
Targeting Androgen, Thyroid Hormone, and Vitamin A and D Receptors to Treat Prostate Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nuclear Hormone Receptors
2.1. The Functionalities
2.2. Phylogenetical Features
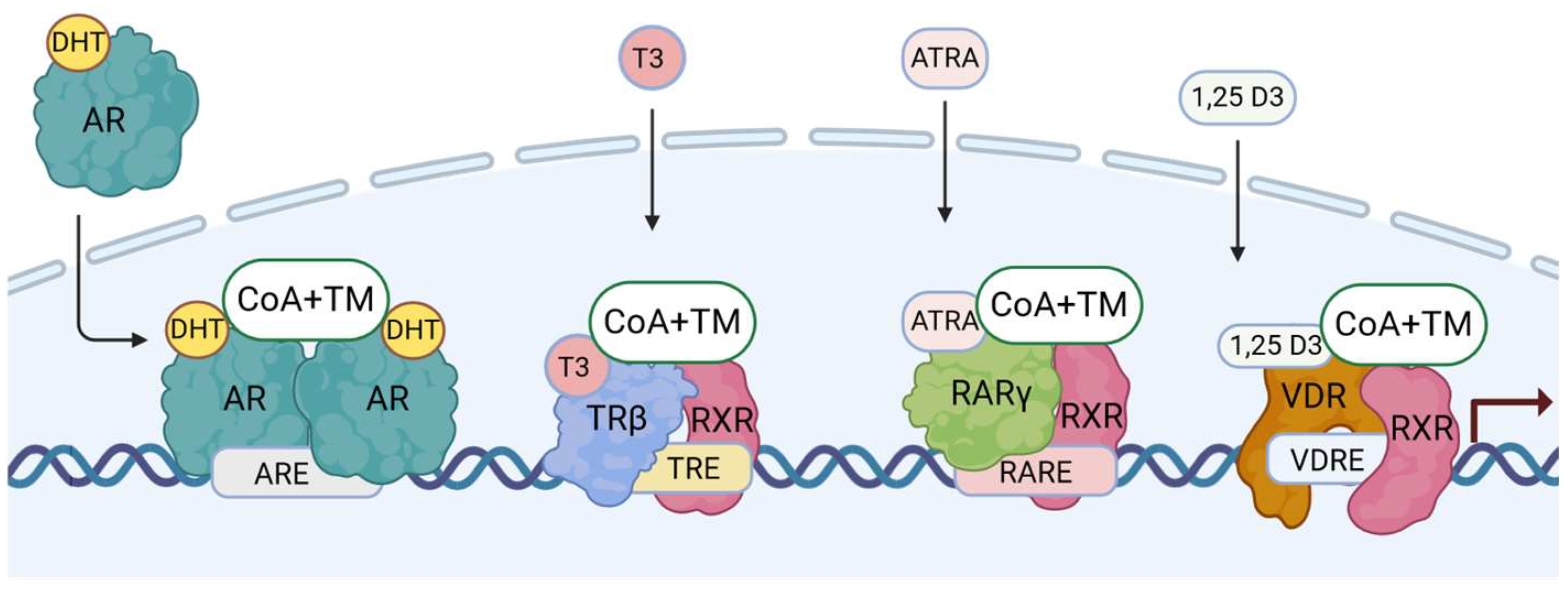
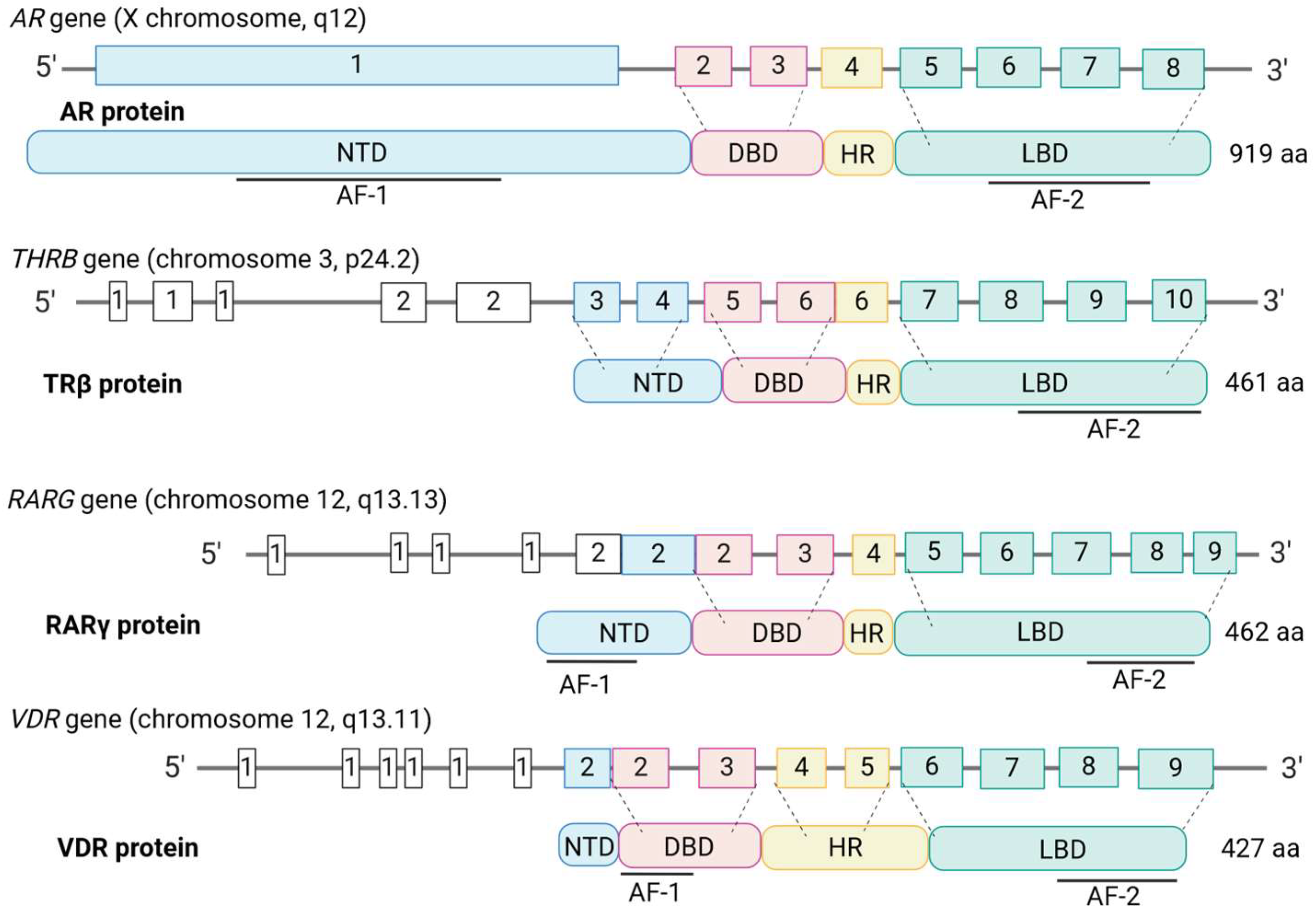
2.3. DNA-Binding Motifs and Gene Expression Regulation
3. Androgen Receptor
3.1. The Functionalities of the AR
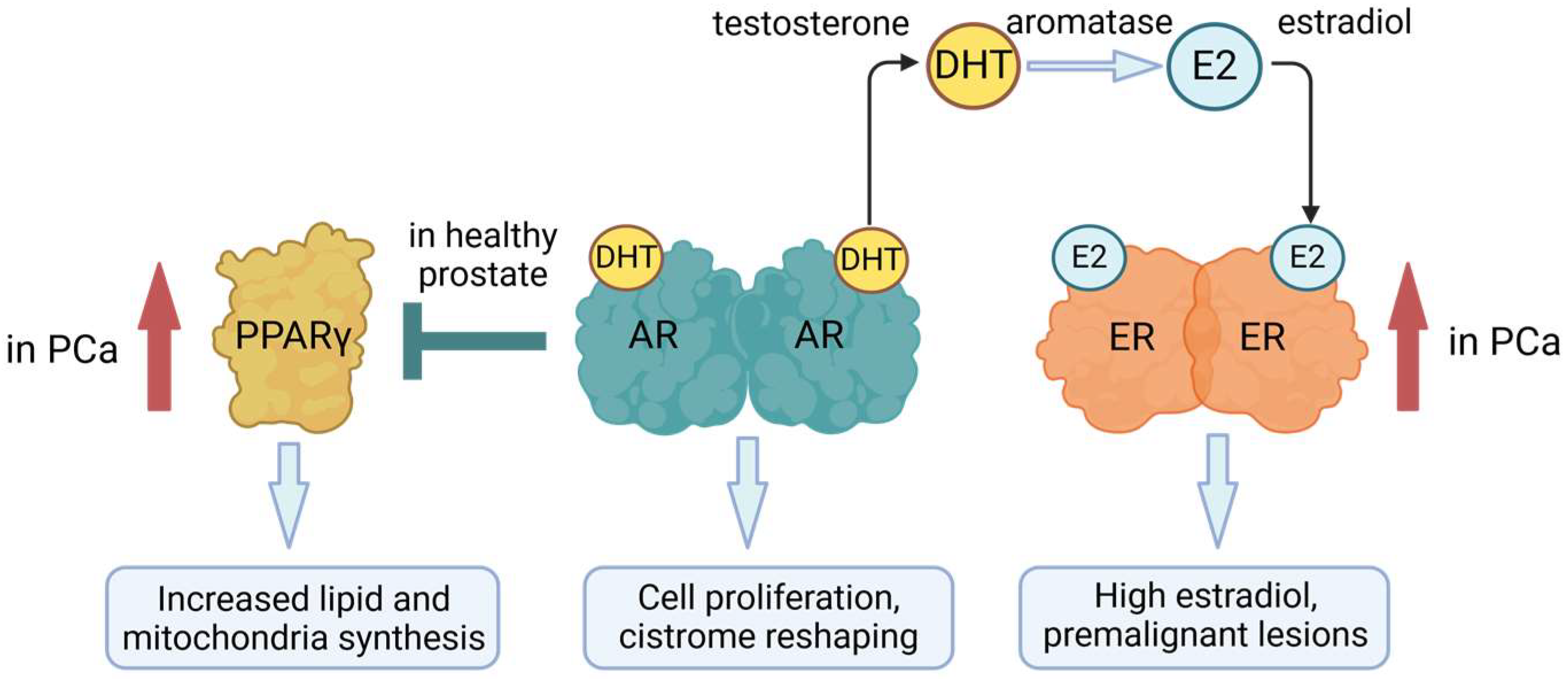
3.2. AR and Prostate Cancer
3.3. Crosstalk between AR and Other NRs/Transcription Factors
4. Thyroid Hormone Receptor
4.1. The Functionalities of TRβ
4.2. THs/TRβ and Prostate Cancer
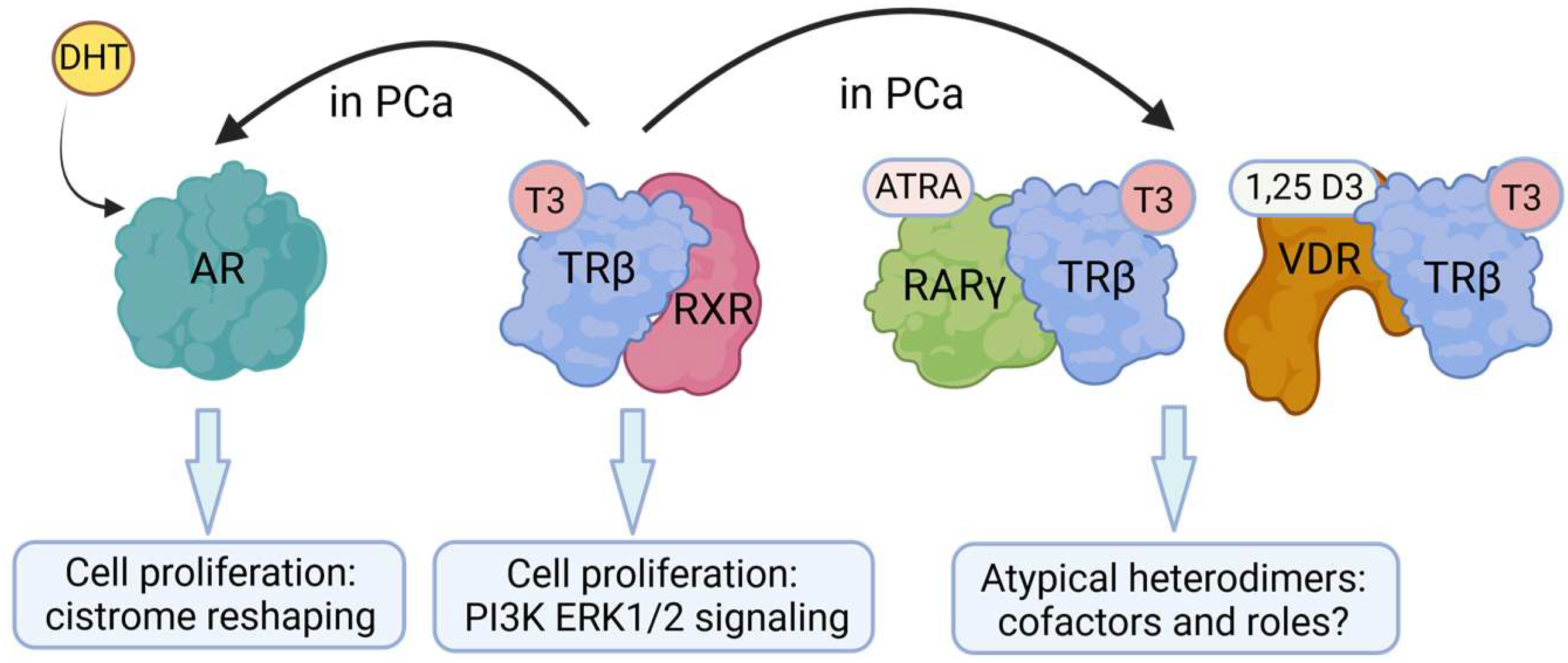
4.3. Crosstalk between TRβ and Other NRs/Transcription Factors
5. Retinoic Acid Receptors
5.1. The Functionalities of RARs
5.2. RARs and Prostate Cancer
5.3. Crosstalk between RARs and Other Transcription Factors
6. Vitamin D Receptor
6.1. The Functionalities of VDRs
6.2. VDRs and Prostate Cancer
6.3. Crosstalk between VDRs and Other NRs/Transcription Factors
7. Drug Combination Therapies Involving NRs
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Huggins, C.; Hodges, C.V. Studies on Prostatic Cancer I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Watson, P.A.; Chen, Y.F.; Balbas, M.D.; Wongvipat, J.; Socci, N.D.; Viale, A.; Kim, K.; Sawyers, C.L. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc. Natl. Acad. Sci. USA 2010, 107, 16759–16765. [Google Scholar] [CrossRef]
- Haile, S.; Sadar, M.D. Androgen receptor and its splice variants in prostate cancer. Cell Mol. Life Sci. 2011, 68, 3971–3981. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Vaishampayan, U.N. Randomized trial of enzalutamide versus bicalutamide in combination with androgen deprivation in metastatic hormone sensitive prostate cancer: A Prostate Cancer Clinical Trials Consortium trial. J. Clin. Oncol. 2018, 36 (Suppl. S6), 190. [Google Scholar] [CrossRef]
- Lai, J.; Myers, S.A.; Lawrence, M.G.; Odorico, D.M.; Clements, J.A. Direct progesterone receptor and indirect androgen receptor interactions with the kallikrein-related peptidase 4 gene promoter in breast and prostate cancer. Mol. Cancer Res. 2009, 7, 129–141. [Google Scholar] [CrossRef]
- Setlur, S.R.; Mertz, K.D.; Hoshida, Y.; Demichelis, F.; Lupien, M.; Perner, S.; Sboner, A.; Pawitan, Y.; Andren, O.; Johnson, L.A.; et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J. Natl. Cancer Inst. 2008, 100, 815–825. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Ploner, C.; Handle, F.; Schaefer, G.; Kroon, J.; Leo, A.; Heidegger, I.; Eder, I.; et al. The Glucocorticoid Receptor Is a Key Player for Prostate Cancer Cell Survival and a Target for Improved Antiandrogen Therapy. Clin. Cancer Res. 2018, 24, 927–938. [Google Scholar] [CrossRef]
- Zhao, L.; Zhou, S.; Gustafsson, J.A. Nuclear Receptors: Recent Drug Discovery for Cancer Therapies. Endocr. Rev. 2019, 40, 1207–1249. [Google Scholar] [CrossRef]
- Leach, D.A.; Powell, S.M.; Bevan, C.L. WOMEN IN CANCER THEMATIC REVIEW: New roles for nuclear receptors in prostate cancer. Endocr. Relat. Cancer 2016, 23, T85–T108. [Google Scholar] [CrossRef] [PubMed]
- Roshan-Moniri, M.; Hsing, M.; Butler, M.S.; Cherkasov, A.; Rennie, P.S. Orphan nuclear receptors as drug targets for the treatment of prostate and breast cancers. Cancer Treat. Rev. 2014, 40, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Means, A.R.; Comstock, J.P.; Rosenfeld, G.C.; O’Malley, B.W. Ovalbumin messenger RNA of chick oviduct: Partial characterization, estrogen dependence, and translation in vitro. Proc. Natl. Acad. Sci. USA 1972, 69, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Frigo, D.E.; Bondesson, M.; Williams, C. Nuclear receptors: From molecular mechanisms to therapeutics. Essays Biochem. 2021, 65, 847–856. [Google Scholar] [CrossRef]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef]
- Green, S.; Walter, P.; Greene, G.; Krust, A.; Goffin, C.; Jensen, E.; Scrace, G.; Waterfield, M.; Chambon, P. Cloning of the human oestrogen receptor cDNA. J. Steroid Biochem. 1986, 24, 77–83. [Google Scholar] [CrossRef]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schutz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Jimenez-Panizo, A.; Perez, P.; Rojas, A.M.; Fuentes-Prior, P.; Estebanez-Perpina, E. Non-canonical dimerization of the androgen receptor and other nuclear receptors: Implications for human disease. Endocr. Relat. Cancer 2019, 26, R479–R497. [Google Scholar] [CrossRef]
- Sever, R.; Glass, C.K. Signaling by nuclear receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a016709. [Google Scholar] [CrossRef]
- Porter, B.A.; Ortiz, M.A.; Bratslavsky, G.; Kotula, L. Structure and Function of the Nuclear Receptor Superfamily and Current Targeted Therapies of Prostate Cancer. Cancers 2019, 11, 1852. [Google Scholar] [CrossRef] [PubMed]
- Helsen, C.; Kerkhofs, S.; Clinckemalie, L.; Spans, L.; Laurent, M.; Boonen, S.; Vanderschueren, D.; Claessens, F. Structural basis for nuclear hormone receptor DNA binding. Mol. Cell Endocrinol. 2012, 348, 411–417. [Google Scholar] [CrossRef]
- Lazar, M.A. Maturing of the nuclear receptor family. J. Clin. Investig. 2017, 127, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Khorasanizadeh, S.; Rastinejad, F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem. Sci. 2001, 26, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription factor-DNA binding: Beyond binding site motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef]
- Bhimsaria, D.; Rodriguez-Martinez, J.A.; Mendez-Johnson, J.L.; Ghoshdastidar, D.; Varadarajan, A.; Bansal, M.; Daniels, D.L.; Ramanathan, P.; Ansari, A.Z. Hidden modes of DNA binding by human nuclear receptors. Nat. Commun. 2023, 14, 4179. [Google Scholar] [CrossRef] [PubMed]
- De Bosscher, K.; Desmet, S.J.; Clarisse, D.; Estebanez-Perpina, E.; Brunsveld, L. Nuclear receptor crosstalk-defining the mechanisms for therapeutic innovation. Nat. Rev. Endocrinol. 2020, 16, 363–377. [Google Scholar] [CrossRef]
- Font-Diaz, J.; Jimenez-Panizo, A.; Caelles, C.; Vivanco, M.D.; Perez, P.; Aranda, A.; Estebanez-Perpina, E.; Castrillo, A.; Ricote, M.; Valledor, A.F. Nuclear receptors: Lipid and hormone sensors with essential roles in the control of cancer development. Semin. Cancer Biol. 2021, 73, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a030452. [Google Scholar] [CrossRef]
- Culig, Z.; Santer, F.R. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev. 2014, 33, 413–427. [Google Scholar] [CrossRef]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R.; Hammes, S.R. Nuclear receptors outside the nucleus: Extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 2016, 17, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Augello, M.A.; Liu, D.; Deonarine, L.D.; Robinson, B.D.; Huang, D.; Stelloo, S.; Blattner, M.; Doane, A.S.; Wong, E.W.P.; Chen, Y.; et al. CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell 2019, 35, 817–819. [Google Scholar] [CrossRef]
- Sonnenschein, C.; Olea, N.; Pasanen, M.E.; Soto, A.M. Negative controls of cell proliferation: Human prostate cancer cells and androgens. Cancer Res. 1989, 49, 3474–3481. [Google Scholar] [PubMed]
- Langeler, E.G.; van Uffelen, C.J.; Blankenstein, M.A.; van Steenbrugge, G.J.; Mulder, E. Effect of culture conditions on androgen sensitivity of the human prostatic cancer cell line LNCaP. Prostate 1993, 23, 213–223. [Google Scholar] [CrossRef]
- Denmeade, S.; Antonarakis, E.S.; Markowski, M.C. Bipolar androgen therapy (BAT): A patient’s guide. Prostate 2022, 82, 753–762. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Han, D.; Labaf, M.; Zhao, Y.; Owiredu, J.; Zhang, S.; Patel, K.; Venkataramani, K.; Steinfeld, J.S.; Han, W.; Li, M.; et al. Androgen receptor splice variants drive castration-resistant prostate cancer metastasis by activating distinct transcriptional programs. J. Clin. Investig. 2024, 134, e168649. [Google Scholar] [CrossRef]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taules, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388. [Google Scholar] [CrossRef]
- Li, M.T.; Richter, F.; Chang, C.; Irwin, R.J.; Huang, H. Androgen and retinoic acid interaction in LNCaP cells, effects on cell proliferation and expression of retinoic acid receptors and epidermal growth factor receptor. BMC Cancer 2002, 2, 16. [Google Scholar] [CrossRef]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Ellem, S.J.; Risbridger, G.P. Aromatase and regulating the estrogen:androgen ratio in the prostate gland. J. Steroid Biochem. Mol. Biol. 2010, 118, 246–251. [Google Scholar] [CrossRef]
- Ross, R.K.; Bernstein, L.; Lobo, R.A.; Shimizu, H.; Stanczyk, F.Z.; Pike, M.C.; Henderson, B.E. 5-alpha-reductase activity and risk of prostate cancer among Japanese and US white and black males. Lancet 1992, 339, 887–889. [Google Scholar] [CrossRef]
- Nelles, J.L.; Hu, W.Y.; Prins, G.S. Estrogen action and prostate cancer. Expert. Rev. Endocrinol. Metab. 2011, 6, 437–451. [Google Scholar] [CrossRef]
- Belluti, S.; Imbriano, C.; Casarini, L. Nuclear Estrogen Receptors in Prostate Cancer: From Genes to Function. Cancers 2023, 15, 4653. [Google Scholar] [CrossRef]
- Weigel, N.L. Interactions between vitamin D and androgen receptor signaling in prostate cancer cells. Nutr. Rev. 2007, 65 Pt 2, S116–S117. [Google Scholar] [CrossRef]
- Mooso, B.; Madhav, A.; Johnson, S.; Roy, M.; Moore, M.E.; Moy, C.; Loredo, G.A.; Mehta, R.G.; Vaughan, A.T.; Ghosh, P.M. Androgen Receptor regulation of Vitamin D receptor in response of castration-resistant prostate cancer cells to 1alpha-Hydroxyvitamin D5-a calcitriol analog. Genes. Cancer 2010, 1, 927–940. [Google Scholar] [CrossRef]
- Murthy, S.; Agoulnik, I.U.; Weigel, N.L. Androgen receptor signaling and vitamin D receptor action in prostate cancer cells. Prostate 2005, 64, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Erzurumlu, Y.; Aydogdu, E.; Dogan, H.K.; Catakli, D.; Muhammed, M.T.; Buyuksandic, B. 1,25(OH)(2) D(3) induced vitamin D receptor signaling negatively regulates endoplasmic reticulum-associated degradation (ERAD) and androgen receptor signaling in human prostate cancer cells. Cell Signal 2023, 103, 110577. [Google Scholar] [CrossRef]
- Rogenhofer, S.; Ellinger, J.; Kahl, P.; Stoehr, C.; Hartmann, A.; Engehausen, D.; Wieland, W.F.; Muller, S.C.; Hofstadter, F.; Walter, B. Enhanced expression of peroxisome proliferate-activated receptor gamma (PPAR-gamma) in advanced prostate cancer. Anticancer. Res. 2012, 32, 3479–3483. [Google Scholar] [PubMed]
- Forootan, F.S.; Forootan, S.S.; Malki, M.I.; Chen, D.; Li, G.; Lin, K.; Rudland, P.S.; Foster, C.S.; Ke, Y. The expression of C-FABP and PPARgamma and their prognostic significance in prostate cancer. Int. J. Oncol. 2014, 44, 265–275. [Google Scholar] [CrossRef]
- Hartley, A.; Ahmad, I. The role of PPARgamma in prostate cancer development and progression. Br. J. Cancer 2023, 128, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Pakula, H.; Xiang, D.; Li, Z. A Tale of Two Signals: AR and WNT in Development and Tumorigenesis of Prostate and Mammary Gland. Cancers 2017, 9, 14. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Q.; Xu, H. Wnt/beta-catenin signal transduction pathway in prostate cancer and associated drug resistance. Discov. Oncol. 2021, 12, 40. [Google Scholar] [CrossRef]
- Hosseinzadeh, L.; Kikhtyak, Z.; Laven-Law, G.; Pederson, S.M.; Puiu, C.G.; D’Santos, C.S.; Lim, E.; Carroll, J.S.; Tilley, W.D.; Dwyer, A.R.; et al. The androgen receptor interacts with GATA3 to transcriptionally regulate a luminal epithelial cell phenotype in breast cancer. Genome Biol. 2024, 25, 44. [Google Scholar] [CrossRef]
- Crockford, S.J. Evolutionary roots of iodine and thyroid hormones in cell-cell signaling. Integr. Comp. Biol. 2009, 49, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.; Heyland, A. Evolution of thyroid hormone signaling in animals: Non-genomic and genomic modes of action. Mol. Cell Endocrinol. 2017, 459, 14–20. [Google Scholar] [CrossRef]
- Heyland, A.; Hodin, J.; Reitzel, A.M. Hormone signaling in evolution and development: A non-model system approach. Bioessays 2005, 27, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Heyland, A.; Moroz, L.L. Cross-kingdom hormonal signaling: An insight from thyroid hormone functions in marine larvae. J. Exp. Biol. 2005, 208 Pt 23, 4355–4361. [Google Scholar] [CrossRef]
- Weinberger, C.; Thompson, C.C.; Ong, E.S.; Lebo, R.; Gruol, D.J.; Evans, R.M. The c-erb-A gene encodes a thyroid hormone receptor. Nature 1986, 324, 641–646. [Google Scholar] [CrossRef]
- Glass, C.K.; Lipkin, S.M.; Devary, O.V.; Rosenfeld, M.G. Positive and negative regulation of gene transcription by a retinoic acid-thyroid hormone receptor heterodimer. Cell 1989, 59, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.Y. Thyroid hormone receptor mutations and disease: Beyond thyroid hormone resistance. Trends Endocrinol. Metab. 2005, 16, 176–182. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Leonard, J.L.; Davis, P.J. Molecular aspects of thyroid hormone actions. Endocr. Rev. 2010, 31, 139–170. [Google Scholar] [CrossRef]
- Chiamolera, M.I.; Sidhaye, A.R.; Matsumoto, S.; He, Q.; Hashimoto, K.; Ortiga-Carvalho, T.M.; Wondisford, F.E. Fundamentally distinct roles of thyroid hormone receptor isoforms in a thyrotroph cell line are due to differential DNA binding. Mol. Endocrinol. 2012, 26, 926–939. [Google Scholar] [CrossRef]
- Putcha, B.D.; Fernandez, E.J. Direct interdomain interactions can mediate allosterism in the thyroid receptor. J. Biol. Chem. 2009, 284, 22517–22524. [Google Scholar] [CrossRef]
- Aranda, A.; Martinez-Iglesias, O.; Ruiz-Llorente, L.; Garcia-Carpizo, V.; Zambrano, A. Thyroid receptor: Roles in cancer. Trends Endocrinol. Metab. 2009, 20, 318–324. [Google Scholar] [CrossRef]
- Flamant, F.; Cheng, S.Y.; Hollenberg, A.N.; Moeller, L.C.; Samarut, J.; Wondisford, F.E.; Yen, P.M.; Refetoff, S. Thyroid Hormone Signaling Pathways: Time for a More Precise Nomenclature. Endocrinology 2017, 158, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Flamant, F. Futures Challenges in Thyroid Hormone Signaling Research. Front. Endocrinol. 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Jouravel, N.; Sablin, E.; Togashi, M.; Baxter, J.D.; Webb, P.; Fletterick, R.J. Molecular basis for dimer formation of TRbeta variant D355R. Proteins 2009, 75, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Tovo-Neto, A.; da Silva Rodrigues, M.; Habibi, H.R.; Nobrega, R.H. Thyroid hormone actions on male reproductive system of teleost fish. Gen. Comp. Endocrinol. 2018, 265, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.S.; Tovo-Neto, A.; Rosa, I.F.; Doretto, L.B.; Fallah, H.P.; Habibi, H.R.; Nobrega, R.H. Thyroid Hormones Deficiency Impairs Male Germ Cell Development: A Cross Talk between Hypothalamic-Pituitary-Thyroid, and-Gonadal Axes in Zebrafish. Front. Cell Dev. Biol. 2022, 10, 865948. [Google Scholar] [CrossRef]
- Nabi, G.; Hao, Y.; Liu, X.; Sun, Y.; Wang, Y.; Jiang, C.; Li, J.; Wu, Y.; Li, D. Hypothalamic-Pituitary-Thyroid Axis Crosstalk With the Hypothalamic-Pituitary-Gonadal Axis and Metabolic Regulation in the Eurasian Tree Sparrow During Mating and Non-mating Periods. Front. Endocrinol. 2020, 11, 303. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Shekhar, S.; Dhole, B. Thyroid and male reproduction. Indian. J. Endocrinol. Metab. 2014, 18, 23–31. [Google Scholar] [CrossRef]
- Quartuccio, M.; Fazio, E.; Medica, P.; Cristarella, S.; Emmanuele, G.; Sinagra, L.; Liotta, L. Correlation between sperm parameters and circulating thyroid hormones and testosterone concentrations in Labrador Retriever dog. Ital. J. Anim. Sci. 2021, 20, 947–954. [Google Scholar] [CrossRef]
- Castaneda Cortes, D.C.; Langlois, V.S.; Fernandino, J.I. Crossover of the hypothalamic pituitary-adrenal/interrenal, -thyroid, and -gonadal axes in testicular development. Front. Endocrinol. 2014, 5, 139. [Google Scholar] [CrossRef]
- Ren, B.; Zhu, Y. A New Perspective on Thyroid Hormones: Crosstalk with Reproductive Hormones in Females. Int. J. Mol. Sci. 2022, 23, 2708. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.S.; Wajner, S.M.; Maia, A.L. The role of thyroid hormone in testicular development and function. J. Endocrinol. 2008, 199, 351–365. [Google Scholar] [CrossRef]
- Aruldhas, M.M.; Ramalingam, N.; Jaganathan, A.; John Sashi, A.M.; Stanley, J.A.; Nagappan, A.S.; Vasavan, J.; Kannan, A.; Seshadri, V.N. Gestational and neonatal-onset hypothyroidism alters androgen receptor status in rat prostate glands at adulthood. Prostate 2010, 70, 689–700. [Google Scholar] [CrossRef]
- Choudhury, S.; Chainy, G.B.; Mishro, M.M. Experimentally induced hypo- and hyper-thyroidism influence on the antioxidant defence system in adult rat testis. Andrologia 2003, 35, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.; Kennedy, L. Testosterone replacement therapy: Role of pituitary and thyroid in diagnosis and treatment. Transl. Androl. Urol. 2016, 5, 850–858. [Google Scholar] [CrossRef]
- Sengupta, P.D.S. Thyroid Disorders and Semen Quality. Biomed. Pharmacol. J. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Goemann, I.M.; Romitti, M.; Meyer, E.L.S.; Wajner, S.M.; Maia, A.L. Role of thyroid hormones in the neoplastic process: An overview. Endocr. Relat. Cancer 2017, 24, R367–R385. [Google Scholar] [CrossRef]
- Hercbergs, A. Clinical Implications and Impact of Discovery of the Thyroid Hormone Receptor on Integrin alphavbeta3-A Review. Front. Endocrinol. 2019, 10, 565. [Google Scholar] [CrossRef]
- Anguiano, B.; Lopez, A.; Delgado, G.; Romero, C.; Aceves, C. Deiodinase type 1 activity is expressed in the prostate of pubescent rats and is modulated by thyroid hormones, prolactin and sex hormones. J. Endocrinol. 2006, 190, 363–371. [Google Scholar] [CrossRef]
- Anguiano, B.; Montes de Oca, C.; Delgado-Gonzalez, E.; Aceves, C. Prostate gland as a target organ of thyroid hormones: Advances and controversies. Endocr. Connect. 2022, 11, e210581. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.R.; Chaker, L.; Ruiter, R.; Aerts, J.G.; Hofman, A.; Dehghan, A.; Franco, O.H.; Stricker, B.H.; Peeters, R.P. Thyroid Function and Cancer Risk: The Rotterdam Study. J. Clin. Endocrinol. Metab. 2016, 101, 5030–5036. [Google Scholar] [CrossRef]
- Mondul, A.M.; Weinstein, S.J.; Bosworth, T.; Remaley, A.T.; Virtamo, J.; Albanes, D. Circulating thyroxine, thyroid-stimulating hormone, and hypothyroid status and the risk of prostate cancer. PLoS ONE 2012, 7, e47730. [Google Scholar] [CrossRef]
- Voutsadakis, I.A. The TSH/Thyroid Hormones Axis and Breast Cancer. J. Clin. Med. 2022, 11, 687. [Google Scholar] [CrossRef]
- Hellevik, A.I.; Asvold, B.O.; Bjoro, T.; Romundstad, P.R.; Nilsen, T.I.; Vatten, L.J. Thyroid function and cancer risk: A prospective population study. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Moeller, L.C.; Fuhrer, D. Thyroid hormone, thyroid hormone receptors, and cancer: A clinical perspective. Endocr. Relat. Cancer 2013, 20, R19–R29. [Google Scholar] [CrossRef]
- Krashin, E.; Piekielko-Witkowska, A.; Ellis, M.; Ashur-Fabian, O. Thyroid Hormones and Cancer: A Comprehensive Review of Preclinical and Clinical Studies. Front. Endocrinol. 2019, 10, 59. [Google Scholar] [CrossRef]
- Liu, Y.C.; Yeh, C.T.; Lin, K.H. Molecular Functions of Thyroid Hormone Signaling in Regulation of Cancer Progression and Anti-Apoptosis. Int. J. Mol. Sci. 2019, 20, 4986. [Google Scholar] [CrossRef] [PubMed]
- Atkins, M.B.; Mier, J.W.; Parkinson, D.R.; Gould, J.A.; Berkman, E.M.; Kaplan, M.M. Hypothyroidism after treatment with interleukin-2 and lymphokine-activated killer cells. N. Engl. J. Med. 1988, 318, 1557–1563. [Google Scholar] [CrossRef]
- Hercbergs, A. Hypothyroidism and tumor regression. N. Engl. J. Med. 1988, 319, 1351–1352. [Google Scholar] [CrossRef]
- Miro, C.; Di Cicco, E.; Ambrosio, R.; Mancino, G.; Di Girolamo, D.; Cicatiello, A.G.; Sagliocchi, S.; Nappi, A.; De Stefano, M.A.; Luongo, C.; et al. Thyroid hormone induces progression and invasiveness of squamous cell carcinomas by promoting a ZEB-1/E-cadherin switch. Nat. Commun. 2019, 10, 5410. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Suzuki, N.; Mori, J.; Oshima, A.; Usami, S.; Hashizume, K. micro-Crystallin as an intracellular 3,5,3’-triiodothyronine holder in vivo. Mol. Endocrinol. 2007, 21, 885–894. [Google Scholar] [CrossRef]
- Aksoy, O.; Pencik, J.; Hartenbach, M.; Moazzami, A.A.; Schlederer, M.; Balber, T.; Varady, A.; Philippe, C.; Baltzer, P.A.; Mazumder, B.; et al. Thyroid and androgen receptor signaling are antagonized by mu-Crystallin in prostate cancer. Int. J. Cancer 2021, 148, 731–747. [Google Scholar] [CrossRef]
- Malinowska, K.; Cavarretta, I.T.; Susani, M.; Wrulich, O.A.; Uberall, F.; Kenner, L.; Culig, Z. Identification of mu-crystallin as an androgen-regulated gene in human prostate cancer. Prostate 2009, 69, 1109–1118. [Google Scholar] [CrossRef]
- Mousses, S.; Bubendorf, L.; Wagner, U.; Hostetter, G.; Kononen, J.; Cornelison, R.; Goldberger, N.; Elkahloun, A.G.; Willi, N.; Koivisto, P.; et al. Clinical validation of candidate genes associated with prostate cancer progression in the CWR22 model system using tissue microarrays. Cancer Res. 2002, 62, 1256–1260. [Google Scholar]
- Aksoy, O.; Hantusch, B.; Kenner, L. Emerging role of T3-binding protein mu-crystallin (CRYM) in health and disease. Trends Endocrinol. Metab. 2022, 33, 804–816. [Google Scholar] [CrossRef]
- Kim, W.G.; Cheng, S.Y. Thyroid hormone receptors and cancer. Biochim. Biophys. Acta 2013, 1830, 3928–3936. [Google Scholar] [CrossRef]
- Muscat, G.E.; Eriksson, N.A.; Byth, K.; Loi, S.; Graham, D.; Jindal, S.; Davis, M.J.; Clyne, C.; Funder, J.W.; Simpson, E.R.; et al. Research resource: Nuclear receptors as transcriptome: Discriminant and prognostic value in breast cancer. Mol. Endocrinol. 2013, 27, 350–365. [Google Scholar] [CrossRef]
- Gu, G.; Gelsomino, L.; Covington, K.R.; Beyer, A.R.; Wang, J.; Rechoum, Y.; Huffman, K.; Carstens, R.; Ando, S.; Fuqua, S.A. Targeting thyroid hormone receptor beta in triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 150, 535–545. [Google Scholar] [CrossRef]
- Park, J.W.; Zhao, L.; Willingham, M.; Cheng, S.Y. Oncogenic mutations of thyroid hormone receptor beta. Oncotarget 2015, 6, 8115–8131. [Google Scholar] [CrossRef]
- Rosen, M.D.; Privalsky, M.L. Thyroid hormone receptor mutations in cancer and resistance to thyroid hormone: Perspective and prognosis. J. Thyroid. Res. 2011, 2011, 361304. [Google Scholar] [CrossRef]
- Davidson, C.D.; Gillis, N.E.; Carr, F.E. Thyroid Hormone Receptor Beta as Tumor Suppressor: Untapped Potential in Treatment and Diagnostics in Solid Tumors. Cancers 2021, 13, 4254. [Google Scholar] [CrossRef]
- Martinez-Iglesias, O.; Garcia-Silva, S.; Tenbaum, S.P.; Regadera, J.; Larcher, F.; Paramio, J.M.; Vennstrom, B.; Aranda, A. Thyroid hormone receptor beta1 acts as a potent suppressor of tumor invasiveness and metastasis. Cancer Res. 2009, 69, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Horkko, T.T.; Tuppurainen, K.; George, S.M.; Jernvall, P.; Karttunen, T.J.; Makinen, M.J. Thyroid hormone receptor beta1 in normal colon and colorectal cancer-association with differentiation, polypoid growth type and K-ras mutations. Int. J. Cancer 2006, 118, 1653–1659. [Google Scholar] [CrossRef]
- Schnoell, J.; Kotowski, U.; Jank, B.J.; Stoiber, S.; Gurnhofer, E.; Schlederer, M.; Heiduschka, G.; Kenner, L.; Kadletz-Wanke, L. Prognostic Relevance of Thyroid-Hormone-Associated Proteins in Adenoid Cystic Carcinoma of the Head and Neck. J. Pers. Med. 2021, 11, 1352. [Google Scholar] [CrossRef]
- Shao, W.; Kuhn, C.; Mayr, D.; Ditsch, N.; Kailuweit, M.; Wolf, V.; Harbeck, N.; Mahner, S.; Jeschke, U.; Cavailles, V.; et al. Cytoplasmic and Nuclear Forms of Thyroid Hormone Receptor beta1 Are Inversely Associated with Survival in Primary Breast Cancer. Int. J. Mol. Sci. 2020, 21, 330. [Google Scholar] [CrossRef]
- Baumann, C.T.; Maruvada, P.; Hager, G.L.; Yen, P.M. Nuclear cytoplasmic shuttling by thyroid hormone receptors. multiple protein interactions are required for nuclear retention. J. Biol. Chem. 2001, 276, 11237–11245. [Google Scholar] [CrossRef]
- Shiota, M.; Fujimoto, N.; Kashiwagi, E.; Eto, M. The Role of Nuclear Receptors in Prostate Cancer. Cells 2019, 8, 602. [Google Scholar] [CrossRef]
- Hercbergs, A.H.; Ashur-Fabian, O.; Garfield, D. Thyroid hormones and cancer: Clinical studies of hypothyroidism in oncology. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 432–436. [Google Scholar] [CrossRef]
- Davis, P.J.; Tang, H.Y.; Hercbergs, A.; Lin, H.Y.; Keating, K.A.; Mousa, S.A. Bioactivity of Thyroid Hormone Analogs at Cancer Cells. Front. Endocrinol. 2018, 9, 739. [Google Scholar] [CrossRef]
- Hall, L.C.; Salazar, E.P.; Kane, S.R.; Liu, N. Effects of thyroid hormones on human breast cancer cell proliferation. J. Steroid Biochem. Mol. Biol. 2008, 109, 57–66. [Google Scholar] [CrossRef]
- Moriggi, G.; Verga Falzacappa, C.; Mangialardo, C.; Michienzi, S.; Stigliano, A.; Brunetti, E.; Toscano, V.; Misiti, S. Thyroid hormones (T3 and T4): Dual effect on human cancer cell proliferation. Anticancer. Res. 2011, 31, 89–96. [Google Scholar]
- Chen, C.Y.; Tsai, M.M.; Chi, H.C.; Lin, K.H. Biological significance of a thyroid hormone-regulated secretome. Biochim. Biophys. Acta 2013, 1834, 2271–2284. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Matak, D.; Szymanski, L.; Czarnecka, K.H.; Lewicki, S.; Zdanowski, R.; Brzezianska-Lasota, E.; Szczylik, C. Triiodothyronine regulates cell growth and survival in renal cell cancer. Int. J. Oncol. 2016, 49, 1666–1678. [Google Scholar] [CrossRef]
- Theodossiou, C.; Skrepnik, N.; Robert, E.G.; Prasad, C.; Axelrad, T.W.; Schapira, D.V.; Hunt, J.D. Propylthiouracil-induced hypothyroidism reduces xenograft tumor growth in athymic nude mice. Cancer 1999, 86, 1596–1601. [Google Scholar] [CrossRef]
- Martinez, M.B.; Ruan, M.; Fitzpatrick, L.A. Altered response to thyroid hormones by prostate and breast cancer cells. Cancer Chemother. Pharmacol. 2000, 45, 93–102. [Google Scholar] [CrossRef]
- Zhu, W.; Young, C.Y. Androgen-Dependent transcriptional regulation of the prostate-specific antigen gene by thyroid hormone 3,5,3’-L-triiodothyronine. J. Androl. 2001, 22, 136–141. [Google Scholar] [CrossRef]
- Esquenet, M.; Swinnen, J.V.; Heyns, W.; Verhoeven, G. Triiodothyronine modulates growth, secretory function and androgen receptor concentration in the prostatic carcinoma cell line LNCaP. Mol. Cell Endocrinol. 1995, 109, 105–111. [Google Scholar] [CrossRef]
- Zhang, S.; Hsieh, M.L.; Zhu, W.; Klee, G.G.; Tindall, D.J.; Young, C.Y. Interactive effects of triiodothyronine and androgens on prostate cell growth and gene expression. Endocrinology 1999, 140, 1665–1671. [Google Scholar] [CrossRef]
- Kotolloshi, R.; Mirzakhani, K.; Ahlburg, J.; Kraft, F.; Pungsrinont, T.; Baniahmad, A. Thyroid hormone induces cellular senescence in prostate cancer cells through induction of DEC1. J. Steroid Biochem. Mol. Biol. 2020, 201, 105689. [Google Scholar] [CrossRef]
- Chen, R.N.; Huang, Y.H.; Lin, Y.C.; Yeh, C.T.; Liang, Y.; Chen, S.L.; Lin, K.H. Thyroid hormone promotes cell invasion through activation of furin expression in human hepatoma cell lines. Endocrinology 2008, 149, 3817–3831. [Google Scholar] [CrossRef]
- Tai, P.J.; Huang, Y.H.; Shih, C.H.; Chen, R.N.; Chen, C.D.; Chen, W.J.; Wang, C.S.; Lin, K.H. Direct regulation of androgen receptor-associated protein 70 by thyroid hormone and its receptors. Endocrinology 2007, 148, 3485–3495. [Google Scholar] [CrossRef]
- Wu, S.M.; Huang, Y.H.; Yeh, C.T.; Tsai, M.M.; Liao, C.H.; Cheng, W.L.; Chen, W.J.; Lin, K.H. Cathepsin H regulated by the thyroid hormone receptors associate with tumor invasion in human hepatoma cells. Oncogene 2011, 30, 2057–2069. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.H.; Privalsky, M.L. Isoform-specific transcriptional activity of overlapping target genes that respond to thyroid hormone receptors alpha1 and beta1. Mol. Endocrinol. 2009, 23, 1758–1775. [Google Scholar] [CrossRef]
- Dong, H.; Yauk, C.L.; Rowan-Carroll, A.; You, S.H.; Zoeller, R.T.; Lambert, I.; Wade, M.G. Identification of thyroid hormone receptor binding sites and target genes using ChIP-on-chip in developing mouse cerebellum. PLoS ONE 2009, 4, e4610. [Google Scholar] [CrossRef]
- Zambrano, A.; Garcia-Carpizo, V.; Gallardo, M.E.; Villamuera, R.; Gomez-Ferreria, M.A.; Pascual, A.; Buisine, N.; Sachs, L.M.; Garesse, R.; Aranda, A. The thyroid hormone receptor beta induces DNA damage and premature senescence. J. Cell Biol. 2014, 204, 129–146. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, D.; Ng, C.F.; Teoh, J.Y.; Yu, S.; Wang, Y.; Chan, F.L. Nuclear receptor profiling in prostatospheroids and castration-resistant prostate cancer. Endocr. Relat. Cancer 2018, 25, 35–50. [Google Scholar] [CrossRef]
- Delgado-Gonzalez, E.; Sanchez-Tusie, A.A.; Morales, G.; Aceves, C.; Anguiano, B. Triiodothyronine Attenuates Prostate Cancer Progression Mediated by beta-Adrenergic Stimulation. Mol. Med. 2016, 22, 1–11. [Google Scholar] [CrossRef]
- Davis, P.J.; Lin, H.Y.; Mousa, S.A.; Luidens, M.K.; Hercbergs, A.A.; Wehling, M.; Davis, F.B. Overlapping nongenomic and genomic actions of thyroid hormone and steroids. Steroids 2011, 76, 829–833. [Google Scholar] [CrossRef]
- Hoermann, R.; Cheung, A.S.; Milne, M.; Grossmann, M. Hypothalamic-Pituitary-Thyroid Axis Set Point Alterations Are Associated With Body Composition in Androgen-Deprived Men. J. Endocr. Soc. 2017, 1, 874–885. [Google Scholar] [CrossRef]
- Miro, C.; Di Giovanni, A.; Murolo, M.; Cicatiello, A.G.; Nappi, A.; Sagliocchi, S.; Di Cicco, E.; Morra, F.; Celetti, A.; Pacifico, F.; et al. Thyroid hormone and androgen signals mutually interplay and enhance inflammation and tumorigenic activation of tumor microenvironment in prostate cancer. Cancer Lett. 2022, 532, 215581. [Google Scholar] [CrossRef]
- Flood, D.E.; Fernandino, J.I.; Langlois, V.S. Thyroid hormones in male reproductive development: Evidence for direct crosstalk between the androgen and thyroid hormone axes. Gen. Comp. Endocrinol. 2013, 192, 2–14. [Google Scholar] [CrossRef]
- Campbell, D.E.K.; Langlois, V.S. Thyroid hormones and androgens differentially regulate gene expression in testes and ovaries of sexually mature Silurana tropicalis. Gen. Comp. Endocrinol. 2018, 267, 172–182. [Google Scholar] [CrossRef]
- Torabinejad, S.; Miro, C.; Barone, B.; Imbimbo, C.; Crocetto, F.; Dentice, M. The androgen-thyroid hormone crosstalk in prostate cancer and the clinical implications. Eur. Thyroid. J. 2023, 12, e220228. [Google Scholar] [CrossRef]
- Cruz, M.A.D.; Lund, D.; Szekeres, F.; Karlsson, S.; Faresjo, M.; Larsson, D. Cis-regulatory elements in conserved non-coding sequences of nuclear receptor genes indicate for crosstalk between endocrine systems. Open Med. 2021, 16, 640–650. [Google Scholar] [CrossRef]
- Rosen, E.D.; Beninghof, E.G.; Koenig, R.J. Dimerization interfaces of thyroid hormone, retinoic acid, vitamin D, and retinoid X receptors. J. Biol. Chem. 1993, 268, 11534–11541. [Google Scholar] [CrossRef]
- Yen, P.M.; Sugawara, A.; Chin, W.W. Triiodothyronine (T3) differentially affects T3-receptor/retinoic acid receptor and T3-receptor/retinoid X receptor heterodimer binding to DNA. J. Biol. Chem. 1992, 267, 23248–23252. [Google Scholar] [CrossRef]
- Schrader, M.; Carlberg, C. Thyroid hormone and retinoic acid receptors form heterodimers with retinoid X receptors on direct repeats, palindromes, and inverted palindromes. DNA Cell Biol. 1994, 13, 333–341. [Google Scholar] [CrossRef]
- Lee, S.; Privalsky, M.L. Heterodimers of retinoic acid receptors and thyroid hormone receptors display unique combinatorial regulatory properties. Mol. Endocrinol. 2005, 19, 863–878. [Google Scholar] [CrossRef]
- Schilthuis, J.G.; Gann, A.A.; Brockes, J.P. Chimeric retinoic acid/thyroid hormone receptors implicate RAR-alpha 1 as mediating growth inhibition by retinoic acid. EMBO J. 1993, 12, 3459–3466. [Google Scholar] [CrossRef]
- Ludwig, M.G.; Basset, P.; Anglard, P. Multiple regulatory elements in the murine stromelysin-3 promoter. Evidence for direct control by CCAAT/enhancer-binding protein beta and thyroid and retinoid receptors. J. Biol. Chem. 2000, 275, 39981–39990. [Google Scholar] [CrossRef]
- Davis, K.D.; Lazar, M.A. Selective antagonism of thyroid hormone action by retinoic acid. J. Biol. Chem. 1992, 267, 3185–3189. [Google Scholar] [CrossRef]
- Gil-Ibanez, P.; Bernal, J.; Morte, B. Thyroid hormone regulation of gene expression in primary cerebrocortical cells: Role of thyroid hormone receptor subtypes and interactions with retinoic acid and glucocorticoids. PLoS ONE 2014, 9, e91692. [Google Scholar] [CrossRef]
- Khanim, F.L.; Gommersall, L.M.; Wood, V.H.; Smith, K.L.; Montalvo, L.; O’Neill, L.P.; Xu, Y.; Peehl, D.M.; Stewart, P.M.; Turner, B.M.; et al. Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene 2004, 23, 6712–6725. [Google Scholar] [CrossRef]
- Girault, I.; Lerebours, F.; Amarir, S.; Tozlu, S.; Tubiana-Hulin, M.; Lidereau, R.; Bieche, I. Expression analysis of estrogen receptor alpha coregulators in breast carcinoma: Evidence that NCOR1 expression is predictive of the response to tamoxifen. Clin. Cancer Res. 2003, 9, 1259–1266. [Google Scholar]
- Long, M.D.; Jacobi, J.J.; Singh, P.K.; Llimos, G.; Wani, S.A.; Rowsam, A.M.; Rosario, S.R.; Hoogstraat, M.; Linder, S.; Kirk, J.; et al. Reduced NCOR2 expression accelerates androgen deprivation therapy failure in prostate cancer. Cell Rep. 2021, 37, 110109. [Google Scholar] [CrossRef]
- Schrader, M.; Muller, K.M.; Carlberg, C. Specificity and flexibility of vitamin D signaling. Modulation of the activation of natural vitamin D response elements by thyroid hormone. J. Biol. Chem. 1994, 269, 5501–5504. [Google Scholar] [CrossRef]
- Schrader, M.; Muller, K.M.; Nayeri, S.; Kahlen, J.P.; Carlberg, C. Vitamin D3-thyroid hormone receptor heterodimer polarity directs ligand sensitivity of transactivation. Nature 1994, 370, 382–386. [Google Scholar] [CrossRef]
- Garcia-Villalba, P.; Jimenez-Lara, A.M.; Aranda, A. Vitamin D interferes with transactivation of the growth hormone gene by thyroid hormone and retinoic acid. Mol. Cell Biol. 1996, 16, 318–327. [Google Scholar] [CrossRef]
- Raval-Pandya, M.; Freedman, L.P.; Li, H.; Christakos, S. Thyroid hormone receptor does not heterodimerize with the vitamin D receptor but represses vitamin D receptor-mediated transactivation. Mol. Endocrinol. 1998, 12, 1367–1379. [Google Scholar] [CrossRef]
- Thompson, P.D.; Hsieh, J.C.; Whitfield, G.K.; Haussler, C.A.; Jurutka, P.W.; Galligan, M.A.; Tillman, J.B.; Spindler, S.R.; Haussler, M.R. Vitamin D receptor displays DNA binding and transactivation as a heterodimer with the retinoid X receptor, but not with the thyroid hormone receptor. J. Cell Biochem. 1999, 75, 462–480. [Google Scholar] [CrossRef]
- Schneider, L.; El-Yazidi, C.; Dace, A.; Maraninchi, M.; Planells, R.; Margotat, A.; Torresani, J. Expression of the 1,25-(OH)2 vitamin D3 receptor gene during the differentiation of mouse Ob17 preadipocytes and cross talk with the thyroid hormone receptor signalling pathway. J. Mol. Endocrinol. 2005, 34, 221–235. [Google Scholar] [CrossRef]
- Gloesmann, M.; Ertl, R.; Himmel, T.; Kummer, S.; Reichart, U. Thyroid Hormone Regulates mRNA Expression of the Vitamin D Receptor in Mouse Photoreceptors. Investig. Ophthalmol. Vis. Sci. 2016, 57, 564. [Google Scholar]
- Barrera-Hernandez, G.; Zhan, Q.; Wong, R.; Cheng, S.Y. Thyroid hormone receptor is a negative regulator in p53-mediated signaling pathways. DNA Cell Biol. 1998, 17, 743–750. [Google Scholar] [CrossRef]
- Singh, B.K.; Sinha, R.A.; Tripathi, M.; Mendoza, A.; Ohba, K.; Sy, J.A.C.; Xie, S.Y.; Zhou, J.; Ho, J.P.; Chang, C.Y.; et al. Thyroid hormone receptor and ERRalpha coordinately regulate mitochondrial fission, mitophagy, biogenesis, and function. Sci. Signal 2018, 11, eaam5855. [Google Scholar] [CrossRef] [PubMed]
- Araki, O.; Ying, H.; Furuya, F.; Zhu, X.; Cheng, S.Y. Thyroid hormone receptor beta mutants: Dominant negative regulators of peroxisome proliferator-activated receptor gamma action. Proc. Natl. Acad. Sci. USA 2005, 102, 16251–16256. [Google Scholar] [CrossRef]
- Guigon, C.J.; Cheng, S.Y. Novel oncogenic actions of TRbeta mutants in tumorigenesis. IUBMB Life 2009, 61, 528–536. [Google Scholar] [CrossRef]
- Hashimoto, K.; Mori, M. Crosstalk of thyroid hormone receptor and liver X receptor in lipid metabolism and beyond [Review]. Endocr. J. 2011, 58, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Guigon, C.J.; Kim, D.W.; Zhu, X.; Zhao, L.; Cheng, S.Y. Tumor suppressor action of liganded thyroid hormone receptor beta by direct repression of beta-catenin gene expression. Endocrinology 2010, 151, 5528–5536. [Google Scholar] [CrossRef] [PubMed]
- Skah, S.; Uchuya-Castillo, J.; Sirakov, M.; Plateroti, M. The thyroid hormone nuclear receptors and the Wnt/beta-catenin pathway: An intriguing liaison. Dev. Biol. 2017, 422, 71–82. [Google Scholar] [CrossRef]
- Zhang, X.K.; Wills, K.N.; Husmann, M.; Hermann, T.; Pfahl, M. Novel pathway for thyroid hormone receptor action through interaction with jun and fos oncogene activities. Mol. Cell Biol. 1991, 11, 6016–6025. [Google Scholar] [CrossRef]
- Favre-Young, H.; Dif, F.; Roussille, F.; Demeneix, B.A.; Kelly, P.A.; Edery, M.; de Luze, A. Cross-talk between signal transducer and activator of transcription (Stat5) and thyroid hormone receptor-beta 1 (TRbeta1) signaling pathways. Mol. Endocrinol. 2000, 14, 1411–1424. [Google Scholar] [CrossRef]
- Guigon, C.J.; Kim, D.W.; Willingham, M.C.; Cheng, S.Y. Mutation of thyroid hormone receptor-beta in mice predisposes to the development of mammary tumors. Oncogene 2011, 30, 3381–3390. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.P.; Marron Fernandez de Velasco, E.; Mizuno, F.; Scappini, E.L.; Gloss, B.; Erxleben, C.; Williams, J.G.; Stapleton, H.M.; Gentile, S.; Armstrong, D.L. A rapid cytoplasmic mechanism for PI3 kinase regulation by the nuclear thyroid hormone receptor, TRbeta, and genetic evidence for its role in the maturation of mouse hippocampal synapses in vivo. Endocrinology 2014, 155, 3713–3724. [Google Scholar] [CrossRef] [PubMed]
- Teixido, N.; Soler, M.; Rivera, N.; Bernues, J.; Meseguer, A. CCAAT/enhancer binding protein-mediated role of thyroid hormone in the developmental expression of the kidney androgen-regulated protein gene in proximal convoluted tubules. Mol. Endocrinol. 2006, 20, 389–404. [Google Scholar] [CrossRef]
- Selva, D.M.; Hammond, G.L. Thyroid hormones act indirectly to increase sex hormone-binding globulin production by liver via hepatocyte nuclear factor-4alpha. J. Mol. Endocrinol. 2009, 43, 19–27. [Google Scholar] [CrossRef]
- Gonzalez-Sancho, J.M.; Garcia, V.; Bonilla, F.; Munoz, A. Thyroid hormone receptors/THR genes in human cancer. Cancer Lett. 2003, 192, 121–132. [Google Scholar] [CrossRef]
- Ichijo, S.; Furuya, F.; Shimura, H.; Hayashi, Y.; Takahashi, K.; Ohta, K.; Kobayashi, T.; Kitamura, K. Activation of the RhoB signaling pathway by thyroid hormone receptor beta in thyroid cancer cells. PLoS ONE 2014, 9, e116252. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. The nuclear receptor superfamily: A personal retrospect on the first two decades. Mol. Endocrinol. 2005, 19, 1418–1428. [Google Scholar] [CrossRef]
- Giguere, V.; Shago, M.; Zirngibl, R.; Tate, P.; Rossant, J.; Varmuza, S. Identification of a novel isoform of the retinoic acid receptor gamma expressed in the mouse embryo. Mol. Cell Biol. 1990, 10, 2335–2340. [Google Scholar] [CrossRef]
- Kastner, P.; Krust, A.; Mendelsohn, C.; Garnier, J.M.; Zelent, A.; Leroy, P.; Staub, A.; Chambon, P. Murine isoforms of retinoic acid receptor gamma with specific patterns of expression. Proc. Natl. Acad. Sci. USA 1990, 87, 2700–2704. [Google Scholar] [CrossRef]
- Leroy, P.; Krust, A.; Zelent, A.; Mendelsohn, C.; Garnier, J.M.; Kastner, P.; Dierich, A.; Chambon, P. Multiple isoforms of the mouse retinoic acid receptor alpha are generated by alternative splicing and differential induction by retinoic acid. EMBO J. 1991, 10, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Zelent, A.; Mendelsohn, C.; Kastner, P.; Krust, A.; Garnier, J.M.; Ruffenach, F.; Leroy, P.; Chambon, P. Differentially expressed isoforms of the mouse retinoic acid receptor beta generated by usage of two promoters and alternative splicing. EMBO J. 1991, 10, 71–81. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Hoffmann, B.; Pfahl, M. Genomic organization of the retinoic acid receptor gamma gene. Nucleic Acids Res. 1991, 19, 573–578. [Google Scholar] [CrossRef]
- Giguere, V.; Ong, E.S.; Segui, P.; Evans, R.M. Identification of a receptor for the morphogen retinoic acid. Nature 1987, 330, 624–629. [Google Scholar] [CrossRef]
- Petkovich, M.; Brand, N.J.; Krust, A.; Chambon, P. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature 1987, 330, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Chatagnon, A.; Veber, P.; Morin, V.; Bedo, J.; Triqueneaux, G.; Semon, M.; Laudet, V.; d’Alche-Buc, F.; Benoit, G. RAR/RXR binding dynamics distinguish pluripotency from differentiation associated cis-regulatory elements. Nucleic Acids Res. 2015, 43, 4833–4854. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes. Dev. 2000, 14, 121–141. [Google Scholar] [CrossRef]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of co-repressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef]
- Kastner, P.; Mark, M.; Ghyselinck, N.; Krezel, W.; Dupe, V.; Grondona, J.M.; Chambon, P. Genetic evidence that the retinoid signal is transduced by heterodimeric RXR/RAR functional units during mouse development. Development 1997, 124, 313–326. [Google Scholar] [CrossRef]
- Lohnes, D.; Mark, M.; Mendelsohn, C.; Dolle, P.; Dierich, A.; Gorry, P.; Gansmuller, A.; Chambon, P. Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 1994, 120, 2723–2748. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.; Lohnes, D.; Decimo, D.; Lufkin, T.; LeMeur, M.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar] [CrossRef] [PubMed]
- Dolle, P.; Ruberte, E.; Leroy, P.; Morriss-Kay, G.; Chambon, P. Retinoic acid receptors and cellular retinoid binding proteins. I. A systematic study of their differential pattern of transcription during mouse organogenesis. Development 1990, 110, 1133–1151. [Google Scholar] [CrossRef]
- Dolle, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal 2009, 7, e006. [Google Scholar] [CrossRef]
- Crowe, D.L.; Hu, L.; Gudas, L.J.; Rheinwald, J.G. Variable expression of retinoic acid receptor (RAR beta) mRNA in human oral and epidermal keratinocytes; relation to keratin 19 expression and keratinization potential. Differentiation 1991, 48, 199–208. [Google Scholar] [CrossRef]
- Hale, L.A.; Tallafuss, A.; Yan, Y.L.; Dudley, L.; Eisen, J.S.; Postlethwait, J.H. Characterization of the retinoic acid receptor genes raraa, rarab and rarg during zebrafish development. Gene Expr. Patterns 2006, 6, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Ruberte, E.; Dolle, P.; Krust, A.; Zelent, A.; Morriss-Kay, G.; Chambon, P. Specific spatial and temporal distribution of retinoic acid receptor gamma transcripts during mouse embryogenesis. Development 1990, 108, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Heyworth, C.M.; Glasow, A.; Huang, Q.H.; Petrie, K.; Lanotte, M.; Benoit, G.; Gallagher, R.; Waxman, S.; Enver, T.; et al. Lineage restriction of the RARalpha gene expression in myeloid differentiation. Blood 2001, 98, 2563–2567. [Google Scholar] [CrossRef]
- Purton, L.E.; Dworkin, S.; Olsen, G.H.; Walkley, C.R.; Fabb, S.A.; Collins, S.J.; Chambon, P. RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med. 2006, 203, 1283–1293. [Google Scholar] [CrossRef]
- Breitman, T.R.; Selonick, S.E.; Collins, S.J. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc. Natl. Acad. Sci. USA 1980, 77, 2936–2940. [Google Scholar] [CrossRef]
- Brown, G.; Marchwicka, A.; Cunningham, A.; Toellner, K.M.; Marcinkowska, E. Antagonizing Retinoic Acid Receptors Increases Myeloid Cell Production by Cultured Human Hematopoietic Stem Cells. Arch. Immunol. Ther. Exp. 2017, 65, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Gratas, C.; Menot, M.L.; Dresch, C.; Chomienne, C. Retinoid acid supports granulocytic but not erythroid differentiation of myeloid progenitors in normal bone marrow cells. Leukemia 1993, 7, 1156–1162. [Google Scholar]
- Wang, Z.Y.; Chen, Z. Acute promyelocytic leukemia: From highly fatal to highly curable. Blood 2008, 111, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Kastner, P.; Chan, S. Function of RARalpha during the maturation of neutrophils. Oncogene 2001, 20, 7178–7185. [Google Scholar] [CrossRef] [PubMed]
- Wai, H.A.; Kawakami, K.; Wada, H.; Muller, F.; Vernallis, A.B.; Brown, G.; Johnson, W.E. The development and growth of tissues derived from cranial neural crest and primitive mesoderm is dependent on the ligation status of retinoic acid receptor gamma: Evidence that retinoic acid receptor gamma functions to maintain stem/progenitor cells in the absence of retinoic acid. Stem Cells Dev. 2015, 24, 507–519. [Google Scholar] [CrossRef]
- Kashyap, V.; Laursen, K.B.; Brenet, F.; Viale, A.J.; Scandura, J.M.; Gudas, L.J. RARgamma is essential for retinoic acid induced chromatin remodeling and transcriptional activation in embryonic stem cells. J. Cell Sci. 2013, 126 Pt 4, 999–1008. [Google Scholar] [CrossRef]
- Kashyap, V.; Gudas, L.J.; Brenet, F.; Funk, P.; Viale, A.; Scandura, J.M. Epigenomic reorganization of the clustered Hox genes in embryonic stem cells induced by retinoic acid. J. Biol. Chem. 2011, 286, 3250–3260. [Google Scholar] [CrossRef]
- Handberg-Thorsager, M.; Gutierrez-Mazariegos, J.; Arold, S.T.; Kumar Nadendla, E.; Bertucci, P.Y.; Germain, P.; Tomancak, P.; Pierzchalski, K.; Jones, J.W.; Albalat, R.; et al. The ancestral retinoic acid receptor was a low-affinity sensor triggering neuronal differentiation. Sci. Adv. 2018, 4, eaao1261. [Google Scholar] [CrossRef]
- Cruz, F.D.; Matushansky, I. Solid tumor differentiation therapy-is it possible? Oncotarget 2012, 3, 559–567. [Google Scholar] [CrossRef]
- Richter, F.; Joyce, A.; Fromowitz, F.; Wang, S.; Watson, J.; Watson, R.; Irwin, R.J., Jr.; Huang, H.F. Immunohistochemical localization of the retinoic Acid receptors in human prostate. J. Androl. 2002, 23, 830–838. [Google Scholar] [CrossRef]
- Hammond, L.A.; Van Krinks, C.H.; Durham, J.; Tomkins, S.E.; Burnett, R.D.; Jones, E.L.; Chandraratna, R.A.; Brown, G. Antagonists of retinoic acid receptors (RARs) are potent growth inhibitors of prostate carcinoma cells. Br. J. Cancer 2001, 85, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Ossowski, L.; Ferrari, A.C. Activation of Rb and decline in androgen receptor protein precede retinoic acid-induced apoptosis in androgen-dependent LNCaP cells and their androgen-independent derivative. J. Cell Physiol. 1999, 179, 336–346. [Google Scholar] [CrossRef]
- Gao, T.; He, B.; Pan, Y.; Li, R.; Xu, Y.; Chen, L.; Nie, Z.; Gu, L.; Wang, S. The association of retinoic acid receptor beta2(RARbeta2) methylation status and prostate cancer risk: A systematic review and meta-analysis. PLoS ONE 2013, 8, e62950. [Google Scholar] [CrossRef]
- Dou, M.; Zhou, X.; Fan, Z.; Ding, X.; Li, L.; Wang, S.; Xue, W.; Wang, H.; Suo, Z.; Deng, X. Clinical Significance of Retinoic Acid Receptor Beta Promoter Methylation in Prostate Cancer: A Meta-Analysis. Cell Physiol. Biochem. 2018, 45, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Park, S.; Uskokovic, M.R.; Dawson, M.I.; Koeffler, H.P. Expression of retinoic acid receptor-beta sensitizes prostate cancer cells to growth inhibition mediated by combinations of retinoids and a 19-nor hexafluoride vitamin D3 analog. Endocrinology 1998, 139, 1972–1980. [Google Scholar] [CrossRef]
- Pasquali, D.; Thaller, C.; Eichele, G. Abnormal level of retinoic acid in prostate cancer tissues. J. Clin. Endocrinol. Metab. 1996, 81, 2186–2191. [Google Scholar] [CrossRef]
- Idres, N.; Marill, J.; Flexor, M.A.; Chabot, G.G. Activation of retinoic acid receptor-dependent transcription by all-trans-retinoic acid metabolites and isomers. J. Biol. Chem. 2002, 277, 31491–31498. [Google Scholar] [CrossRef]
- Petrie, K.; Urban-Wojciuk, Z.; Sbirkov, Y.; Graham, A.; Hamann, A.; Brown, G. Retinoic acid receptor gamma is a therapeutically targetable driver of growth and survival in prostate cancer. Cancer Rep. 2020, 3, e1284. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.L.; Luo, Q.; Rui, G.; Zhang, W.; Zhang, Q.Y.; Chen, Q.X.; Shen, D.Y. Oncogenic activity of retinoic acid receptor gamma is exhibited through activation of the Akt/NF-kappaB and Wnt/beta-catenin pathways in cholangiocarcinoma. Mol. Cell Biol. 2013, 33, 3416–3425. [Google Scholar] [CrossRef]
- Yamakawa, K.; Koyanagi-Aoi, M.; Machinaga, A.; Kakiuchi, N.; Hirano, T.; Kodama, Y.; Aoi, T. Blockage of retinoic acid signaling via RARgamma suppressed the proliferation of pancreatic cancer cells by arresting the cell cycle progression of the G1-S phase. Cancer Cell Int. 2023, 23, 94. [Google Scholar] [CrossRef]
- Keedwell, R.G.; Zhao, Y.; Hammond, L.A.; Wen, K.; Qin, S.; Atangan, L.I.; Shurland, D.L.; Wallace, D.M.; Bird, R.; Reitmair, A.; et al. An antagonist of retinoic acid receptors more effectively inhibits growth of human prostate cancer cells than normal prostate epithelium. Br. J. Cancer 2004, 91, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Colucci, M.; Zumerle, S.; Bressan, S.; Gianfanti, F.; Troiani, M.; Valdata, A.; D’Ambrosio, M.; Pasquini, E.; Varesi, A.; Cogo, F.; et al. Retinoic acid receptor activation reprograms senescence response and enhances anti-tumor activity of natural killer cells. Cancer Cell 2024, 42, 646–661.e9. [Google Scholar] [CrossRef]
- Brown, G. Deregulation of All-Trans Retinoic Acid Signaling and Development in Cancer. Int. J. Mol. Sci. 2023, 24, 89. [Google Scholar] [CrossRef]
- Barrett, A.; Shi, J.Y.; Howell, L.; Sbirkov, Y.; Brown, G.; Zelent, A.; Petrie, K. Expression of retinoic acid receptor gamma is regulated by miR-30a. Klin. Pädiatrie 2023, 235, 2. [Google Scholar]
- Zeng, W.; Zhang, C.; Cheng, H.; Wu, Y.L.; Liu, J.; Chen, Z.; Huang, J.G.; Ericksen, R.E.; Chen, L.; Zhang, H.; et al. Targeting to the non-genomic activity of retinoic acid receptor-gamma by acacetin in hepatocellular carcinoma. Sci. Rep. 2017, 7, 348. [Google Scholar] [CrossRef] [PubMed]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor gamma in the mouse. Cell 1993, 73, 643–658. [Google Scholar] [CrossRef]
- Chung, S.S.; Wang, X.; Roberts, S.S.; Griffey, S.M.; Reczek, P.R.; Wolgemuth, D.J. Oral administration of a retinoic Acid receptor antagonist reversibly inhibits spermatogenesis in mice. Endocrinology 2011, 152, 2492–2502. [Google Scholar] [CrossRef]
- Schulze, G.E.; Clay, R.J.; Mezza, L.E.; Bregman, C.L.; Buroker, R.A.; Frantz, J.D. BMS-189453, a novel retinoid receptor antagonist, is a potent testicular toxin. Toxicol. Sci. 2001, 59, 297–308. [Google Scholar] [CrossRef]
- Long, M.D.; Singh, P.K.; Russell, J.R.; Llimos, G.; Rosario, S.; Rizvi, A.; van den Berg, P.R.; Kirk, J.; Sucheston-Campbell, L.E.; Smiraglia, D.J.; et al. The miR-96 and RARgamma signaling axis governs androgen signaling and prostate cancer progression. Oncogene 2019, 38, 421–444. [Google Scholar] [CrossRef]
- Nicholson, R.C.; Mader, S.; Nagpal, S.; Leid, M.; Rochette-Egly, C.; Chambon, P. Negative regulation of the rat stromelysin gene promoter by retinoic acid is mediated by an AP1 binding site. EMBO J. 1990, 9, 4443–4454. [Google Scholar] [CrossRef]
- Yasuhara, R.; Yuasa, T.; Williams, J.A.; Byers, S.W.; Shah, S.; Pacifici, M.; Iwamoto, M.; Enomoto-Iwamoto, M. Wnt/beta-catenin and retinoic acid receptor signaling pathways interact to regulate chondrocyte function and matrix turnover. J. Biol. Chem. 2010, 285, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Konsavage, W.M., Jr.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/beta-catenin signaling regulates Yes-associated protein (YAP) gene expression in colorectal carcinoma cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef] [PubMed]
- Bauzone, M.; Souidi, M.; Dessein, A.F.; Wisztorski, M.; Vincent, A.; Gimeno, J.P.; Monte, D.; Van Seuningen, I.; Gespach, C.; Huet, G. Cross-talk between YAP and RAR-RXR Drives Expression of Stemness Genes to Promote 5-FU Resistance and Self-Renewal in Colorectal Cancer Cells. Mol. Cancer Res. 2021, 19, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Ou, C.; Sun, Z.; Li, S.; Li, G.; Li, X.; Ma, J. Dual roles of yes-associated protein (YAP) in colorectal cancer. Oncotarget 2017, 8, 75727–75741. [Google Scholar] [CrossRef]
- Hindley, C.J.; Condurat, A.L.; Menon, V.; Thomas, R.; Azmitia, L.M.; Davis, J.A.; Pruszak, J. The Hippo pathway member YAP enhances human neural crest cell fate and migration. Sci. Rep. 2016, 6, 23208. [Google Scholar] [CrossRef]
- Marchwicka, A.; Cebrat, M.; Laszkiewicz, A.; Sniezewski, L.; Brown, G.; Marcinkowska, E. Regulation of vitamin D receptor expression by retinoic acid receptor alpha in acute myeloid leukemia cells. J. Steroid Biochem. Mol. Biol. 2016, 159, 121–130. [Google Scholar] [CrossRef]
- Schrader, M.; Muller, K.M.; Becker-Andre, M.; Carlberg, C. Response element selectivity for heterodimerization of vitamin D receptors with retinoic acid and retinoid X receptors. J. Mol. Endocrinol. 1994, 12, 327–339. [Google Scholar] [CrossRef]
- Holick, M.F.; Uskokovic, M.; Henley, J.W.; MacLaughlin, J.; Holick, S.A.; Potts, J.T., Jr. The photoproduction of 1 alpha,25-dihydroxyvitamin D3 in skin: An approach to the therapy of vitamin-D-resistant syndromes. N. Engl. J. Med. 1980, 303, 349–354. [Google Scholar] [CrossRef]
- Baker, A.R.; McDonnell, D.P.; Hughes, M.; Crisp, T.M.; Mangelsdorf, D.J.; Haussler, M.R.; Pike, J.W.; Shine, J.; O’Malley, B.W. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 3294–3298. [Google Scholar] [CrossRef]
- Crofts, L.A.; Hancock, M.S.; Morrison, N.A.; Eisman, J.A. Multiple promoters direct the tissue-specific expression of novel N-terminal variant human vitamin D receptor gene transcripts. Proc. Natl. Acad. Sci. USA 1998, 95, 10529–10534. [Google Scholar] [CrossRef]
- Zella, L.A.; Meyer, M.B.; Nerenz, R.D.; Lee, S.M.; Martowicz, M.L.; Pike, J.W. Multifunctional enhancers regulate mouse and human vitamin D receptor gene transcription. Mol. Endocrinol. 2010, 24, 128–147. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Kesterson, R.A.; Yamamoto, H.; Taketani, Y.; Nishiwaki, E.; Tatsumi, S.; Inoue, Y.; Morita, K.; Takeda, E.; Pike, J.W. Structural organization of the human vitamin D receptor chromosomal gene and its promoter. Mol. Endocrinol. 1997, 11, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Sunn, K.L.; Cock, T.A.; Crofts, L.A.; Eisman, J.A.; Gardiner, E.M. Novel N-terminal variant of human VDR. Mol. Endocrinol. 2001, 15, 1599–1609. [Google Scholar] [CrossRef]
- Fleet, J.C. The role of vitamin D in the endocrinology controlling calcium homeostasis. Mol. Cell Endocrinol. 2017, 453, 36–45. [Google Scholar] [CrossRef]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar] [CrossRef]
- Carlberg, C. Vitamin D and Its Target Genes. Nutrients 2022, 14, 1354. [Google Scholar] [CrossRef]
- Fleet, J.C.; DeSmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar] [CrossRef]
- Cheskis, B.J.; Freedman, L.P.; Nagpal, S. Vitamin D receptor ligands for osteoporosis. Curr. Opin. Investig. Drugs 2006, 7, 906–911. [Google Scholar]
- Fendler, A.; Stephan, C.; Ralla, B.; Jung, K. Discordant Health Implications and Molecular Mechanisms of Vitamin D in Clinical and Preclinical Studies of Prostate Cancer: A Critical Appraisal of the Literature Data. Int. J. Mol. Sci. 2024, 25, 5286. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Liu, M.D.; Yao, K.; Xu, S.; Yu, D.X.; Xie, D.D.; Xu, D.X. Vitamin D deficiency aggravates growth and metastasis of prostate cancer through promoting EMT in two beta-catenin-related mechanisms. J. Nutr. Biochem. 2023, 111, 109177. [Google Scholar] [CrossRef] [PubMed]
- Muindi, J.R.; Yu, W.D.; Ma, Y.; Engler, K.L.; Kong, R.X.; Trump, D.L.; Johnson, C.S. CYP24A1 inhibition enhances the antitumor activity of calcitriol. Endocrinology 2010, 151, 4301–4312. [Google Scholar] [CrossRef] [PubMed]
- Blutt, S.E.; McDonnell, T.J.; Polek, T.C.; Weigel, N.L. Calcitriol-induced apoptosis in LNCaP cells is blocked by overexpression of Bcl-2. Endocrinology 2000, 141, 10–17. [Google Scholar] [CrossRef]
- Krishnan, A.V.; Shinghal, R.; Raghavachari, N.; Brooks, J.D.; Peehl, D.M.; Feldman, D. Analysis of vitamin D-regulated gene expression in LNCaP human prostate cancer cells using cDNA microarrays. Prostate 2004, 59, 243–251. [Google Scholar] [CrossRef]
- Bao, B.Y.; Yeh, S.D.; Lee, Y.F. 1alpha,25-dihydroxyvitamin D3 inhibits prostate cancer cell invasion via modulation of selective proteases. Carcinogenesis 2006, 27, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Abu El Maaty, M.A.; Alborzinia, H.; Khan, S.J.; Buttner, M.; Wolfl, S. 1,25(OH)(2)D(3) disrupts glucose metabolism in prostate cancer cells leading to a truncation of the TCA cycle and inhibition of TXNIP expression. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1618–1630. [Google Scholar] [CrossRef]
- Giangreco, A.A.; Vaishnav, A.; Wagner, D.; Finelli, A.; Fleshner, N.; Van der Kwast, T.; Vieth, R.; Nonn, L. Tumor suppressor microRNAs, miR-100 and -125b, are regulated by 1,25-dihydroxyvitamin D in primary prostate cells and in patient tissue. Cancer Prev. Res. 2013, 6, 483–494. [Google Scholar] [CrossRef]
- Bao, B.Y.; Ting, H.J.; Hsu, J.W.; Lee, Y.F. Protective role of 1 alpha, 25-dihydroxyvitamin D3 against oxidative stress in nonmalignant human prostate epithelial cells. Int. J. Cancer 2008, 122, 2699–2706. [Google Scholar] [CrossRef]
- Kovalenko, P.L.; Zhang, Z.; Cui, M.; Clinton, S.K.; Fleet, J.C. 1,25 dihydroxyvitamin D-mediated orchestration of anticancer, transcript-level effects in the immortalized, non-transformed prostate epithelial cell line, RWPE1. BMC Genomics 2010, 11, 26. [Google Scholar] [CrossRef]
- McCray, T.; Pacheco, J.V.; Loitz, C.C.; Garcia, J.; Baumann, B.; Schlicht, M.J.; Valyi-Nagy, K.; Abern, M.R.; Nonn, L. Vitamin D sufficiency enhances differentiation of patient-derived prostate epithelial organoids. iScience 2021, 24, 101974. [Google Scholar] [CrossRef]
- Maestro, M.A.; Molnar, F.; Carlberg, C. Vitamin D and Its Synthetic Analogs. J. Med. Chem. 2019, 62, 6854–6875. [Google Scholar] [CrossRef] [PubMed]
- Maestro, M.A.; Seoane, S. The Centennial Collection of VDR Ligands: Metabolites, Analogs, Hybrids and Non-Secosteroidal Ligands. Nutrients 2022, 14, 4927. [Google Scholar] [CrossRef] [PubMed]
- Baurska, H.; Klopot, A.; Kielbinski, M.; Chrobak, A.; Wijas, E.; Kutner, A.; Marcinkowska, E. Structure-function analysis of vitamin D(2) analogs as potential inducers of leukemia differentiation and inhibitors of prostate cancer proliferation. J. Steroid Biochem. Mol. Biol. 2011, 126, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Trynda, J.; Turlej, E.; Milczarek, M.; Pietraszek, A.; Chodynski, M.; Kutner, A.; Wietrzyk, J. Antiproliferative Activity and in Vivo Toxicity of Double-Point Modified Analogs of 1,25-Dihydroxyergocalciferol. Int. J. Mol. Sci. 2015, 16, 24873–24894. [Google Scholar] [CrossRef]
- Wietrzyk, J.; Nevozhay, D.; Milczarek, M.; Filip, B.; Kutner, A. Toxicity and antitumor activity of the vitamin D analogs PRI-1906 and PRI-1907 in combined treatment with cyclophosphamide in a mouse mammary cancer model. Cancer Chemother. Pharmacol. 2008, 62, 787–797. [Google Scholar] [CrossRef]
- Milczarek, M.; Chodynski, M.; Pietraszek, A.; Stachowicz-Suhs, M.; Yasuda, K.; Sakaki, T.; Wietrzyk, J.; Kutner, A. Synthesis, CYP24A1-Dependent Metabolism and Antiproliferative Potential against Colorectal Cancer Cells of 1,25-Dihydroxyvitamin D(2) Derivatives Modified at the Side Chain and the A-Ring. Int. J. Mol. Sci. 2020, 21, 642. [Google Scholar] [CrossRef]
- Banwell, C.M.; Singh, R.; Stewart, P.M.; Uskokovic, M.R.; Campbell, M.J. Antiproliferative signalling by 1,25(OH)2D3 in prostate and breast cancer is suppressed by a mechanism involving histone deacetylation. Recent. Results Cancer Res. 2003, 164, 83–98. [Google Scholar] [CrossRef]
- Trump, D.L.; Aragon-Ching, J.B. Vitamin D in prostate cancer. Asian J. Androl. 2018, 20, 244–252. [Google Scholar] [CrossRef]
- Xu, Y.; Shao, X.; Yao, Y.; Xu, L.; Chang, L.; Jiang, Z.; Lin, Z. Positive association between circulating 25-hydroxyvitamin D levels and prostate cancer risk: New findings from an updated meta-analysis. J. Cancer Res. Clin. Oncol. 2014, 140, 1465–1477. [Google Scholar] [CrossRef]
- Gilbert, R.; Martin, R.M.; Beynon, R.; Harris, R.; Savovic, J.; Zuccolo, L.; Bekkering, G.E.; Fraser, W.D.; Sterne, J.A.; Metcalfe, C. Associations of circulating and dietary vitamin D with prostate cancer risk: A systematic review and dose-response meta-analysis. Cancer Causes Control. 2011, 22, 319–340. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef]
- Manson, J.E.; Bassuk, S.S.; Buring, J.E.; Group, V.R. Principal results of the VITamin D and OmegA-3 TriaL (VITAL) and updated meta-analyses of relevant vitamin D trials. J. Steroid Biochem. Mol. Biol. 2020, 198, 105522. [Google Scholar] [CrossRef] [PubMed]
- Aranow, C. Vitamin D and the immune system. J. Investig. Med. 2011, 59, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Glaser, R.; Mrowietz, U. Vitamin D(3) and analogues modulate the expression of CSF-1 and its receptor in human dendritic cells. Biochem. Biophys. Res. Commun. 2002, 297, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, X.; Bai, G.; Qiang, D.; Zhang, L.; Liu, H.; He, Y.; Tang, Y.; Li, L. Vitamin D deficiency leads to the abnormal activation of the complement system. Immunol. Res. 2023, 71, 29–38. [Google Scholar] [CrossRef]
- Szeles, L.; Keresztes, G.; Torocsik, D.; Balajthy, Z.; Krenacs, L.; Poliska, S.; Steinmeyer, A.; Zuegel, U.; Pruenster, M.; Rot, A.; et al. 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J. Immunol. 2009, 182, 2074–2083. [Google Scholar] [CrossRef]
- Iho, S.; Takahashi, T.; Kura, F.; Sugiyama, H.; Hoshino, T. The effect of 1,25-dihydroxyvitamin D3 on in vitro immunoglobulin production in human B cells. J. Immunol. 1986, 136, 4427–4431. [Google Scholar] [CrossRef]
- Cantorna, M.T.; Snyder, L.; Lin, Y.D.; Yang, L. Vitamin D and 1,25(OH)2D regulation of T cells. Nutrients 2015, 7, 3011–3021. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes. Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef]
- Gilad, L.A.; Bresler, T.; Gnainsky, J.; Smirnoff, P.; Schwartz, B. Regulation of vitamin D receptor expression via estrogen-induced activation of the ERK 1/2 signaling pathway in colon and breast cancer cells. J. Endocrinol. 2005, 185, 577–592. [Google Scholar] [CrossRef]
- Santos-Martinez, N.; Diaz, L.; Ordaz-Rosado, D.; Garcia-Quiroz, J.; Barrera, D.; Avila, E.; Halhali, A.; Medina-Franco, H.; Ibarra-Sanchez, M.J.; Esparza-Lopez, J.; et al. Calcitriol restores antiestrogen responsiveness in estrogen receptor negative breast cancer cells: A potential new therapeutic approach. BMC Cancer 2014, 14, 230. [Google Scholar] [CrossRef]
- Alimirah, F.; Peng, X.; Yuan, L.; Mehta, R.R.; von Knethen, A.; Choubey, D.; Mehta, R.G. Crosstalk between the peroxisome proliferator-activated receptor gamma (PPARgamma) and the vitamin D receptor (VDR) in human breast cancer cells: PPARgamma binds to VDR and inhibits 1alpha,25-dihydroxyvitamin D3 mediated transactivation. Exp. Cell Res. 2012, 318, 2490–2497. [Google Scholar] [CrossRef]
- Wood, R.J. Vitamin D and adipogenesis: New molecular insights. Nutr. Rev. 2008, 66, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, T.W.; Vaisanen, S.; Frank, C.; Molnar, F.; Sinkkonen, L.; Carlberg, C. The human peroxisome proliferator-activated receptor delta gene is a primary target of 1alpha,25-dihydroxyvitamin D3 and its nuclear receptor. J. Mol. Biol. 2005, 349, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, M.; Guo, Y.; Song, Z.; Liu, B. 1,25-Dihydroxyvitamin D(3) Promotes High Glucose-Induced M1 Macrophage Switching to M2 via the VDR-PPARgamma Signaling Pathway. Biomed. Res. Int. 2015, 2015, 157834. [Google Scholar] [CrossRef]
- Sertznig, P.; Dunlop, T.; Seifert, M.; Tilgen, W.; Reichrath, J. Cross-talk between vitamin D receptor (VDR)- and peroxisome proliferator-activated receptor (PPAR)-signaling in melanoma cells. Anticancer. Res. 2009, 29, 3647–3658. [Google Scholar]
- Chen, K.; O’Brien, J.; McVey, A.; Jenjitranant, P.; Kelly, B.D.; Kasivisvanathan, V.; Lawrentschuk, N.; Murphy, D.G.; Azad, A.A. Combination treatment in metastatic prostate cancer: Is the bar too high or have we fallen short? Nat. Rev. Urol. 2023, 20, 116–123. [Google Scholar] [CrossRef]
- Jacob, A.; Raj, R.; Allison, D.B.; Myint, Z.W. Androgen Receptor Signaling in Prostate Cancer and Therapeutic Strategies. Cancers 2021, 13, 5417. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Khalaf, D.J.; Annala, M.; Taavitsainen, S.; Finch, D.L.; Oja, C.; Vergidis, J.; Zulfiqar, M.; Sunderland, K.; Azad, A.A.; Kollmannsberger, C.K.; et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: A multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol. 2019, 20, 1730–1739. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Borre, M.; Gurney, H.; Loriot, Y.; Andresen-Daniil, C.; Kalleda, R.; Pham, T.; Taplin, M.E.; PLATO collaborators. Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer With Rising Prostate-Specific Antigen During Enzalutamide Treatment. J. Clin. Oncol. 2018, 36, 2639–2646. [Google Scholar] [CrossRef]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Flechon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Liang, J.; Shao, Y.; Xiong, S.; Feng, S.; Li, X. Evaluation of the Efficacy of PARP Inhibitors in Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis. Front. Pharmacol. 2021, 12, 777663. [Google Scholar] [CrossRef] [PubMed]
- Cleutjens, C.B.; Steketee, K.; van Eekelen, C.C.; van der Korput, J.A.; Brinkmann, A.O.; Trapman, J. Both androgen receptor and glucocorticoid receptor are able to induce prostate-specific antigen expression, but differ in their growth-stimulating properties of LNCaP cells. Endocrinology 1997, 138, 5293–5300. [Google Scholar] [CrossRef]
- Du, X.; Eksterowicz, J.; Zhou, H.; Rew, Y.; Zhu, L.; Yan, X.; Medina, J.C.; Huang, T.; Chen, X.; Sutimantanapi, D.; et al. Discovery of a Potent Steroidal Glucocorticoid Receptor Antagonist with Enhanced Selectivity against the Progesterone and Androgen Receptors (OP-3633). J. Med. Chem. 2019, 62, 6751–6764. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Thakkar, A.; Wang, B.; Picon-Ruiz, M.; Buchwald, P.; Ince, T.A. Vitamin D and androgen receptor-targeted therapy for triple-negative breast cancer. Breast Cancer Res. Treat. 2016, 157, 77–90. [Google Scholar] [CrossRef]
- Fahlen, M.; Zhang, H.; Löfgren, L.; Masironi, B.; Von Schoultz, E.; Von Schoultz, B.O.; Sahlin, L. Expression of Progesterone and Androgen Receptors in the Breast of Premenopausal Women, Considering Menstrual Phase. Anticancer. Res. 2018, 38, 1499–1510. [Google Scholar] [CrossRef]
- Klijn, J.G.; Setyono-Han, B.; Foekens, J.A. Progesterone antagonists and progesterone receptor modulators in the treatment of breast cancer. Steroids 2000, 65, 825–830. [Google Scholar] [CrossRef]
- Horwitz, K.B.; Sartorius, C.A. 90 YEARS OF PROGESTERONE: Progesterone and progesterone receptors in breast cancer: Past, present, future. J. Mol. Endocrinol. 2020, 65, T49–T63. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hantusch, B.; Kenner, L.; Stanulović, V.S.; Hoogenkamp, M.; Brown, G. Targeting Androgen, Thyroid Hormone, and Vitamin A and D Receptors to Treat Prostate Cancer. Int. J. Mol. Sci. 2024, 25, 9245. https://doi.org/10.3390/ijms25179245
Hantusch B, Kenner L, Stanulović VS, Hoogenkamp M, Brown G. Targeting Androgen, Thyroid Hormone, and Vitamin A and D Receptors to Treat Prostate Cancer. International Journal of Molecular Sciences. 2024; 25(17):9245. https://doi.org/10.3390/ijms25179245
Chicago/Turabian StyleHantusch, Brigitte, Lukas Kenner, Vesna S. Stanulović, Maarten Hoogenkamp, and Geoffrey Brown. 2024. "Targeting Androgen, Thyroid Hormone, and Vitamin A and D Receptors to Treat Prostate Cancer" International Journal of Molecular Sciences 25, no. 17: 9245. https://doi.org/10.3390/ijms25179245