
Spatial Transcriptomics Identifies Cellular and Molecular Characteristics of Scleroderma Skin Lesions: Pilot Study in Juvenile Scleroderma
 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Biological Meaningful Anatomical Structures (Spatial Domains) of Skin Can Be Identified from Spatial Gene Expression Data
2.1.1. Quality Control for Sequencing and Elimination of Technical Batch Effect
2.1.2. Biologically Meaningful Anatomical Structures (Spatial Domains) Were Identified by Unsupervised Clustering Based on Gene Expression Data of Spots
2.2. Joint Analysis of Paired scRNA Data and ST Data from Same Patients Reveals the Cell Type Composition of Spatial Domains
2.2.1. Combining Spatial Transcriptomics and scRNA Data Validates the Comparable Accuracy between the Two Platforms
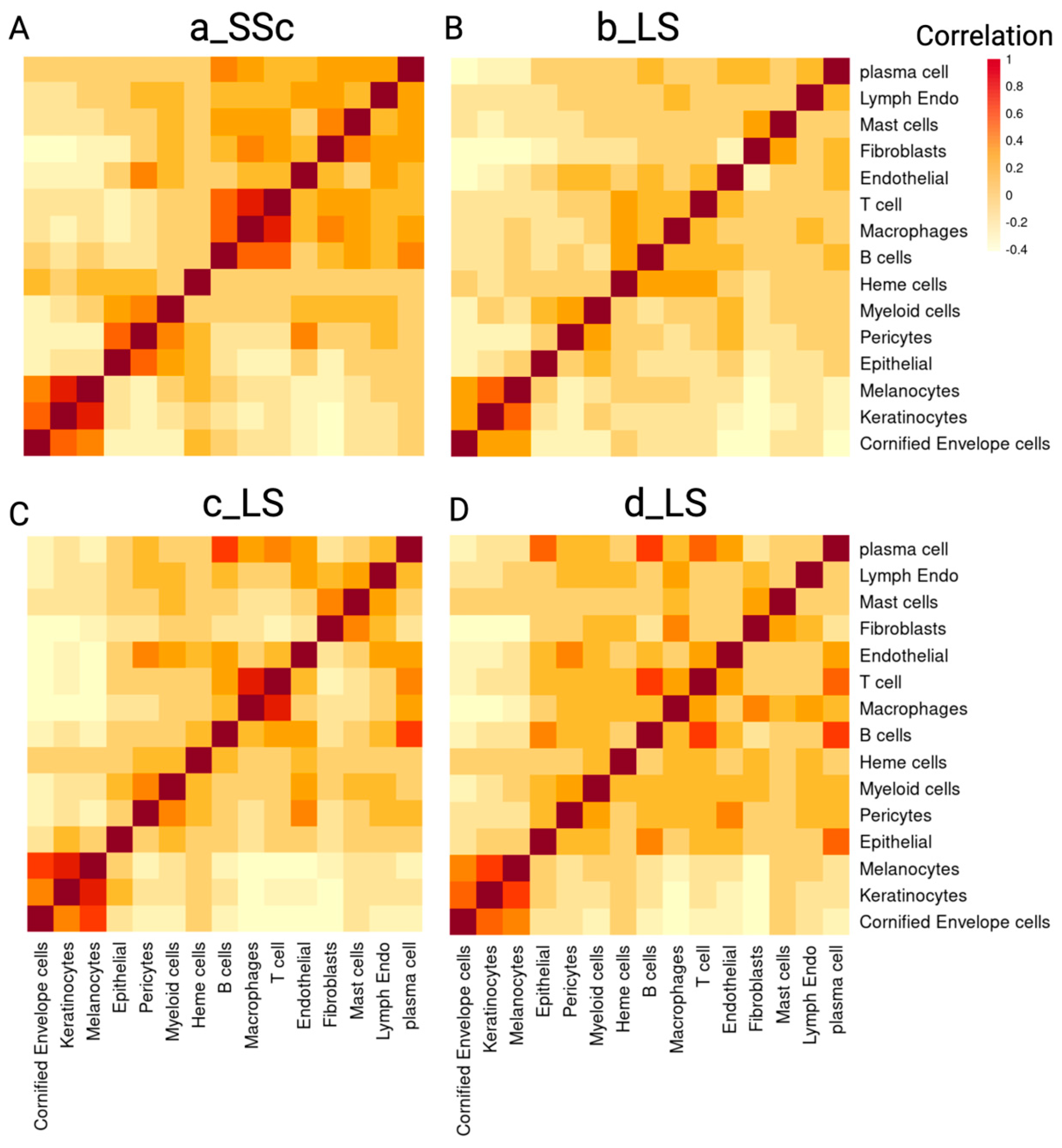
2.2.2. Colocalization Analysis Based on the Cell Type Composition Reveals Spatial Relationships between Immune Cells in Scleroderma Skin
2.3. Pathologist Annotation-Guided Analysis of Normal and Inflammatory Infiltrated Regions in Scleroderma Skin Lesions
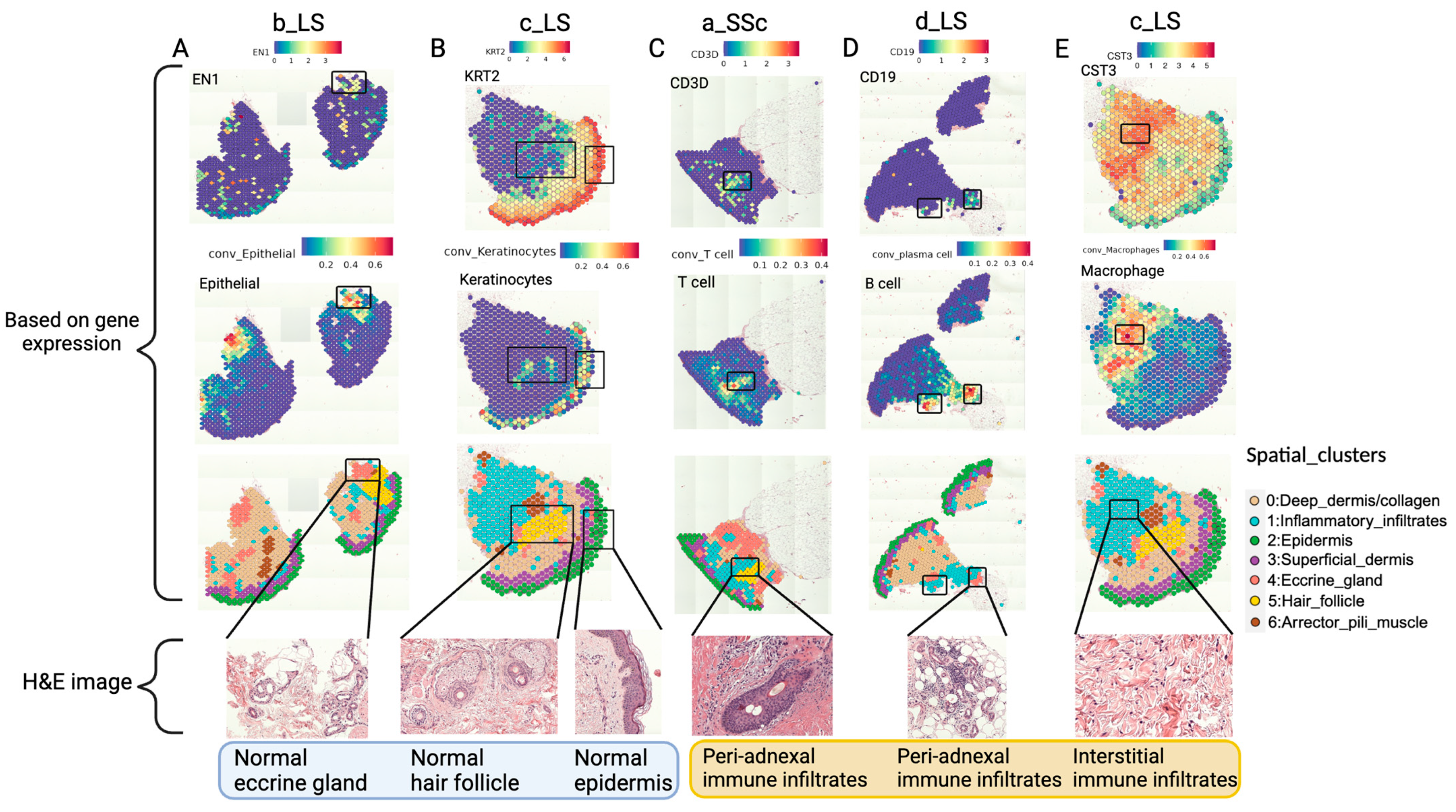
2.3.1. Spatial Transcriptomics Data Can Identify Normal Anatomical Structures and Areas of Inflammatory Infiltrates on H&E Slides of Scleroderma Skin Tissue
2.3.2. Combining the Pathologist-Annotated H&E Images with ST Data Reveals the Spatial Distribution of Immune Cells and Their Relationship with Other Anatomical Structures in Scleroderma Skin
3. Discussion
4. Materials and Methods
4.1. Human Patient Skin Sample Collection
4.2. Spatial Transcriptomics Experiment and Data Processing
4.2.1. Skin Sample Processing and Spatial Transcriptomics Sequencing
4.2.2. Visium Spatial Transcriptomics Data Preprocessing
4.3. Single-Cell Sequencing Experiment and Data Processing
4.3.1. Single-Cell RNA Sequencing Experiment
4.3.2. Single-Cell RNA Sequencing Data Processing
4.4. Joint Analysis of scRNA Data and ST Data
4.4.1. Comparing Marker Genes in scRNA Data and ST Data (Multimodal Integration Analysis)
4.4.2. Robust Cell Type Decomposition (RCTD) Deconvolution
4.5. Manual Annotation Based on H&E Image
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, S.C.; Zheng, R.J. Overview of Juvenile localized scleroderma and its management. World J. Pediatr. 2020, 16, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.M.; Torok, K.S.; Li, S.C.; Taber, S.F.; Lu, T.T.; Zulian, F. Immunopathogenesis of Juvenile Systemic Sclerosis. Front. Immunol. 2019, 10, 1352. [Google Scholar] [CrossRef] [PubMed]
- Foeldvari, I.; Klotsche, J.; Hinrichs, B.; Helmus, N.; Kasapcopur, O.; Adrovic, A.; Sztajnbok, F.; Terreri, M.T.; Anton, J.; Smith, V.; et al. Underdetection of Interstitial Lung Disease in Juvenile Systemic Sclerosis. Arthritis Care Res. 2022, 74, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic sclerosis. Nat. Rev. Dis. Primers 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Canizares, N.; Li, S.C. Juvenile localized scleroderma: Updates and differences from adult-onset disease. Rheum. Dis. Clin. 2021, 47, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; Susa, J.S.; Currimbhoy, S.; Jacobe, H. Histopathological changes in morphea and their clinical correlates: Results from the morphea in adults and children cohort V. J. Am. Acad. Dermatol. 2017, 76, 1124–1130. [Google Scholar] [CrossRef]
- Tabib, T.; Huang, M.; Morse, N.; Papazoglou, A.; Behera, R.; Jia, M.; Bulik, M.; Monier, D.E.; Benos, P.V.; Chen, W.; et al. Myofibroblast transcriptome indicates SFRP2(hi) fibroblast progenitors in systemic sclerosis skin. Nat. Commun. 2021, 12, 4384. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Tabib, T.; Morse, C.; Yang, Y.; Domsic, R.T.; Khanna, D.; Lafyatis, R. Expansion of Fcgamma Receptor IIIa-Positive Macrophages, Ficolin 1-Positive Monocyte-Derived Dendritic Cells, and Plasmacytoid Dendritic Cells Associated With Severe Skin Disease in Systemic Sclerosis. Arthritis Rheumatol. 2022, 74, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Gaydosik, A.M.; Tabib, T.; Domsic, R.; Khanna, D.; Lafyatis, R.; Fuschiotti, P. Single-cell transcriptome analysis identifies skin-specific T-cell responses in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Schutt, C.; Mirizio, E.; Salgado, C.; Reyes-Mugica, M.; Wang, X.; Chen, W.; Grunwaldt, L.; Schollaert, K.L.; Torok, K.S. Transcriptomic Evaluation of Juvenile Localized Scleroderma Skin With Histologic and Clinical Correlation. Arthritis Rheumatol. 2021, 73, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Werner, G.; Sanyal, A.; Mirizio, E.; Hutchins, T.; Tabib, T.; Lafyatis, R.; Jacobe, H.; Torok, K.S. Single-Cell Transcriptome Analysis Identifies Subclusters with Inflammatory Fibroblast Responses in Localized Scleroderma. Int. J. Mol. Sci. 2023, 24, 9796. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.; Sathe, A.; Hess, P.R.; Rong, J.; Wu, C.Y.; Furth, E.; Susztak, K.; Levinsohn, J.; Ji, H.P.; Zhang, N. Niche-DE: Niche-differential gene expression analysis in spatial transcriptomics data identifies context-dependent cell-cell interactions. Genome Biol. 2024, 25, 14. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Barkley, D.; Franca, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Van Praet, J.T.; Smith, V.; Haspeslagh, M.; Degryse, N.; Elewaut, D.; De Keyser, F. Histopathological cutaneous alterations in systemic sclerosis: A clinicopathological study. Arthritis Res. Ther. 2011, 13, R35. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, A.J.; Maliga, Z.; Vallius, T.; Quattrochi, B.; Chen, A.A.; Jacobson, C.A.; Pelletier, R.J.; Yapp, C.; Arias-Camison, R.; Chen, Y.A.; et al. The Spatial Landscape of Progression and Immunoediting in Primary Melanoma at Single-Cell Resolution. Cancer Discov. 2022, 12, 1518–1541. [Google Scholar] [CrossRef] [PubMed]
- Thrane, K.; Eriksson, H.; Maaskola, J.; Hansson, J.; Lundeberg, J. Spatially Resolved Transcriptomics Enables Dissection of Genetic Heterogeneity in Stage III Cutaneous Malignant Melanoma. Cancer Res. 2018, 78, 5970–5979. [Google Scholar] [CrossRef] [PubMed]
- Kiuru, M.; Kriner, M.A.; Wong, S.; Zhu, G.; Terrell, J.R.; Li, Q.; Hoang, M.; Beechem, J.; McPherson, J.D. High-Plex Spatial RNA Profiling Reveals Cell Type–Specific Biomarker Expression during Melanoma Development. J. Investig. Dermatol. 2022, 142, 1401–1412.e20. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 1661–1662. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Castillo, R.L.; Sidhu, I.; Dolgalev, I.; Chu, T.; Prystupa, A.; Subudhi, I.; Yan, D.; Konieczny, P.; Hsieh, B.; Haberman, R.H.; et al. Spatial transcriptomics stratifies psoriatic disease severity by emergent cellular ecosystems. Sci. Immunol. 2023, 8, eabq7991. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schabitz, A.; Hillig, C.; Mubarak, M.; Jargosch, M.; Farnoud, A.; Scala, E.; Kurzen, N.; Pilz, A.C.; Bhalla, N.; Thomas, J.; et al. Spatial transcriptomics landscape of lesions from non-communicable inflammatory skin diseases. Nat. Commun. 2022, 13, 7729. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ding, Y.; Ouyang, Z.; Zhang, C.; Zhu, Y.; Xu, Q.; Sun, H.; Qu, J.; Sun, Y. Tyrosine phosphatase SHP2 exacerbates psoriasis-like skin inflammation in mice via ERK5-dependent NETosis. MedComm 2022, 3, e120. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitamura, Y.; Reiger, M.; Kim, J.; Xiao, Y.; Zhakparov, D.; Tan, G.; Ruckert, B.; Rinaldi, A.O.; Baerenfaller, K.; Akdis, M.; et al. Spatial transcriptomics combined with single-cell RNA-sequencing unravels the complex inflammatory cell network in atopic dermatitis. Allergy 2023, 78, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Hughes, T.K.; Teles, R.M.B.; Andrade, P.R.; de Andrade Silva, B.J.; Plazyo, O.; Tsoi, L.C.; Do, T.; Wadsworth, M.H., 2nd; Oulee, A.; et al. The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat. Immunol. 2021, 22, 839–850. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cable, D.M.; Murray, E.; Zou, L.S.; Goeva, A.; Macosko, E.Z.; Chen, F.; Irizarry, R.A. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 2022, 40, 517–526. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Fuschiotti, P.; Larregina, A.T.; Ho, J.; Feghali-Bostwick, C.; Medsger, T.A., Jr. Interleukin-13-producing CD8+ T cells mediate dermal fibrosis in patients with systemic sclerosis. Arthritis Rheum. 2013, 65, 236–246. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mattoo, H.; Mahajan, V.S.; Maehara, T.; Deshpande, V.; Della-Torre, E.; Wallace, Z.S.; Kulikova, M.; Drijvers, J.M.; Daccache, J.; Carruthers, M.N.; et al. Clonal expansion of CD4+ cytotoxic T lymphocytes in patients with IgG4-related disease. J. Allergy Clin. Immunol. 2016, 138, 825–838. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bertolini, M.; McElwee, K.; Gilhar, A.; Bulfone-Paus, S.; Paus, R. Hair follicle immune privilege and its collapse in alopecia areata. Exp. Dermatol. 2020, 29, 703–725. [Google Scholar] [CrossRef] [PubMed]
- Bray, N. Autoimmune disease: Getting to the root of hair loss in alopecia. Nat. Rev. Drug Discov. 2014, 13, 724–725. [Google Scholar] [CrossRef] [PubMed]
- Christmann, R.B.; Sampaio-Barros, P.; Stifano, G.; Borges, C.L.; de Carvalho, C.R.; Kairalla, R.; Parra, E.R.; Spira, A.; Simms, R.; Capellozzi, V.L.; et al. Association of Interferon- and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol. 2014, 66, 714–725. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Higashi-Kuwata, N.; Jinnin, M.; Makino, T.; Fukushima, S.; Inoue, Y.; Muchemwa, F.C.; Yonemura, Y.; Komohara, Y.; Takeya, M.; Mitsuya, H.; et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res. Ther. 2010, 12, R128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mirizio, E.; Tabib, T.; Wang, X.; Chen, W.; Liu, C.; Lafyatis, R.; Jacobe, H.; Torok, K.S. Single-cell transcriptome conservation in a comparative analysis of fresh and cryopreserved human skin tissue: Pilot in localized scleroderma. Arthritis Res. Ther. 2020, 22, 263. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, D.; Schroeder, A.; Yan, H.; Yang, H.; Hu, J.; Lee, M.Y.Y.; Cho, K.S.; Susztak, K.; Xu, G.X.; Feldman, M.D.; et al. Inferring super-resolution tissue architecture by integrating spatial transcriptomics with histology. Nat. Biotechnol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Zhao, E.; Stone, M.R.; Ren, X.; Guenthoer, J.; Smythe, K.S.; Pulliam, T.; Williams, S.R.; Uytingco, C.R.; Taylor, S.E.B.; Nghiem, P.; et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat. Biotechnol. 2021, 39, 1375–1384. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, C.; Xu, Z.; Wang, X.; Tao, S.; MacDonald, W.A.; He, K.; Poholek, A.C.; Chen, K.; Huang, H.; Chen, W. Innovative super-resolution in spatial transcriptomics: A transformer model exploiting histology images and spatial gene expression. Brief. Bioinform. 2024, 25, bbae052. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Benjamin, K.; Bhandari, A.; Kepple, J.D.; Qi, R.; Shang, Z.; Xing, Y.; An, Y.; Zhang, N.; Hou, Y.; Crockford, T.L.; et al. Multiscale topology classifies cells in subcellular spatial transcriptomics. Nature 2024, 630, 943–949. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Janesick, A.; Shelansky, R.; Gottscho, A.D.; Wagner, F.; Williams, S.R.; Rouault, M.; Beliakoff, G.; Morrison, C.A.; Oliveira, M.F.; Sicherman, J.T.; et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 2023, 14, 8353. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, E.D.; Ma, R.; Navarro Negredo, P.; Brunet, A.; Zou, J. TISSUE: Uncertainty-calibrated prediction of single-cell spatial transcriptomics improves downstream analyses. Nat. Methods 2024, 21, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhou, X. Spatially informed cell-type deconvolution for spatial transcriptomics. Nat. Biotechnol. 2022, 40, 1349–1359. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ferreli, C.; Gasparini, G.; Parodi, A.; Cozzani, E.; Rongioletti, F.; Atzori, L. Cutaneous Manifestations of Scleroderma and Scleroderma-Like Disorders: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2017, 53, 306–336. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QC Metrics | a_SSc | b_LS | c_LS | d_LS |
|---|---|---|---|---|
| Number of spots under tissue | 460 | 988 | 564 | 717 |
| Mean UMI counts per spot | 13,373 | 4678 | 9716 | 5990 |
| Mean number of genes per spot | 4589 | 2032 | 3668 | 2765 |
| Percentage of reads mapped to probe set | 96.6% | 96.5% | 96.9% | 96.6% |
| Saturation of sequencing | 94.0% | 95.0% | 95.6% | 95.2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Esencan, D.; Salgado, C.M.; Zhao, C.; Lai, Y.-J.; Hutchins, T.; Sanyal, A.; Chen, W.; Torok, K.S. Spatial Transcriptomics Identifies Cellular and Molecular Characteristics of Scleroderma Skin Lesions: Pilot Study in Juvenile Scleroderma. Int. J. Mol. Sci. 2024, 25, 9182. https://doi.org/10.3390/ijms25179182
Liu T, Esencan D, Salgado CM, Zhao C, Lai Y-J, Hutchins T, Sanyal A, Chen W, Torok KS. Spatial Transcriptomics Identifies Cellular and Molecular Characteristics of Scleroderma Skin Lesions: Pilot Study in Juvenile Scleroderma. International Journal of Molecular Sciences. 2024; 25(17):9182. https://doi.org/10.3390/ijms25179182
Chicago/Turabian StyleLiu, Tianhao, Deren Esencan, Claudia M. Salgado, Chongyue Zhao, Ying-Ju Lai, Theresa Hutchins, Anwesha Sanyal, Wei Chen, and Kathryn S. Torok. 2024. "Spatial Transcriptomics Identifies Cellular and Molecular Characteristics of Scleroderma Skin Lesions: Pilot Study in Juvenile Scleroderma" International Journal of Molecular Sciences 25, no. 17: 9182. https://doi.org/10.3390/ijms25179182
APA StyleLiu, T., Esencan, D., Salgado, C. M., Zhao, C., Lai, Y.-J., Hutchins, T., Sanyal, A., Chen, W., & Torok, K. S. (2024). Spatial Transcriptomics Identifies Cellular and Molecular Characteristics of Scleroderma Skin Lesions: Pilot Study in Juvenile Scleroderma. International Journal of Molecular Sciences, 25(17), 9182. https://doi.org/10.3390/ijms25179182