R.L.; Mermelstein, P.G. Calcineurin regulation of neuronal plasticity. Biochem. Biophys. Res. Commun. 2003, 311, 1159–1171. [Google Scholar] [CrossRef]
- Hemenway, C.S.; Heitman, J. Calcineurin Structure, Function, and Inhibition. Cell Biochem. Biophys. 1999, 30, 115–151. [Google Scholar] [CrossRef]
- Tarasova, E.O.; Gaydukov, A.E.; Balezina, O.P. Calcineurin and Its Role in Synaptic Transmission. Biochemistry 2018, 83, 674–689. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Mobley, W.C. Alzheimer disease pathogenesis: Insights from molecular and cellular biology studies of oligomeric Aβ and tau species. Front. Neurosci. 2019, 13, 659. [Google Scholar] [CrossRef]
- Ghag, G.; Bhatt, N.; Cantu, D.V.; Guerrero-Munoz, M.J.; Ellsworth, A.; Sengupta, U.; Kayed, R. Soluble tau aggregates, not large fibrils, are the toxic species that display seeding and cross-seeding behavior. Protein Sci. 2018, 27, 1901–1909. [Google Scholar] [CrossRef]
- Penke, B.; Szűcs, M.; Bogár, F. Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: Their role in Alzheimer’s pathogenesis. Molecules 2020, 25, 1659. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion channel formation by amyloid-β42 oligomers but not amyloid-β40 in cellular membranes. J. Biol. Chem. 2017, 292, 144–1413. [Google Scholar] [CrossRef]
- Drolle, E.; Negoda, A.; Hammond, K.; Pavlov, E.; Leonenko, Z. Changes in lipid membranes may trigger amyloid toxicity in Alzheimer’s disease. PLoS ONE 2017, 12, e0182194. [Google Scholar] [CrossRef]
- Williams, T.L.; Serpell, L.C. Membrane and surface interactions of Alzheimer’s Aβ peptide-Insights into the mechanism of cytotoxicity. FEBS J. 2011, 278, 3905–3917. [Google Scholar] [CrossRef]
- Yasumoto, T.; Takamura, Y.; Tsuji, M.; Watanabe-Nakayama, T.; Imamura, K.; Inoue, H.; Nakamura, S.; Inoue, T.; Kimura, A.; Yano, S.; et al. High molecular weight amyloid β1-42 oligomers induce neurotoxicity via plasma membrane damage. FASEB J. 2019, 33, 9220–9234. [Google Scholar] [CrossRef]
- Popugaeva, E.; Pchitskaya, E.; Bezprozvanny, I. Dysregulation of neuronal calcium homeostasis in Alzheimer’s disease—A therapeutic opportunity? Biochem. Biophys. Res. Commun. 2017, 483, 998–1004. [Google Scholar] [CrossRef]
- Abdul, H.M.; Sama, M.A.; Furman, J.L.; Mathis, D.M.; Beckett, T.L.; Weidner, A.M.; Patel, E.S.; Baig, I.; Murphy, M.P.; LeVine, H.; et al. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. 2009, 29, 12957–12969. [Google Scholar] [CrossRef]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Hussain, T.; Zhao, D.; Yang, L. A central role for calcineurin in protein misfolding neurodegenerative diseases. Cell. Mol. Life Sci. 2017, 74, 1061–1074. [Google Scholar] [CrossRef]
- Cui, K.; Xu, L.; Tao, T.; Huang, L.; Li, J.; Hong, J.; Li, H.; Chi, Y. Human tau accumulation promotes glycogen synthase kinase-3b acetylation and thus upregulates the kinase: A vicious cycle in Alzheimer neurodegeneration. EBioMedicine 2022, 78, 103970. [Google Scholar] [CrossRef]
- Avila, J.; León-Espinosa, G.; García, E.; García-Escudero, V.; Hernández, F.; DeFelipe, J. Tau phosphorylation by GSK3 in different conditions. Int. J. Alzheimer’s Dis. 2012, 2012, 578373. [Google Scholar] [CrossRef]
- Singh, A.; Allen, D.; Fracassi, A.; Tumurbaatar, B.; Natarajan, C.; Scaduto, P.; Woltjer, R.; Kayed, R.; Limon, A.; Krishnan, B.; et al. Functional Integrity of Synapses in the Central Nervous System of Cognitively Intact Individuals with High Alzheimer’s Disease Neuropathology Is Associated with Absence of Synaptic Tau Oligomers. J. Alzheimer’s Dis. 2020, 78, 1661–1678. [Google Scholar] [CrossRef]
- Uddin, M.S.; Stachowiak, A.; Al Mamun, A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Shekari, A.; Fahnestock, M. Cholinergic neurodegeneration in Alzheimer disease mouse models. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2021; Volume 182, pp. 191–209. [Google Scholar]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Source | Dilution | Ref. | Supplier | RRID | Application |
|---|---|---|---|---|---|---|
| Calcineurin | Rabbit polyclonal | 1:300 | ab3673 | abcam, Cambridge, UK | AB_303991 | IF; WB |
| Human Tau | Chicken | 1:100 | ab75714 | abcam | AB_1310734 | IF |
| pTau (AT8) | Mouse | 1:100 | MN1020 | Thermo fisher | AB_223647 | IF; WB |
| GSK3beta | Mouse | 1:200 | ab93926 | abcam | AB_10563643 | IF |
| IgG | Goat anti-rabbit Alexa Fluor 594 | 1:400 | A-11012 | Invitrogen, Carlsbad, CA, USA | AB_2534079 | IF |
| IgG | Goat anti-mouse Alexa Fluor 488 | 1:400 | A-11001 | Invitrogen | AB_2534069 | IF |
| IgG | Goat anti-mouse Alexa Fluor 594 | 1.400 | A-11032 | Invitrogen | AB_2534091 | IF |
| IgY | Goat anti-chicken 488 | 1:400 | A-11039 | Invitrogen | AB_2534096 | IF |
| IgY | Goat anti-chicken 594 | 1:400 | A-11042 | Invitrogen | AB_2534099 | IF |
| T18 | Rabbit polyclonal | 1:1000 | in-house | WB | ||
| β-actin | Rabbit polyclonal | 1:5000 | ab8227 | abcam | AB_2305186 | WB |
| P62 | Rabbit monoclonal | 1:1000 | 8025 | Cell signaling, Danvers, MA, USA | AB_10859911 | WB |
| LC3A/B | Rabbit monoclonal | 1:1000 | ab228525 | abcam | AB_2827794 | WB |
| Atg3 | Rabbit polyclonal | 1:1000 | 3415 | Cell signaling | AB_2059244 | WB |
| Atg5 | Rabbit monoclonal | 1:1000 | Ab228525 | abcam | AB_2650499 | WB |
| Atg9 | Rabbit monoclonal | 1:1000 | Ab228525 | abcam | AB_10863880 | WB |
| Atg16L1 | Rabbit polyclonal | 1:1000 | Ab228525 | abcam | WB | |
| Beclin 1 | Rabbit monoclonal | 1:1000 | Ab228525 | abcam | AB_2692326 | WB |
| 6E10 | Mouse monoclonal | 1:1000 | 803002 | Biolegend, San Diego, CA, USA | AB_2564654 | WB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcatti, M.; Tumurbaatar, B.; Borghi, M.; Guptarak, J.; Zhang, W.-R.; Krishnan, B.; Kayed, R.; Fracassi, A.; Taglialatela, G. Inhibition of Calcineurin with FK506 Reduces Tau Levels and Attenuates Synaptic Impairment Driven by Tau Oligomers in the Hippocampus of Male Mouse Models. Int. J. Mol. Sci. 2024, 25, 9092. https://doi.org/10.3390/ijms25169092
Marcatti M, Tumurbaatar B, Borghi M, Guptarak J, Zhang W-R, Krishnan B, Kayed R, Fracassi A, Taglialatela G. Inhibition of Calcineurin with FK506 Reduces Tau Levels and Attenuates Synaptic Impairment Driven by Tau Oligomers in the Hippocampus of Male Mouse Models. International Journal of Molecular Sciences. 2024; 25(16):9092. https://doi.org/10.3390/ijms25169092
Chicago/Turabian StyleMarcatti, Michela, Batbayar Tumurbaatar, Michela Borghi, Jutatip Guptarak, Wen-Ru Zhang, Balaji Krishnan, Rakez Kayed, Anna Fracassi, and Giulio Taglialatela. 2024. "Inhibition of Calcineurin with FK506 Reduces Tau Levels and Attenuates Synaptic Impairment Driven by Tau Oligomers in the Hippocampus of Male Mouse Models" International Journal of Molecular Sciences 25, no. 16: 9092. https://doi.org/10.3390/ijms25169092