Abstract
Newts are excellent vertebrate models for investigating tissue regeneration due to their remarkable regenerative capabilities. To investigate the mRNA and microRNAs (miRNAs) profiles within the blastema niche of regenerating newt limbs, we amputated the limbs of Chinese fire belly newts (Cynops orientalis) and conducted comprehensive analyses of the transcriptome and microRNA profiles at five distinct time points post-amputation (0 hours, 1 day, 5 days 10 days and 20 days). We identified 24 significantly differentially expressed (DE) genes and 20 significantly DE miRNAs. Utilizing weighted gene co-expression network analysis (WGCNA) and gene ontology (GO) enrichment analysis, we identified four genes likely to playing crucial roles in the early stages of limb regeneration: Cemip, Rhou, Gpd2 and Pcna. Moreover, mRNA–miRNA integration analysis uncovered seven human miRNAs (miR-19b-1, miR-19b-2, miR-21-5p, miR-127-5p, miR-150-5p, miR-194-5p, and miR-210-5p) may regulate the expression of these four key genes. The temporal expression patterns of these key genes and miRNAs further validated the robustness of the identified mRNA-miRNA landscape. Our study successfully identified candidate key genes and elucidated a portion of the genetic regulatory mechanisms involved in newt limb regeneration. These findings offer valuable insights for further exploration of the intricate processes of tissue regeneration.
1. Introduction
Many organisms in nature exhibit varying degrees of regenerative capacity, with newts, a group of vertebrates, standing out for their remarkable proficiency in this regard. Newts possess the ability to regenerate a diverse array of organs and tissues, including the limbs, eyes (comprising the lens and retina), and optic nerves, among others [1]. The process of limb regeneration is a highly intricate biological phenomenon that involves the growth and development of multiple tissues such as the epidermis, dermis, limb bone, and skeletal muscle [2]. Broadly, limb regeneration can be categorized into the following phases: wound healing (0–5 days), blastema formation and pattern differentiation, and the differentiation (6 days–20 days), and growth of new cells to reconstruct the limb (21 days–40 days) [1,3]. The unique nature of external limb injuries introduces additional factors, including the immune system response, inflammatory reactions, and cell migration, all of which play crucial roles in the regeneration process [4,5]. Throughout limb regeneration, dynamic changes occur in tissue repair, growth, and other biological processes, collectively forming a complex regulatory network that ensures the normal progression of regeneration [6]. The temporal and differential expression of genes emerges as a crucial factor in regulating the body’s response to external stimuli and the processes of tissue regeneration and development. Consequently, genetic research and analysis of the intricate phenomenon of newt limb regeneration [7,8] represents a prudent approach and a vital avenue for exploring its underlying molecular mechanisms.
MicroRNAs (miRNAs), a class of short, non-coding RNAs, play a crucial role by binding to the 3’ or 5’ end of target mRNAs, thereby inhibiting normal transcription and leading to the reduction or silencing of gene expression [9]. This distinctive feature positions miRNAs as vital regulators in various biological processes. In recent years, significant progress has been achieved in the exploration of co-expression of miRNA and genetic targets in cancer [10,11]. Tumors, characterized by continuous cell division, share certain features with the regenerative processes observed in salamander, including migration, invasion, and extracellular matrix (ECM) remodeling [11,12,13]. Notably, signaling pathways, such as Wnt, Notch, and TGF-β, in both cancer and regeneration, play pivotal roles [6,14,15,16,17]. However, distinctions exist, with some studies suggesting that while both processes share biological processes, regeneration is more ordered and converged compared to the disordered development of cancer [1,18]. Previous studies have aptly described this phenomenon as “similar pathways, different outputs” [19,20]. Considering the critical role of miRNAs in regulating gene expression and their involvement in molecular mechanisms, a comprehensive investigation of limb regeneration through the perspective of miRNA is essential. Previous study has highlighted a significant upregulation of miR-21 expression during axolotl regeneration, underscoring its indispensable role [2,21]. A thorough analysis of differentially expressed miRNAs and their target genes at various regeneration time points is crucial for gaining a deeper understanding of the genetic interactions and regulatory mechanisms underlying this complex biological process.
In this study, we investigated the mRNA and miRNA profiles within the blastema niche during the early stages of limb regeneration in the Chinese fire belly newt (Cynops orientalis). Transcriptome sequencing was conducted on the healing tissues at 0 hours, 1 day, 5 days, 10 days, and 20 days post-amputation of the right front limb (Figure 1). Concurrently, miRNA sequencing was carried out to comprehensively capture the molecular changes and regulatory patterns associated with the healing progress. Leveraging the generated sequencing data, we conducted differential expression analysis of genes across different time points, followed by mRNA-miRNA co-expression analysis. Our primary objective is to identify key genes exhibiting differential expression at distinct time points, along with the miRNAs regulating them. By extrapolating from the genetic analysis results, we aim to speculate on the potential significance of these key genes in orchestrating the intricate biological processes involved in newt limb regeneration.

Figure 1.
Schematic overview of the blastemal time course experiment and study strategy. Tissues collected from five distinct time points following the amputation experiment of the Chinese fire belly newt were utilized for RNA extraction. Subsequently, transcriptome and microRNAome sequencing was conducted. A comprehensive bioinformatics integration analysis was performed on these data, leading to the identification of key genes and targeting miRNAs that potentially exert significant regulatory functions during the early stages of newt limb regeneration.
2. Results
2.1. Identification of Differentially Expressed Genes and miRNAs
Differential expression analysis was conducted across the time course by comparing each time point to the zero-hour control. The analysis revealed 24 significantly differentially expressed (DE) genes and 20 DE miRNAs. Of these, 21 genes and 14 miRNAs were upregulated, while 3 genes and 6 miRNAs were downregulated, as depicted in Figure 2 and Figure 4. Notably, the genes Cnp, Sycn, Rbbp8nl, Slc6a3, Cemip, and Rnf207 were consistently differentially expressed across multiple time points, with both Rbbp8nl and Slc6a3 showing significant differential expression in three comparisons (0 h vs. 5 d, 0 h vs. 10 d and 0 h vs. 20 d). This may suggest their significant roles in the early stages of regeneration due to their persistently elevated expression levels following amputation. The gene Rbbp8nl plays an indispensable role in double-strand breaks (DSB) [22], while Slc6a3 is known to mediates dopamine binding and transport.
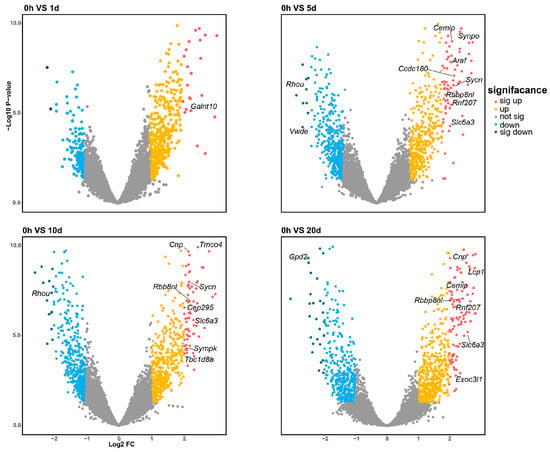
Figure 2.
Four volcano plots illustrate the differential expression profiles of mRNA at 1, 5, 10, and 20 days post-amputation compared to the 0-hour control. The differentially expressed genes are color-coded according to their level of differential expression. The number of significantly DE genes increases with the time elapsed following amputation.
2.2. Co-Expression Modules and Enrichment Analysis
Weighted Gene Co-expression Network Analysis (WGCNA) was employed to identify clusters of genes exhibiting highly interconnected relationships, referred to as modules. Following the normalization and absolute value transformation of gene expression data, genes displaying similar expression trends were grouped into clusters, each representing a distinct module. In this WGCNA analysis, all DE genes were categorized into six co-expression modules based on their expression pattern similarities, each module containing 2 to 5 gene members, represented by distinct colors (Figure 3a). Notably, the blue and green modules demonstrated the strongest significant associations with the time course. However, due to the limitations in non-reference transcriptome annotation, no enriched terms were identified in the green module. All three functional enrichments corresponding to the blue module showed robust statistical significance. Among these, the myoblast migration process, regulated by Net1 and Six4, plays a crucial role in muscle injury and recovery by mediating muscle cells migration [23]. The hyaluronan catabolic process, mediated by Cd44 and Cemip, is crucial for maintaining tissue lubrication, normal structure, and function, and is indispensable in the early stages of tissue repair [24]. Interesting, Cemip, also known as Hybid or Kiaa1199, was significantly upregulated at both 5 and 20 days post-amputation. Cemip has been demonstrated to bind hyaluronic acid [25,26], a major component of the extracellular matrix essential for wound repair, cell migration, and skin healing. The ceramide transport process, mediated by Abca2 and Cptp, primarily functions in intercellular signal transduction and apoptosis regulation, modulating the cellular microenvironment in response to external stimuli [27]. These processes create favorable biochemical conditions for limb regeneration post-injury. The statistical significance of their positive correlation with temporal changes further underscores the unique biological significance of this module. Within the yellow module, “cellular extravasation” emerged as the most significant annotation, involving Bst1, Ccl25 and Crk. This enrichment is also noteworthy, as cellular extravasation regulates the migration of white blood cells during the inflammatory response and plays a role in lymphocyte activation, setting the stage for the subsequent migration of healing substances, cell growth, and apoptosis [28,29]. The GO enrichment results are shown in Figure 3b, with key genes involved in each biological process provided in Table 1 for a detailed breakdown of the results.
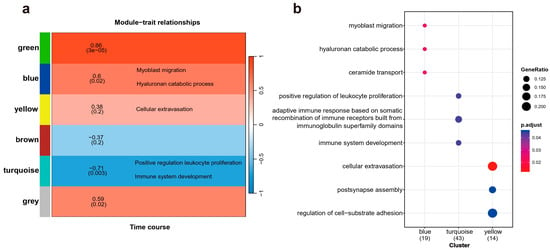
Figure 3.
The relationship between gene module clustering and temporal characteristics, along with the biological processes enriched by these modules. (a) A total of six modules are annotated, with a gradient from red to blue indicating the varying strength of the correlation between each module and temporal traits. (b) A dot plot illustrating Gene Ontology (GO) enrichment displays all biological process entries surpassing the statistical significance threshold (p < 0.05).

Table 1.
Based on the module separation by WGCNA and the list of genes enriched by GO, distinct biological processes are enriched by the genes corresponding to each module, as outlined in the table.
2.3. Target Genes of DE miRNAs
The correlation between expression levels of the aforementioned DE genes and miRNAs at various time points is depicted in Figure 4a. Following bidirectional target prediction between miRNA and mRNA, we performed a Pearson correlation test on the DE miRNAs and DE mRNAs to confirm the statistical significance of their correlation. Pairs that met the criteria under the set threshold were selected, resulting in the prediction of 16 DE miRNAs targeting a total of 13 genes. Since miRNAs typically negatively regulate the expression of their target genes, we further refined the predicted target genes. After filtering, only 8 of the predicted target genes met the conditions of being present in the mRNA expression data and exhibiting a significant negatively correlation with DE miRNAs. A correlation network is shown in Figure 4b,c.
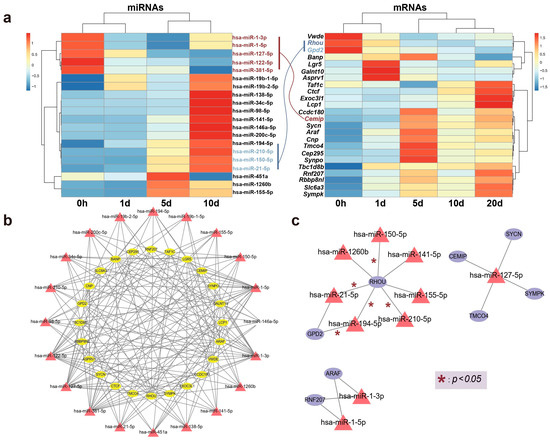
Figure 4.
The relationship between candidate genes and miRNAs. (a) A heatmap illustrating the correlation between the relative expression levels of genes and miRNAs over time, with connections between key genes and miRNAs colored according to the direction of expression. (b) Establishment of a shared network expression relationship through the identification of interacting pairs. (c) A co-expression network of selected key candidate genes and their regulating miRNAs is illustrated, with edges marked with * denoting statistically significant interactions (* p < 0.05).
Remarkably, one gene, Rhou, was targeted by 7 miRNAs, including hsa-miR-21-5p, hsa-miR-210-5p, hsa-miR-150-5p, hsa-miR-194-5p, hsa-miR-141-5p, hsa-miR-1260b, and hsa-miR-155-5p. Notably, Rhou, as a non-canonical Wnt-induced gene, has been shown to reduce cell apoptosis and increase proliferation when knocked down [30]. This is attributed to the molecular function of Rhou, which regulates cell adhesion, reduces excessive migration, promotes the proper cellular arrangement during regeneration, and regulates cell differentiation and morphological regeneration. Besides Rhou, the miRNAs hsa-miR-194-5p and hsa-miR-21-5p also regulate the Gpd2 gene. The downregulation of Gpd2 contributes to the reduction of mitochondrial activity and may mediate a metabolic shift from oxidative phosphorylation to glycolysis, which is critical for regenerative tissues [31]. Figure 5 illustrates the potential effects of limb regeneration following the inhibition of expression of Rhou and Gpd2 regulated by specific miRNAs. Consistent with our findings, upregulation of hsa-miR-21-5p expression has also been reported during tissue regeneration processes in the Mexican axolotl (Ambystoma Mexicanum) [2]. Through the detection and analysis of target pairs, we constructed a comprehensive co-expression landscape, providing a more intuitive interpretation of the interaction between genes and miRNAs. This serves as data basis for further exploration of the role of miRNAs in newt limb regeneration.
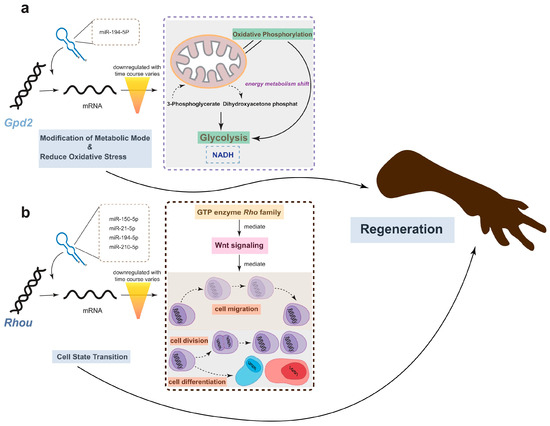
Figure 5.
The potential effects of limb regeneration following the inhibition of expression of Gpd2 and Rhou. (a) Inhibition of Gpd2 affects mitochondrial aerobic metabolism, reducing NADH production efficiency through glycolysis, and mitigating the effects of oxidative stress. (b) As a member of the GTPase family, Rhou is involved in cytoskeleton dynamics and cell migration. The low expression of this gene ensures that cells cease migrating at the appropriate time and commence differentiation into the necessary cell types for regeneration. The combined low expression of both genes promotes regeneration.
2.4. Temporal Dynamics of Expression
As mentioned in the WGCNA analysis, genes with similar functions can be clustered into the same module, which may influence complex biological phenomena. The temporal expression patterns of these genes are also significant interest. Clusters of genes with different temporal expression patterns suggest their activity trends in biological processes during the early stages of regeneration. To enhance clarity in illustrating the temporal changes of key genes, all transcriptome data were categorized into eight clusters, and miRNA data were classified into four categories. Among the three key genes highlighted earlier, Cemip was assigned to cluster 3, while Rhou and Gpd2 were associated with cluster 6. The graphical representation (Figure 6a) depicts the temporal dynamics and correlation of these gene and miRNA clusters, underscoring a significant concordance with the observed expression pattern changes of the specified key genes. We also queried the Pcna gene, which plays a significant role in several cellular processes related to DNA repair and cell cycle regulation, and serves as a proliferative marker. Pcna, which exhibited a potentially periodic expression pattern (though not statistically significant according transcriptome data), was classified into cluster 4 [32]. In addition, our quantitative real-time PCR (qPCR) validations for Pcna demonstrated a periodic expression pattern that aligned with transcriptome data, but with significantly reduced expression at both 1 and 10 days post-amputation (Figure 6b).
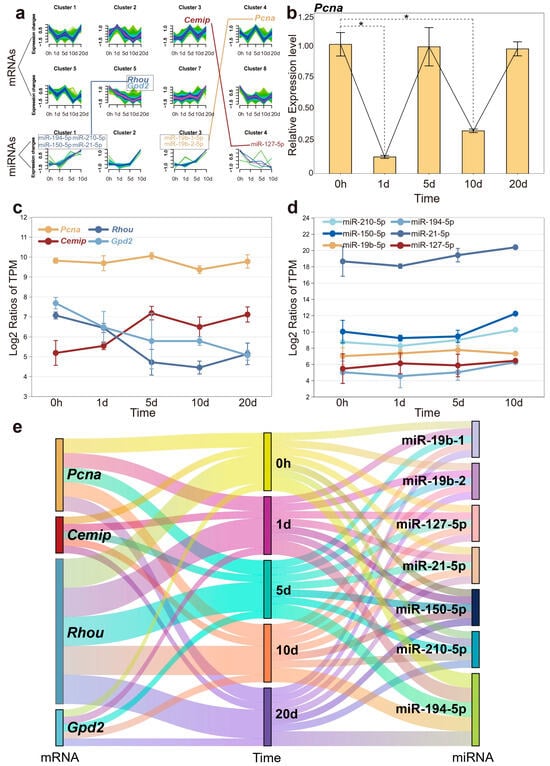
Figure 6.
The expression dynamics of mRNA and miRNA over the time course. (a) Different clusters represent distinct expression trends of mRNA and miRNA, with connecting lines indicating their respective relationships. (b) Quantitative real-time PCR results for Pcna, with significant differences indicated (* p < 0.05). (c) The expression profiles of four candidate genes are depicted across different time points post-amputation. (d) The expression trends of pivotal miRNAs regulating candidate genes. (e) A river plot to illustrate the correlation among miRNAs, mRNAs, and time points. Each river represents a molecule, and the flow of these rivers visually demonstrates the interaction of mRNA and miRNA at different time points.
After analyzing the expression trends of key genes and miRNAs individually, we found that the results were highly consistent with the performance of their respective gene clusters. Interestingly, the expression level of Pcna was generally higher than that of other genes, while miR-21-5p even reached twice the expression level of other members (Figure 6c,d). As mentioned earlier, miR-21-5p was upregulated during tissue regeneration, which may suggest its role as a fundamental regulatory factor for wound repair and growth. The high expression level ensures its stable negative regulation of target genes, maintaining the normal progress of biological processes. Figure 6e illustrates the correlations among miRNAs, mRNAs, and the various time points.
3. Discussion
Salamanders, as a distinct group of vertebrates, exhibit remarkable tissue regeneration capabilities. Historically, significant strides have been made in studying the axolotl (Ambystoma mexicanum), exploring tissue regeneration at the omics level, as well as physiological processes [33,34,35]. Furthermore, non-model organisms also prove valuable for investigating tissue regeneration. Compared with salamanders, newts possess a superior regenerative ability [36], primarily reflected in the fact that newts can regenerate tissues throughout their lifespan, whereas salamanders retain this ability only during early life stages [37,38]. Therefore, genetic research on newts may uncover the genetic basis of more unique tissue regeneration phenomena compared with salamanders. In this study, we present a comprehensive analysis of mRNA and miRNA expression patterns during the early stages of limb regeneration in the Chinese fire belly newt. Our research identified 24 significantly differentially expressed mRNAs and 20 miRNAs. Enrichment analysis revealed numerous biological processes intricately linked with limb regeneration. Notably, the Cemip gene, associated with the hyaluronan catabolic process category, emerged as a highly differentially expressed gene. Employing stringent criteria for strong mRNA-miRNA interactions, our analysis successfully matched the identified candidate key genes (Cemip, Rhou, Gpd2 and Pcna) with their respective regulated miRNAs. These findings reinforce our understanding and elucidation of the early expression patterns during limb regeneration in newts.
The traumatic limb injury caused by external forces differs significantly, from programmed cell apoptosis during early embryonic development, involving numerous unconventional biological processes that regulate the growth of new tissues [39]. During the initial stages of limb regeneration, the primary focus is on wound healing, bud formation, and growth. Our GO enrichment results highlight cellular extravasation as the most significant biological process, enriched by Bst1, Ccl25, and Crk genes. Cellular extravasation, the process of cells escaping from blood vessels or tissues, typically occurs in situations of inflammation, infection, or injury, where immune cells such as leukocytes [40] enter surrounding tissues through blood vessel walls to participate in immune responses or tissue repair. This crucial biological process aids the body in fighting infection or injury, promoting tissue regeneration and repair [35,41,42]. Previous studies on the mentioned genes have successfully validated their significant role in regulating cell adhesion at different levels, controlling the timing and rate of cell migration [23,43,44]. Consistent with expectations, we identified the regulation of cell-substrate adhesion in the yellow module, which includes cellular extravasation. These results suggest that in the early stages of limb regeneration, cell adhesion may play an important role in wound healing and bud formation. Simultaneously, cellular extravasation interacts with immune responses and inflammatory reactions to regulate the process. Interestingly, our research findings show that the clusters in the module negatively correlated with time-related traits are all immune-related biological processes (Figure 2). The relatively low immune capacity can maintain macrophages [40,45] at a relatively stable stage, preventing them from excessively attacking the injured limbs and ensuring the programmed differentiation and proliferation of cells.
Cemip, a gene present among significantly DE genes and associated with enriched biological processes related to hyaluronan catabolism, stands out as a highly intriguing and promising gene for further exploration. Cemip, plays a crucial role in encoding hyaluronic acid (HA), a major component of the extracellular matrix (ECM) [46]. Research has revealed that Cemip is regulated by the Wnt/β-catenin and TGF-β signaling pathways [26,47]. These pathways, extensively studied and confirmed, play crucial roles in embryonic development, cell cycle regulation, and cell proliferation [14,17,48,49]. Hyaluronic acid, a major component of the extracellular matrix, is widely recognized as a natural substance bound by Cemip. HA creates a moist environment that promotes cell growth and exhibits anti-inflammatory and antibacterial properties, thereby significantly contributing to the wound healing process [46,50]. Two down-regulated genes, Rhou and Gpd2, are noteworthy. Rhou, encoding a Ras family protein and belonging to the Rho GTPase family, regulates various signaling pathways critical for cell processes such as morphology, migration, adhesion, and matrix remodeling [30,51]. It also plays a regulatory role in the Wnt signaling pathway [52]. During the early stages of limb regeneration, Rhou primarily participates in the processes of cell proliferation and differentiation. The downregulation of this gene ensures that cells suspend migration and maintain a relatively primitive morphology while simultaneously reducing cell adhesion to promote proper alignment and organization of cells [53]. On the other hand, the downregulation of Gpd2 can effectively diminish mitochondrial activity, resulting in a reduced generation of reactive oxygen species (ROS) [31]. Additionally, it modulates the oxidative metabolism pathway within the cell. This metabolic shift facilitates the provision of the essential energy and biosynthetic precursors required for rapidly dividing cells during the process of tissue regeneration. Consequently, the downregulation of GPD2 supports the proliferation and survival of progenitor or blastemal cells [54], which are crucial for the successful regeneration of tissues. Interestingly, our WGCNA and GO results show that immune-related processes also exhibit a negative correlation with the early stage of regeneration, which supports the hypothesis that a mild inflammatory response and a lower level of immunity, both regulated by genes, interact to provide a relatively stable environment for tissue regeneration. Our analysis, particularly qPCR, demonstrated a periodic pattern of Pcna expression This gene is highly capable of mediating double-strand breaks (DSB) and directing their repair, a process essential for DNA repair, cell cycle regulation, and potentially closely linked to longevity and aging [55,56]. Salamander Pcna has been shown to evolve at an unprecedented rate among vertebrates, according to the previous study, which may be associated with their unique regenerative ability [57]. Given the Chinese fire belly newt’s remarkable regenerative capacity, periodic expression of Pcna may be crucial for the regeneration process.
In addition to the four genes we identified, exploring the miRNAs that target them is crucial for a comprehensive understanding of the molecular mechanisms involved. Notably, miR-21-5p emerges as a prominent candidate, exhibiting a significant increase in expression during limb regeneration in Mexican axolotls [2,58,59,60]. The expression level results mentioned earlier also indicated that the expression level of miR-21-5p was significantly higher than other candidates. Our findings further support this observation, strengthening the link between miR-21-5p and the regenerative process. This miRNA is frequently implicated in cancer research, serving as a detectable target for various cancer types [61,62,63]. Elevated expression levels of miR-21-5p are indicative of cancer, suggesting potential molecular parallels between cancer and tissue regeneration that merit exploration. Among the other key miRNAs (miR-19b-1, miR-19b-2, miR-127-5p, miR-150-5p, miR-194-5p, and miR-210-5p) we discovered, their primary function remains a class of cancer detection markers [64,65,66,67]. While current miRNA research primarily centers on their target genes, the unique molecular mechanisms of miRNAs pose challenges for direct functional studies. Given that one miRNA can target multiple genes, constructing an mRNA-miRNA regulatory network emerges as an effective research approach. As a result, the interaction landscape we have constructed makes the relationship between the two clearly visible. The existence of multiple targeted nodes also serves as a priority for further exploring molecular functions, providing insights into the future exploration of limb regeneration.
4. Materials and Methods
4.1. Sample Preparation and Transcriptome Sequencing
We performed right forelimb amputations on Chinese fire-bellied newts (Cynops orientalis) at the midstylopod level. Proximal healing tissues were collected at 0 hours, 1 day, 5 days, 10 days, and 20 days post-amputation. corresponding to the early stages of regeneration, which include wound healing, blastema formation, and differentiation process. Three biological replicates were conducted at each time point. RNA extraction was carried out using the Trizol protocol (Invitrogen, Carlsbad, CA, USA). Total RNA concentration and integrity were assessed using agarose gel electrophoresis, a NanoPhotometer spectrophotometer (IMPLEN, Westlake Village, CA, USA), and an Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA). cDNA libraries were constructed and subsequently sequenced on an Illumina HiSeq4000 platform (Illumina, San Diego, CA, USA), generating approximately 4 Gb of raw data for each transcriptome. miRNA libraries were constructed with a small RNA Sample Pre Kit and subsequently sequenced on an Illumina HiSeq2500 platform (Illumina, San Diego, CA, USA), generating approximately 10 Mb raw reads for each sample.
4.2. Construction of RNA-Seq Assembly and Counts Matrix of Expression
The sequencing data underwent quality control using FastQC v0.12.0 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, accessed on 18 August 2024), and adaptor contaminants were filtered using Cutadapt v4.4 (https://cutadapt.readthedocs.io/en/stable/, accessed on 18 August 2024). Subsequently, transcript sequences were assembled using Trinity v2.15.1 [68]. To achieve higher-quality transcript sequences, TransPS v1.1.0 [69,70] was employed to perform re-scaffolding on the assembly data obtained from Trinity. We selected the axolotl (Ambystoma mexicanum) as a closely related species and utilized its protein sequence data for alignment and de-redundancy through the BLAST+ v2.2.26 [71]. Transcript abundance for each sample was estimated in a genome-free manner using Kallisto v0.50.0 [72], and an expression matrix of counts was constructed. To enhance the robustness of our analysis, we filtered out lowly expressed transcripts (count < 30) and explored relationships among all samples and biological replicates to eliminate potential confounders. For the functional annotation of the transcripts, we utilized Trinotate v4.0.2 (https://github.com/Trinotate/Trinotate/, accessed on 18 August 2024), which enabled the addition of functional annotations to facilitate subsequent analysis.
4.3. Identification of Known and Novel miRNAs
Quality control on miRNA reads was performed using the FastQC and Cutadapt, as previously described. The clean reads were aligned with the miRbase v22 database [73] using Bowtie v1.3.1 [74]. Subsequently, miRDeep2 v2.0.1.3 [75] was used to identify both known and novel miRNAs. miRNAs with read count greater than 5 and a true positive probability exceeding 60% were selected for further analysis. A miRNA was considered reliable if it expressed in at least two out of the three individuals at a given time point.
4.4. Differential Expression of mRNAs and miRNAs
Differential expression (DE) analyses were conducted on both mRNA and miRNA matrices across the time course using R package DESeq2 v1.40.2 [76] and edgeR v3.42.4 [77]. We assessed the expression patterns in blastemal samples at each time point relative to the 0-hour control. Genes were deemed significantly expressed if they exhibited a log2 fold-change (LFC) of ≥2 with a p-value < 0.05. For miRNA, due to their overall stable expression levels and relatively small data set compared to transcripts, significance was assigned to those with an LFC of ≥1 and a p-value < 0.05.
4.5. mRNA Co-Expression Network Analysis (WGCNA)
The R package WGCNA v1.72-1 [78] was utilized for mRNA co-expression network analysis, with ‘Time course’ designated as a phenotypic trait. Following a comparison of correlations among different samples, we proceeded with additional analyses using the recommended power β (soft threshold) of 28. Subsequently, gene modules were identified by evaluating the degree of gene expression changes and correlations over the time course. Each color signifies a module comprising a cluster of genes grouped together based on their expression patterns and associations.
4.6. Gene Ontology Enrichment Analysis
To conduct a gene ontology (GO) enrichment analysis, we employed the R package clusterProfiler v4.8.2 [79]. We utilized the org.Hs.eg.db v3.17.0 (https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html, accessed on 18 August 2024) package in R as the reference database. Our main emphasis was on gene ontology (GO) terms associated with “Biological Process (BP)”. The significance of overrepresentation was assessed based on the y FDR-adjusted p-value (<0.05).
4.7. Prediction of miRNA Target Genes and Identification of Potential miRNA–Gene Pairs
To elucidate the target genes of DE miRNAs, we utilized the R package multiMiR v1.22.0 [80], which integrates multiple microRNA-target databases. Bidirectional predictions were conducted using either DE genes or DE miRNAs as input to validate our findings. Subsequently, to identify negatively correlated miRNA–mRNA pairs, we performed a Pearson’s correlation test using DE miRNA and DE mRNA data. MiRNA–mRNA pairs were selected for downstream analyses if they exhibited a negative correlation (Pearson’s coefficient > 0.9). The interaction information was then imported into Cytoscape v3.8.0 [81] for visualization and further analysis.
4.8. Gene Expression Trend Analysis
To investigate the temporal changes in gene expression, we conducted a gene expression trend analysis using the R package Mfuzz v2.60.6 [82]. Given that each sample in our study comprised three biological replicates, we utilized the mRNA expression matrix with averaged values as the input, configuring the number of clusters to 8. For miRNAs, we used the same method, but the number of clusters was set to 4. Upon visualization, we were able to discern the temporal expression patterns of significantly DE genes, shedding light on their potential pivotal role in the limb regeneration process. To validate the expression trends across different time points, we conducted quantitative real-time PCR (qPCR) experiments. To improve accuracy, we introduced three biological replicates and two technical replicates for each time point.
5. Conclusions
In this study, we conducted an integrated analysis of mRNA and miRNA expression profiles at various time points during the early stages of limb regeneration in the Chinese fire-bellied newt. Cluster analysis and GO enrichment were employed, revealing that certain differentially expressed mRNAs are intricately associated with the regeneration process. Concurrently, we identified four candidate genes (Cemip, Rhou, Gpd2 and Pcna) and seven miRNAs (miR-21-5p, miR-127-5p, miR-150-5p, miR-194-5p, miR-210-5p, miR-19b-1, and miR-19b-2) that regulate their expression. Furthermore, we explored their expression patterns at different time points during limb regeneration. These research findings illuminate the expression dynamics of mRNA and miRNA implicated in the genetic aspects of limb regeneration in newts, offering valuable insights for future investigations into the intricate biological processes underlying tissue regeneration.
Author Contributions
Conceptualization, B.L.; methodology, B.L. and Q.Z.; software, Q.Z.; validation, Q.Z. and B.L.; formal analysis, Q.Z.; resources, B.L.; data curation, Q.Z.; writing—original draft preparation, Q.Z.; writing—review and editing, B.L.; visualization, Q.Z.; supervision, B.L.; project administration, B.L.; funding acquisition, B.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, grant numbers 32170432 and 31401961; Western Lights Young Scholars Plan of Chinese Academy of Sciences, grant number 2021XBZG_XBQNXZ_A_005; and Sichuan Science and Technology Program, grant number 18YYJC0171.
Institutional Review Board Statement
All experimental protocols were performed and all animals were handled in strict accordance with the recommendations in the guidelines of the China Council on Animal Care and approved by Chengdu Institute of Biology’s Animal Experiments Ethics Committee (approval code: 20170076).
Informed Consent Statement
Not applicable.
Data Availability Statement
Transcriptome and miRNA sequencing data were deposited in the Genome Sequence Archive (GSA) of the National Genomics Data Center (NGDC) at https://ngdc.cncb.ac.cn/gsa/ (accessed on 18 August 2024) under accession number PRJCA025436.
Acknowledgments
We are grateful to Miaozhe Huo and Hong Jin for their help with the expression experiment.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Joven, A.; Elewa, A.; Simon, A. Model systems for regeneration: Salamanders. Development 2019, 146, dev167700. [Google Scholar] [CrossRef]
- Abo-Al-Ela, H.G.; Burgos-Aceves, M.A. Exploring the role of microRNAs in axolotl regeneration. J. Cell. Physiol. 2021, 236, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Gómez, C.M.A.; Echeverri, K. Salamanders: The molecular basis of tissue regeneration and its relevance to human disease. Curr. Top. Dev. Biol. 2021, 145, 235–275. [Google Scholar]
- Gómez, C.M.A.; Molina, A.G.; Zapata, J.D.; Delgado, J.P. Limb regeneration in a direct-developing terrestrial salamander, Bolitoglossa ramosi (Caudata: Plethodontidae): Limb regeneration in plethodontid salamanders. Regeneration 2017, 4, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Ju, B.G.; Kim, W.S. Endogenous retinoic acid mediates the early events in salamander limb regeneration. Anim. Cells Syst. 2012, 16, 462–468. [Google Scholar] [CrossRef]
- Maden, M. Salamanders as Key Models for Development and Regeneration Research. Methods Mol. Biol. 2023, 2562, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Rascón, C.A.; Tian, S.; Nie, J.; Barry, C.; Chu, L.-F.; Ardalani, H.; Wagner, R.J.; Probasco, M.D.; Bolin, J.M.; et al. Comparative RNA-seq Analysis in the Unsequenced Axolotl: The Oncogene Burst Highlights Early Gene Expression in the Blastema. PLoS Comput. Biol. 2013, 9, e1002936. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, A.F.; Costa, C.M.; Lorena, J.; Moreira, R.N.; Frota-Lima, G.N.; Furtado, C.; Robinson, M.; Amemiya, C.T.; Darnet, S.; Schneider, I. Tetrapod limb and sarcopterygian fin regeneration share a core genetic programme. Nat. Commun. 2016, 7, 13364. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Wnt/β-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Fan, L.; Zhao, L.; Su, Y. The interaction of Notch and Wnt signaling pathways in vertebrate regeneration. Cell Regen. 2021, 10, 1–17. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. THE WNT SIGNALING PATHWAY IN DEVELOPMENT AND DISEASE. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Avraham, T.; Daluvoy, S.; Zampell, J.; Yan, A.; Haviv, Y.S.; Rockson, S.G.; Mehrara, B.J. Blockade of Transforming Growth Factor-β1 Accelerates Lymphatic Regeneration during Wound Repair. Am. J. Pathol. 2010, 177, 3202–3214. [Google Scholar] [CrossRef]
- Yu, Y.; Tang, J.; Su, J.; Cui, J.; Xie, X.; Chen, F. Integrative Analysis of MicroRNAome, Transcriptome, and Proteome during the Limb Regeneration of Cynops orientalis. J. Proteome Res. 2019, 18, 1088–1098. [Google Scholar] [CrossRef]
- Bin, L.; Malley, C.; Taylor, P.; Preethi Boorgula, M.; Chavan, S.; Daya, M.; Mathias, M.; Shankar, G.; Rafaels, N.; Vergara, C.; et al. Whole genome sequencing identifies novel genetic mutations in patients with eczema herpeticum. Allergy 2021, 76, 2510–2523. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.H.; Yeo, M.G.; Oh, H.J.; Song, W.K. v-Crk Induces Rac-dependent Membrane Ruffling and Cell Migration in CAS-deficient Embryonic Fibroblasts. Mol. Cells 2008, 25, 131–137. [Google Scholar] [CrossRef]
- Sundman, E.A.; Cole, B.J.; Karas, V.; Della Valle, C.; Tetreault, M.W.; Mohammed, H.O.; Fortier, L.A. The Anti-inflammatory and Matrix Restorative Mechanisms of Platelet-Rich Plasma in Osteoarthritis. Am. J. Sports Med. 2014, 42, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Fang, X.; Yang, Y.; Song, Y. Silencing of CEMIP suppresses Wnt/β-catenin/Snail signaling transduction and inhibits EMT program of colorectal cancer cells. Acta Histochem. 2018, 120, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Nagaoka, A.; Kusaka-Kikushima, A.; Tobiishi, M.; Kawabata, K.; Sayo, T.; Sakai, S.; Sugiyama, Y.; Enomoto, H.; Okada, Y.; et al. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc. Natl. Acad. Sci. USA 2013, 110, 5612–5617. [Google Scholar] [CrossRef]
- Hanada, K. Intracellular trafficking of ceramide by ceramide transfer protein. Proc. Jpn. Acad. Ser. B 2010, 86, 426–437. [Google Scholar] [CrossRef]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of Blood Vessels and Tissues by a Population of Monocytes with Patrolling Behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef]
- Barreiro, O.; Sánchez-Madrid, F. Bases moleculares de las interacciones leucocito-endotelio durante la respuesta inflamatoria. REC Interv. Cardiol. 2009, 62, 552–562. [Google Scholar] [CrossRef]
- Slaymi, C.; Vignal, E.; Crès, G.; Roux, P.; Blangy, A.; Raynaud, P.; Fort, P. The atypical RhoU/Wrch1 Rho GTPase controls cell proliferation and apoptosis in the gut epithelium. Biol. Cell 2019, 111, 121–141. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Baple, E.L.; Chambers, H.; Cross, H.E.; Fawcett, H.; Nakazawa, Y.; Chioza, B.A.; Harlalka, G.V.; Mansour, S.; Sreekantan-Nair, A.; Patton, M.A.; et al. Hypomorphic PCNA mutation underlies a human DNA repair disorder. J. Clin. Investig. 2014, 124, 3137–3146. [Google Scholar] [CrossRef]
- Nowoshilow, S.; Schloissnig, S.; Fei, J.-F.; Dahl, A.; Pang, A.W.C.; Pippel, M.; Winkler, S.; Hastie, A.R.; Young, G.; Roscito, J.G.; et al. The axolotl genome and the evolution of key tissue formation regulators. Nature 2018, 554, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Guzmán, T.; Wang, H.; Khattak, S.; Schuez, M.; Roensch, K.; Nacu, E.; Tazaki, A.; Joven, A.; Tanaka, E.M.; Simon, A. Fundamental Differences in Dedifferentiation and Stem Cell Recruitment during Skeletal Muscle Regeneration in Two Salamander Species. Cell Stem Cell 2014, 14, 174–187. [Google Scholar] [CrossRef]
- Sousounis, K.; Athippozhy, A.T.; Voss, S.R.; Tsonis, P.A. Plasticity for axolotl lens regeneration is associated with age-related changes in gene expression. Regeneration 2014, 1, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, G.; Eguchi, Y.; Nakamura, K.; Yadav, M.C.; Millán, J.L.; Tsonis, P.A. Regenerative capacity in newts is not altered by repeated regeneration and ageing. Nat. Commun. 2011, 2, 384. [Google Scholar] [CrossRef]
- Torres, M. Limb regrowth takes two. Nature 2016, 533, 328–330. [Google Scholar] [CrossRef][Green Version]
- Nacu, E.; Tanaka, E.M. Limb Regeneration: A New Development? Annu. Rev. Cell Dev. Biol. 2011, 27, 409–440. [Google Scholar] [CrossRef]
- Arnold, L.; Henry, A.; Poron, F.; Baba-Amer, Y.; van Rooijen, N.; Plonquet, A.; Gherardi, R.K.; Chazaud, B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J. Exp. Med. 2007, 204, 1057–1069. [Google Scholar] [CrossRef]
- Wilgus, T.A. Immune cells in the healing skin wound: Influential players at each stage of repair. Pharmacol. Res. 2008, 58, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Haertel, E.; Werner, S.; Schäfer, M. Transcriptional regulation of wound inflammation. Semin. Immunol. 2014, 26, 321–328. [Google Scholar] [CrossRef]
- Ericsson, A.; Kotarsky, K.; Svensson, M.; Sigvardsson, M.; Agace, W. Functional Characterization of the CCL25 Promoter in Small Intestinal Epithelial Cells Suggests a Regulatory Role for Caudal-Related Homeobox (Cdx) Transcription Factors. J. Immunol. 2006, 176, 3642–3651. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, A. Expression and regulation of CCL25 and its role in T cell localization and function within the small intestine. Ph.D. Thesis, Faculty of Medicine, Lund University, Lund, Sweden, 2005. [Google Scholar]
- Godwin, J.W.; Pinto, A.R.; Rosenthal, N.A. Macrophages are required for adult salamander limb regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 9415–9420. [Google Scholar] [CrossRef]
- Spataro, S.; Guerra, C.; Cavalli, A.; Sgrignani, J.; Sleeman, J.; Poulain, L.; Boland, A.; Scapozza, L.; Moll, S.; Prunotto, M. CEMIP (HYBID, KIAA1199): Structure, function and expression in health and disease. FEBS J. 2023, 290, 3946–3962. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Liang, W.; Ye, Y.; Yi, B. PERK-Dependent Activation of the JAK2/STAT3 Pathway Contributes to High Glucose-Induced Extracellular Matrix Deposition in Renal Tubular Epithelial Cells. Int. J. Endocrinol. 2021, 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kispert, A.; Vainio, S.; McMahon, A.P. Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 1998, 125, 4225–4234. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-β Signaling from Cell Membrane to the Nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Neuman, M.G.; Nanau, R.M.; Oruña-Sanchez, L.; Coto, G. Hyaluronic Acid and Wound Healing. J. Pharm. Pharm. Sci. 2015, 18, 53–60. [Google Scholar] [CrossRef]
- Katoh, M. Networking of WNT, FGF, Notch, BMP, and Hedgehog Signaling Pathways during Carcinogenesis. Stem Cell Rev. 2007, 3, 30–38. [Google Scholar] [CrossRef]
- Jaffe, A.B.; Hall, A. RHO GTPASES: Biochemistry and Biology. Annu. Rev. Cell Dev. Biol. 2005, 21, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho GTPases and the control of cell behaviour. Biochem. Soc. Trans. 2005, 33, 891–895. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Dovrat, D.; Stodola, J.L.; Burgers, P.M.; Aharoni, A. Sequential switching of binding partners on PCNA during in vitro Okazaki fragment maturation. Proc. Natl. Acad. Sci. USA 2014, 111, 14118–14123. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, E.; Bartnicki, F.; Fujisawa, R.; Bonarek, P.; Hermanowicz, P.; Tsurimoto, T.; Muszyńska, K.; Strzalka, W. Inhibition of DNA replication by an anti-PCNA aptamer/PCNA complex. Nucleic Acids Res. 2018, 46, 25–41. [Google Scholar] [CrossRef] [PubMed]
- Lu, B. Evolutionary Insights into the Relationship of Frogs, Salamanders, and Caecilians and Their Adaptive Traits, with an Emphasis on Salamander Regeneration and Longevity. Animals 2023, 13, 3449. [Google Scholar] [CrossRef]
- Gearhart, M.D.; Erickson, J.R.; Walsh, A.; Echeverri, K. Identification of Conserved and Novel MicroRNAs during Tail Regeneration in the Mexican Axolotl. Int. J. Mol. Sci. 2015, 16, 22046–22061. [Google Scholar] [CrossRef]
- Holman, E.C.; Campbell, L.J.; Hines, J.; Crews, C.M. Microarray Analysis of microRNA Expression during Axolotl Limb Regeneration. PLoS ONE 2012, 7, e41804. [Google Scholar] [CrossRef]
- King, B.L.; Yin, V.P. A Conserved MicroRNA Regulatory Circuit Is Differentially Controlled during Limb/Appendage Regeneration. PLoS ONE 2016, 11, e0157106. [Google Scholar] [CrossRef]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef]
- Iorio, M.V.; Ferracin, M.; Liu, C.-G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA Gene Expression Deregulation in Human Breast Cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe–Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Mavrakis, K.J.; Wolfe, A.L.; Oricchio, E.; Palomero, T.; de Keersmaecker, K.; McJunkin, K.; Zuber, J.; James, T.; Khan, A.A.; Leslie, C.S.; et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat. Cell Biol. 2010, 12, 372–379. [Google Scholar] [CrossRef]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Kroh, E.; Wood, B.; Arroyo, J.D.; Dougherty, K.J.; Miyaji, M.M.; Tait, J.F.; Tewari, M. Blood Cell Origin of Circulating MicroRNAs: A Cautionary Note for Cancer Biomarker Studies. Cancer Prev. Res. 2012, 5, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Lawrie, C.H.; Gal, S.; Dunlop, H.M.; Pushkaran, B.; Liggins, A.P.; Pulford, K.; Banham, A.H.; Pezzella, F.; Boultwood, J.; Wainscoat, J.S.; et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br. J. Haematol. 2008, 141, 672–675. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Liu, M.; Adelman, Z.N.; Zhang, L. TransPS: A Transcriptome Post Scaffolding Method for Assembling High Quality Contigs. Comput. Biol. J. 2014, 2014, 961823. [Google Scholar] [CrossRef]
- Banerjee, B.; Koner, D.; Hasan, R.; Bhattacharya, S.; Saha, N. Transcriptome analysis reveals novel insights in air-breathing magur catfish (Clarias magur) in response to high environmental ammonia. Gene 2019, 703, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 421. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Grocock, R.J.; Van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Ru, Y.; Kechris, K.J.; Tabakoff, B.; Hoffman, P.; Radcliffe, R.A.; Bowler, R.; Mahaffey, S.; Rossi, S.; Calin, G.A.; Bemis, L.; et al. The multiMiR R package and database: Integration of microRNA–target interactions along with their disease and drug associations. Nucleic Acids Res. 2014, 42, e133. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).