
Inhibitors of NLRP3 Inflammasome Formation: A Cardioprotective Role for the Gasotransmitters Carbon Monoxide, Nitric Oxide, and Hydrogen Sulphide in Acute Myocardial Infarction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Myocardial Ischaemia Reperfusion Injury (IRI)
Inflammatory Responses Following Myocardial IRI
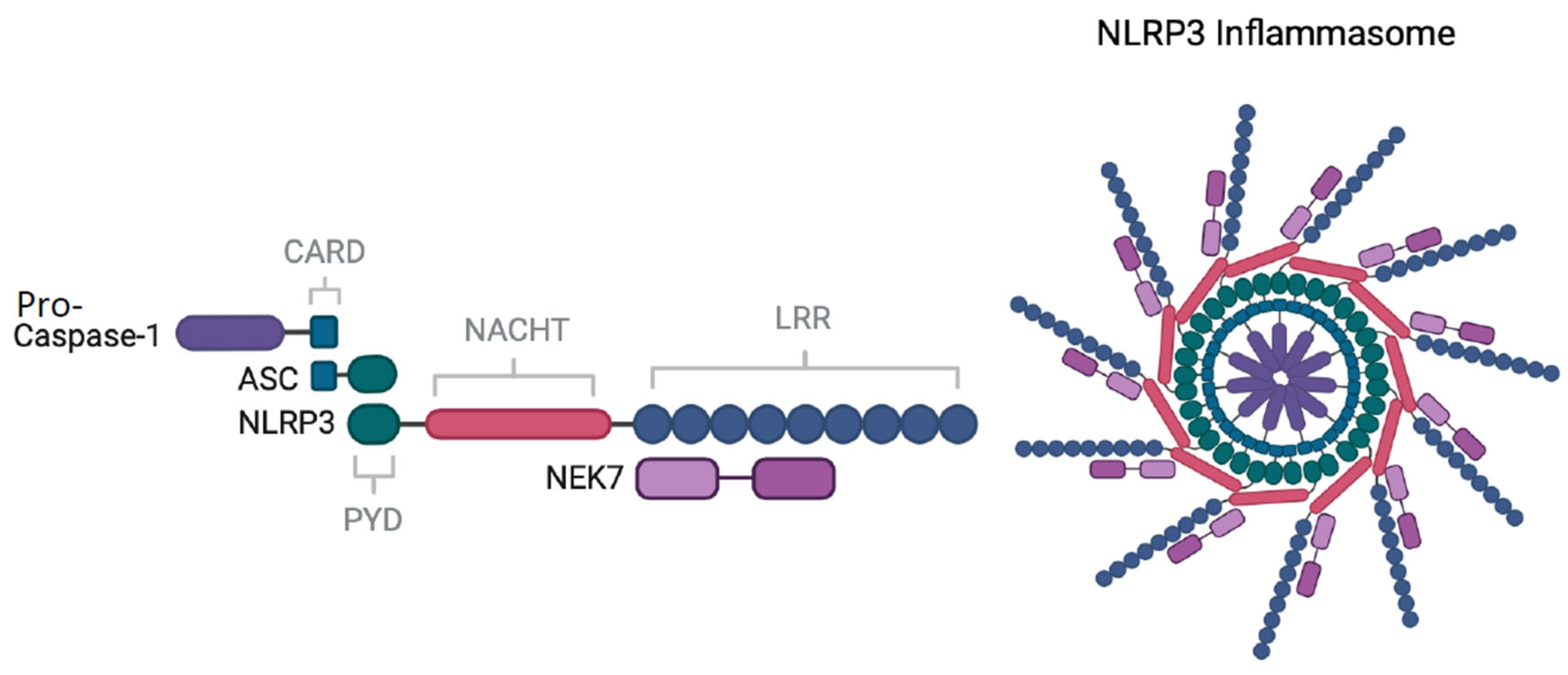
3. The NLRP3 Inflammasome
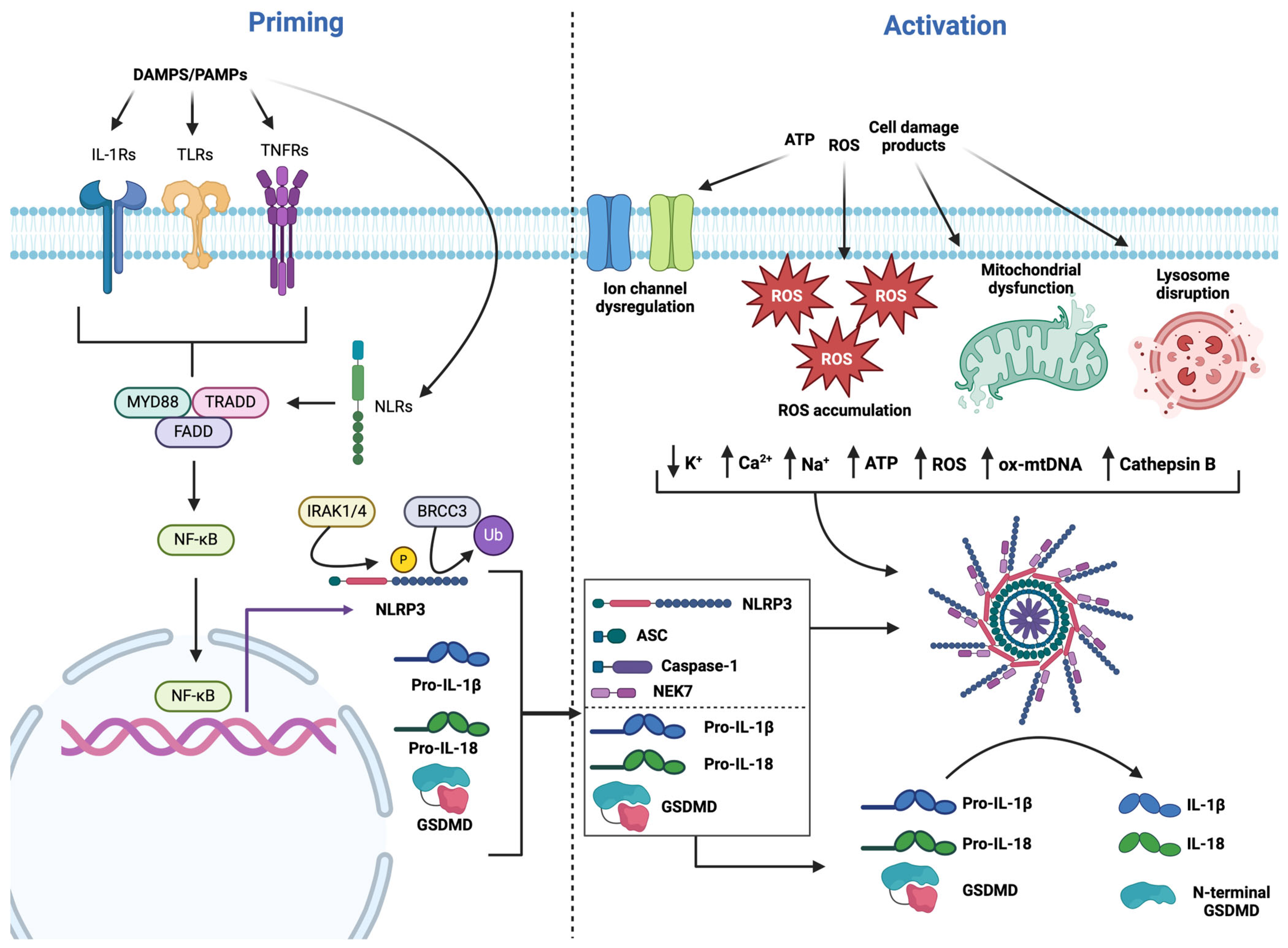
3.1. NLRP3 Priming
3.2. NLRP3 Activation
3.3. Targeted NLRP3 Inhibitors
4. Gasotransmitters as Cardioprotectants
4.1. Endogenous Carbon Monoxide
4.2. Exogenous CO Delivery
4.3. Pleiotropic Effects of CO
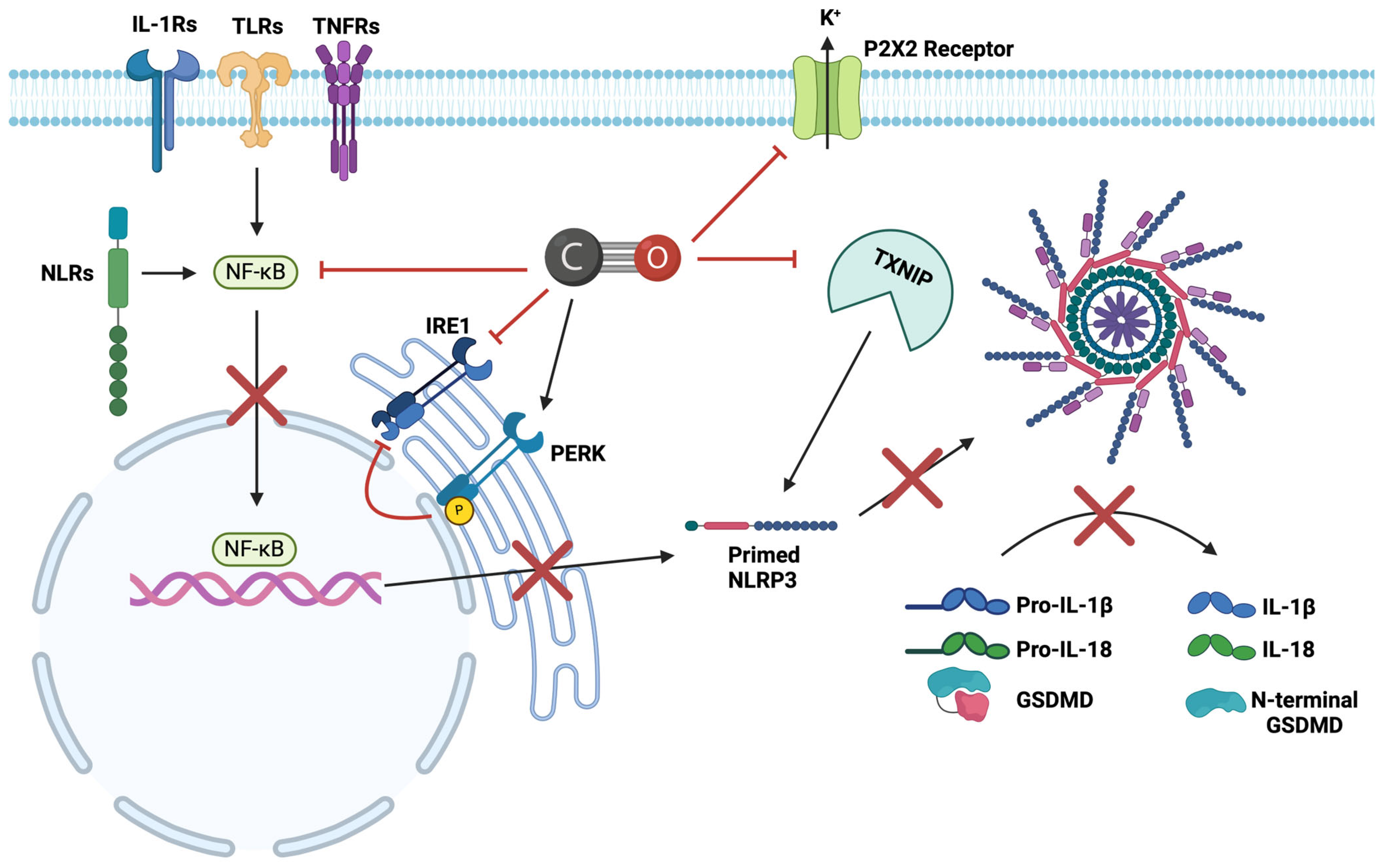
4.4. Inhibitory Effects of CO on NLRP3
4.5. Endogenous Nitric Oxide
4.6. Exogenous NO Application
4.7. Pleiotropic Effects of NO
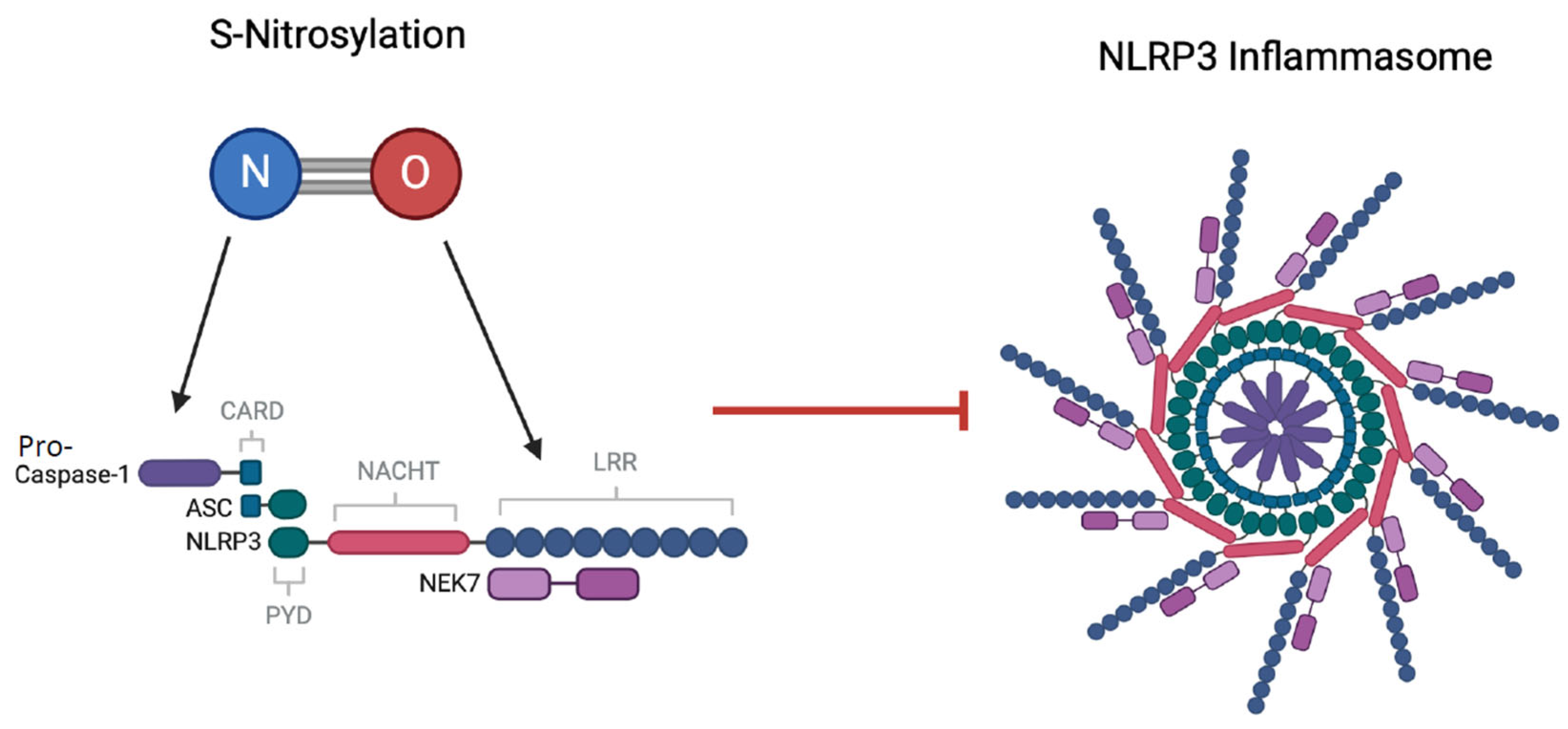
4.8. Inhibitory Effects of NO on NLRP3
4.9. Endogenous Hydrogen Sulphide
4.10. Exogenous H2S Application
4.11. Pleiotropic Effects of H2S
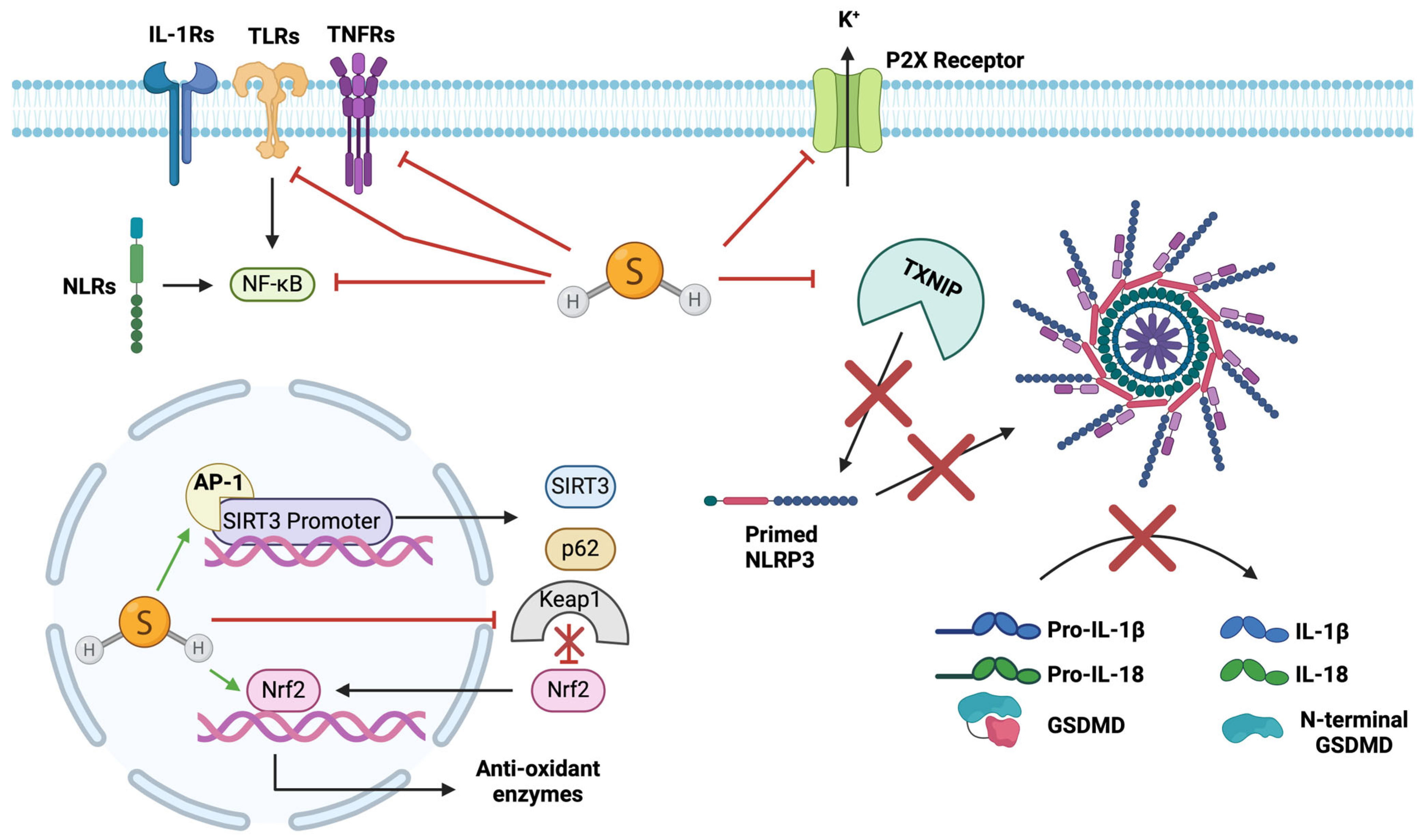
4.12. Inhibitory Effects of H2S on NLRP3
5. Limitations of the Current Literature
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Doenst, T.; Haverich, A.; Serruys, P.; Bonow, R.O.; Kappetein, P.; Falk, V.; Velazquez, E.; Diegeler, A.; Sigusch, H. PCI and CABG for treating stable coronary artery disease: JACC review topic of the week. J. Am. Coll. Cardiol. 2019, 73, 964–976. [Google Scholar] [CrossRef]
- Ruggieri, V.G.; Bounader, K.; Verhoye, J.P.; Onorati, F.; Rubino, A.S.; Gatti, G.; Tauriainen, T.; De Feo, M.; Reichart, D.; Dalén, M. Prognostic impact of prolonged cross-clamp time in coronary artery bypass grafting. Heart Lung Circ. 2018, 27, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.G. Off-pump coronary artery bypass grafting in octogenarians. J. Thorac. Dis. 2016, 8, S799. [Google Scholar] [CrossRef] [PubMed]
- Dobson, G.P.; Faggian, G.; Onorati, F.; Vinten-Johansen, J. Hyperkalemic cardioplegia for adult and pediatric surgery: End of an era? Front. Physiol. 2013, 4, 228. [Google Scholar] [CrossRef]
- Suleiman, M.; Hancock, M.; Shukla, R.; Rajakaruna, C.; Angelini, G. Cardioplegic strategies to protect the hypertrophic heart during cardiac surgery. Perfusion 2011, 26, 48–56. [Google Scholar] [CrossRef]
- Wilson, N. Rheumatic heart disease in indigenous populations—New Zealand experience. Heart Lung Circ. 2010, 19, 282–288. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Garcia-Dorado, D.; Bøtker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Penna, C. Hypertension, hypertrophy, and reperfusion injury. J. Cardiovasc. Med. 2017, 18, 131–135. [Google Scholar] [CrossRef]
- Janier, M.F.; Vanoverschelde, J.-L.; Bergmann, S.R. Ischemic preconditioning stimulates anaerobic glycolysis in the isolated rabbit heart. Am. J. Physiol. Heart Circ. Physiol. 1994, 267, H1353–H1360. [Google Scholar] [CrossRef]
- Jennings, R.B.; Reimer, K.A.; Hill, M.L.; Mayer, S.E. Total ischemia in dog hearts, in vitro. 1. Comparison of high energy phosphate production, utilization, and depletion, and of adenine nucleotide catabolism in total ischemia in vitro vs. severe ischemia in vivo. Circ. Res. 1981, 49, 892–900. [Google Scholar] [CrossRef]
- Cross, H.R.; Opie, L.H.; Radda, G.K.; Clarke, K. Is a high glycogen content beneficial or detrimental to the ischemic rat heart? A controversy resolved. Circ. Res. 1996, 78, 482–491. [Google Scholar] [CrossRef]
- Neely, J.R.; Grotyohann, L.W. Role of glycolytic products in damage to ischemic myocardium. Dissociation of adenosine triphosphate levels and recovery of function of reperfused ischemic hearts. Circ. Res. 1984, 55, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Neely, J.R. Role of intracellular Na+ in Ca2+ overload and depressed recovery of ventricular function of reperfused ischemic rat hearts. Possible involvement of H+-Na+ and Na+-Ca2+ exchange. Circ. Res. 1989, 65, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Khandoudi, N.; Bernard, M.; Cozzone, P.; Feuvray, D. Intracellular pH and role of Na+/H+ exchange during ischaemia and reperfusion of normal and diabetic rat hearts. Cardiovasc. Res. 1990, 24, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Cragoe, E., Jr.; Smith, T. Relations among sodium pump inhibition, Na-Ca and Na-H exchange activities, and Ca-H interaction in cultured chick heart cells. Circ. Res. 1987, 60, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Perlman, M.; London, R.E.; Steenbergen, C. Amiloride delays the ischemia-induced rise in cytosolic free calcium. Circ. Res. 1991, 68, 1250–1258. [Google Scholar] [CrossRef]
- Bigger, J.T., Jr.; Fleiss, J.L.; Kleiger, R.; Miller, J.P.; Rolnitzky, L. The relationships among ventricular arrhythmias, left ventricular dysfunction, and mortality in the 2 years after myocardial infarction. Circulation 1984, 69, 250–258. [Google Scholar] [CrossRef]
- Tani, M.; Asakura, Y.; Hasegawa, H.; Shinmura, K.; Ebihara, Y.; Nakamura, Y. Effect of preconditioning on ryanodine-sensitive Ca2+ release from sarcoplasmic reticulum of rat heart. Am. J. Physiol. Heart Circ. Physiol. 1996, 271, H876–H881. [Google Scholar] [CrossRef]
- Correa, F.; Soto, V.; Zazueta, C. Mitochondrial permeability transition relevance for apoptotic triggering in the post-ischemic heart. Int. J. Biochem. Cell Biol. 2007, 39, 787–798. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Halestrap, A.P. Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase. Implications for the immunosuppressive and toxic effects of cyclosporin. Biochem. J. 1991, 274, 611–614. [Google Scholar] [CrossRef]
- Jennings, R.; Hawkins, H.K.; Lowe, J.E.; Hill, M.L.; Klotman, S.; Reimer, K.A. Relation between high energy phosphate and lethal injury in myocardial ischemia in the dog. Am. J. Pathol. 1978, 92, 187. [Google Scholar]
- Singh, R.B.; Chohan, P.K.; Dhalla, N.S.; Netticadan, T. The sarcoplasmic reticulum proteins are targets for calpain action in the ischemic–reperfused heart. J. Mol. Cell. Cardiol. 2004, 37, 101–110. [Google Scholar] [CrossRef]
- Lindsay, D.P.; Camara, A.K.; Stowe, D.F.; Lubbe, R.; Aldakkak, M. Differential effects of buffer pH on Ca2+-induced ROS emission with inhibited mitochondrial complexes I and III. Front. Physiol. 2015, 6, 58. [Google Scholar] [CrossRef]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia–reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Kim, J.-S.; Jin, Y.; Lemasters, J.J. Reactive oxygen species, but not Ca2+ overloading, trigger pH-and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2024–H2034. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ao, X.; Chu, X.; Wan, Q.; Wang, Y.; Xiao, D.; Yu, W.; Li, M.; Yu, F. ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biol. 2019, 20, 414–426. [Google Scholar] [CrossRef]
- Zhu, P.; Hu, S.; Jin, Q.; Li, D.; Tian, F.; Toan, S.; Li, Y.; Zhou, H.; Chen, Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: A mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018, 16, 157–168. [Google Scholar] [CrossRef]
- Becker, R.C.; Owens, A.P., III; Sadayappan, S. Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. J. Thromb. Thrombolysis 2020, 49, 177–183. [Google Scholar] [CrossRef]
- Rai, V.; Sharma, P.; Agrawal, S.; Agrawal, D.K. Relevance of mouse models of cardiac fibrosis and hypertrophy in cardiac research. Mol. Cell. Biochem. 2017, 424, 123–145. [Google Scholar] [CrossRef]
- Hirai, S. Systemic inflammatory response syndrome after cardiac surgery under cardiopulmonary bypass. Ann. Thorac. Cardiovasc. Surg. 2003, 9, 365–370. [Google Scholar]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef]
- Sandanger, Ø.; Ranheim, T.; Vinge, L.E.; Bliksøen, M.; Alfsnes, K.; Finsen, A.V.; Dahl, C.P.; Askevold, E.T.; Florholmen, G.; Christensen, G. The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia–reperfusion injury. Cardiovasc. Res. 2013, 99, 164–174. [Google Scholar] [CrossRef]
- Westermann, D.; Lindner, D.; Kasner, M.; Zietsch, C.; Savvatis, K.; Escher, F.; Von Schlippenbach, J.; Skurk, C.; Steendijk, P.; Riad, A. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 2011, 4, 44–52. [Google Scholar] [CrossRef]
- Cao, L.; Chen, Y.; Zhang, Z.; Li, Y.; Zhao, P. Endoplasmic reticulum stress-induced NLRP1 inflammasome activation contributes to myocardial ischemia/reperfusion injury. Shock 2019, 51, 511–518. [Google Scholar] [CrossRef]
- Mezzaroma, E.; Toldo, S.; Farkas, D.; Seropian, I.M.; Van Tassell, B.W.; Salloum, F.N.; Kannan, H.R.; Menna, A.C.; Voelkel, N.F.; Abbate, A. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. USA 2011, 108, 19725–19730. [Google Scholar] [CrossRef]
- Toldo, S.; Marchetti, C.; Mauro, A.G.; Chojnacki, J.; Mezzaroma, E.; Carbone, S.; Zhang, S.; Van Tassell, B.; Salloum, F.N.; Abbate, A. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia–reperfusion in the mouse. Int. J. Cardiol. 2016, 209, 215–220. [Google Scholar] [CrossRef]
- Bracey, N.A.; Beck, P.L.; Muruve, D.A.; Hirota, S.A.; Guo, J.; Jabagi, H.; Wright, J.R., Jr.; MacDonald, J.A.; Lees-Miller, J.P.; Roach, D. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Exp. Physiol. 2013, 98, 462–472. [Google Scholar] [CrossRef]
- Willingham, S.B.; Allen, I.C.; Bergstralh, D.T.; Brickey, W.J.; Huang, M.T.-H.; Taxman, D.J.; Duncan, J.A.; Ting, J.P.-Y. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and-independent pathways. J. Immunol. 2009, 183, 2008–2015. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle–Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef]
- Proell, M.; Gerlic, M.; Mace, P.D.; Reed, J.C.; Riedl, S.J. The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 2013, 449, 613–621. [Google Scholar] [CrossRef]
- Shi, H.; Wang, Y.; Li, X.; Zhan, X.; Tang, M.; Fina, M.; Su, L.; Pratt, D.; Bu, C.H.; Hildebrand, S. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol. 2016, 17, 250–258. [Google Scholar] [CrossRef]
- Xiao, L.; Magupalli, V.G.; Wu, H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature 2023, 613, 595–600. [Google Scholar] [CrossRef]
- Sutterwala, F.S.; Ogura, Y.; Szczepanik, M.; Lara-Tejero, M.; Lichtenberger, G.S.; Grant, E.P.; Bertin, J.; Coyle, A.J.; Galán, J.E.; Askenase, P.W. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 2006, 24, 317–327. [Google Scholar] [CrossRef]
- Toldo, S.; Mezzaroma, E.; McGeough, M.D.; Peña, C.A.; Marchetti, C.; Sonnino, C.; Van Tassell, B.W.; Salloum, F.N.; Voelkel, N.F.; Hoffman, H.M. Independent roles of the priming and the triggering of the NLRP3 inflammasome in the heart. Cardiovasc. Res. 2015, 105, 203–212. [Google Scholar] [CrossRef]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.-S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting edge: TNF-α mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 2012, 586, 1022–1026. [Google Scholar] [CrossRef]
- Ermolaeva, M.A.; Michallet, M.-C.; Papadopoulou, N.; Utermöhlen, O.; Kranidioti, K.; Kollias, G.; Tschopp, J.; Pasparakis, M. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat. Immunol. 2008, 9, 1037–1046. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, F.; Zhang, L.; Wang, X.; Lai, X.; Shen, Y.; Zhang, M.; Zhou, B.; Lang, H.; Yu, P. Alpha-lipoic acid preconditioning and ischaemic postconditioning synergistically protect rats from cerebral injury induced by ischemia and reperfusion partly via inhibition TLR4/MyD88/NF-κB signaling pathway. Cell. Physiol. Biochem. 2018, 51, 1448–1460. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Núñez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef]
- Gurung, P.; Anand, P.K.; Malireddi, R.S.; Walle, L.V.; Van Opdenbosch, N.; Dillon, C.P.; Weinlich, R.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.-D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014, 192, 1835–1846. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, C.; Zhang, H.; Zhao, Q.; Liu, Y.; Xu, C.; Xie, Q.; Wu, X.; Yu, X.; Zhang, J. MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep. 2016, 16, 3247–3259. [Google Scholar] [CrossRef] [PubMed]
- Allam, R.; Lawlor, K.E.; Yu, E.C.W.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J. Mitochondrial apoptosis is dispensable for NLRP 3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef]
- Baudino, T.A.; Carver, W.; Giles, W.; Borg, T.K. Cardiac fibroblasts: Friend or foe? Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1015–H1026. [Google Scholar] [CrossRef]
- Liu, Z.; Gan, L.; Xu, Y.; Luo, D.; Ren, Q.; Wu, S.; Sun, C. Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J. Pineal Res. 2017, 63, e12414. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Kang, S.; Anderson, C.; Sagara, J.; Fitzgerald, K.A.; Alnemri, E.S. Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J. Immunol. 2013, 191, 3995–3999. [Google Scholar] [CrossRef]
- Jha, A.; Kumar, V.; Haque, S.; Ayasolla, K.; Saha, S.; Lan, X.; Malhotra, A.; Saleem, M.A.; Skorecki, K.; Singhal, P.C. Alterations in plasma membrane ion channel structures stimulate NLRP3 inflammasome activation in APOL1 risk milieu. FEBS J. 2020, 287, 2000–2022. [Google Scholar] [CrossRef]
- Shen, S.; He, F.; Cheng, C.; Xu, B.; Sheng, J. Uric acid aggravates myocardial ischemia–reperfusion injury via ROS/NLRP3 pyroptosis pathway. Biomed. Pharmacother. 2021, 133, 110990. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Boini, K.M.; Pitzer, A.L.; Gulbins, E.; Zhang, Y.; Li, P.-L. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Walev, I.; Klein, J.; Husmann, M.; Valeva, A.; Strauch, S.; Wirtz, H.; Weichel, O.; Bhakdi, S. Potassium regulates IL-1β processing via calcium-independent phospholipase A2. J. Immunol. 2000, 164, 5120–5124. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Prabhu, S.D.; Reddy, V.S.; Boylston, W.H.; Valente, A.J.; Chandrasekar, B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J. Biol. Chem. 2009, 284, 7853–7865. [Google Scholar] [CrossRef]
- Ding, H.-S.; Yang, J.; Chen, P.; Yang, J.; Bo, S.-Q.; Ding, J.-W.; Yu, Q.-Q. The HMGB1–TLR4 axis contributes to myocardial ischemia/reperfusion injury via regulation of cardiomyocyte apoptosis. Gene 2013, 527, 389–393. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-j.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Billingham, L.K.; Stoolman, J.S.; Vasan, K.; Rodriguez, A.E.; Poor, T.A.; Szibor, M.; Jacobs, H.T.; Reczek, C.R.; Rashidi, A.; Zhang, P. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat. Immunol. 2022, 23, 692–704. [Google Scholar] [CrossRef]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA fragments exit mitochondria via mPTP-and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Cabral, A.; Cabral, J.E.; Wang, A.; Zhang, Y.; Liang, H.; Nikbakht, D.; Corona, L.; Hoffman, H.M.; McNulty, R. Differential Binding of NLRP3 to non-oxidized and Ox-mtDNA mediates NLRP3 Inflammasome Activation. Commun. Biol. 2023, 6, 578. [Google Scholar]
- Chevriaux, A.; Pilot, T.; Derangère, V.; Simonin, H.; Martine, P.; Chalmin, F.; Ghiringhelli, F.; Rébé, C. Cathepsin B is required for NLRP3 inflammasome activation in macrophages, through NLRP3 interaction. Front. Cell Dev. Biol. 2020, 8, 167. [Google Scholar] [CrossRef]
- Bai, H.; Yang, B.; Yu, W.; Xiao, Y.; Yu, D.; Zhang, Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp. Cell Res. 2018, 362, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, P.; Smit-McBride, Z.; Oltjen, S.L.; Hjelmeland, L.M. Regulation of cysteine cathepsin expression by oxidative stress in the retinal pigment epithelium/choroid of the mouse. Exp. Eye Res. 2006, 83, 679–687. [Google Scholar] [CrossRef]
- Jha, S.; Srivastava, S.Y.; Brickey, W.J.; Iocca, H.; Toews, A.; Morrison, J.P.; Chen, V.S.; Gris, D.; Matsushima, G.K.; Ting, J.P.-Y. The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J. Neurosci. 2010, 30, 15811–15820. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Müller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef]
- Lei, Q.; Yi, T.; Chen, C. NF-κB-Gasdermin D (GSDMD) axis couples oxidative stress and NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome-mediated cardiomyocyte pyroptosis following myocardial infarction. Med. Sci. Monit. 2018, 24, 6044. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; de Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef]
- Gao, R.; Shi, H.; Chang, S.; Gao, Y.; Li, X.; Lv, C.; Yang, H.; Xiang, H.; Yang, J.; Xu, L. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int. Immunopharmacol. 2019, 74, 105575. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Van Tassell, B.W.; Mezzaroma, E.; Del Buono, M.G.; Prestamburgo, A.; Potere, N.; Abbate, A. The NLRP3 inflammasome inhibitor, OLT1177 (dapansutrile), reduces infarct size and preserves contractile function after ischemia reperfusion injury in the mouse. J. Cardiovasc. Pharmacol. 2019, 73, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Wohlford, G.F.; Van Tassell, B.W.; Billingsley, H.E.; Kadariya, D.; Carbone, S.; Mihalick, V.L.; Bonaventura, A.; Vecchié, A.; Chiabrando, J.G.; Bressi, E. Phase 1b, randomized, double-blinded, dose escalation, single-center, repeat dose safety and pharmacodynamics study of the oral nlrp3 inhibitor dapansutrile in subjects with nyha ii–iii systolic heart failure. J. Cardiovasc. Pharmacol. 2021, 77, 49–60. [Google Scholar] [CrossRef]
- Gastaldi, S.; Rocca, C.; Gianquinto, E.; Granieri, M.C.; Boscaro, V.; Blua, F.; Rolando, B.; Marini, E.; Gallicchio, M.; De Bartolo, A. Discovery of a novel 1, 3, 4-oxadiazol-2-one-based NLRP3 inhibitor as a pharmacological agent to mitigate cardiac and metabolic complications in an experimental model of diet-induced metaflammation. Eur. J. Med. Chem. 2023, 257, 115542. [Google Scholar] [CrossRef] [PubMed]
- Bertinaria, M.; Gastaldi, S.; Marini, E.; Giorgis, M. Development of covalent NLRP3 inflammasome inhibitors: Chemistry and biological activity. Arch. Biochem. Biophys. 2019, 670, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Penna, C.; Tullio, F.; Femminò, S.; Nigro, D.; Chiazza, F.; Serpe, L.; Collotta, D.; Alloatti, G.; Cocco, M. Pharmacological inhibition of NLRP3 inflammasome attenuates myocardial ischemia/reperfusion injury by activation of RISK and mitochondrial pathways. Oxid. Med. Cell. Longev. 2016, 2016, 5271251. [Google Scholar] [CrossRef]
- Penna, C.; Aragno, M.; Cento, A.S.; Femminò, S.; Russo, I.; Bello, F.D.; Chiazza, F.; Collotta, D.; Alves, G.F.; Bertinaria, M. Ticagrelor conditioning effects are not additive to cardioprotection induced by direct NLRP3 inflammasome inhibition: Role of RISK, NLRP3, and Redox Cascades. Oxid. Med. Cell. Longev. 2020, 2020, 9219825. [Google Scholar] [CrossRef]
- Allen, T.A.; Root, W.S. Partition of carbon monoxide and oxygen between air and whole blood of rats, dogs and men as affected by plasma pH. J. Appl. Physiol. 1957, 10, 186–190. [Google Scholar] [CrossRef]
- Sjöstrand, T. Endogenous formation of carbon monoxide in man under normal and pathological conditions. Scand. J. Clin. Investig. 1949, 1, 201–214. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase: Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D.; Trakshel, G.; Kutty, R.K. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J. Biol. Chem. 1986, 261, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Mccoubrey, W.K., Jr.; Huang, T.; Maines, M.D. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur. J. Biochem. 1997, 247, 725–732. [Google Scholar] [CrossRef]
- Zakhary, R.; Gaine, S.P.; Dinerman, J.L.; Ruat, M.; Flavahan, N.A.; Snyder, S.H. Heme oxygenase 2: Endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc. Natl. Acad. Sci. USA 1996, 93, 795–798. [Google Scholar] [CrossRef]
- Jancsó, G.; Cserepes, B.; Gasz, B.; Benko, L.; Borsiczky, B.; Ferenc, A.; Kürthy, M.; Rácz, B.; Lantos, J.; Gál, J. Expression and Protective Role of Heme Oxygenase-1 in Delayed Myocardial Preconditioning. Ann. N. Y. Acad. Sci. 2007, 1095, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Yet, S.-F.; Tian, R.; Layne, M.D.; Wang, Z.Y.; Maemura, K.; Solovyeva, M.; Ith, B.; Melo, L.G.; Zhang, L.; Ingwall, J.S. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ. Res. 2001, 89, 168–173. [Google Scholar] [CrossRef]
- Nakao, A.; Neto, J.S.; Kanno, S.; Stolz, D.B.; Kimizuka, K.; Liu, F.; Bach, F.H.; Billiar, T.R.; Choi, A.M.; Otterbein, L.E. Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am. J. Transplant. 2005, 5, 282–291. [Google Scholar] [CrossRef]
- Segersvärd, H.; Lakkisto, P.; Hänninen, M.; Forsten, H.; Siren, J.; Immonen, K.; Kosonen, R.; Sarparanta, M.; Laine, M.; Tikkanen, I. Carbon monoxide releasing molecule improves structural and functional cardiac recovery after myocardial injury. Eur. J. Pharmacol. 2018, 818, 57–66. [Google Scholar] [CrossRef]
- Goldbaum, L.; Orellano, T.; Dergal, E. Mechanism of the toxic action of carbon monoxide. Ann. Clin. Lab. Sci. 1976, 6, 372–376. [Google Scholar]
- Stupfel, M.; Bouley, G. Physiological and biochemical effects on rats and mice exposed to small concentrations of carbon monoxide for long periods. Ann. N. Y. Acad. Sci. 1970, 174, 342–368. [Google Scholar] [CrossRef]
- Kueh, J.T.B.; Stanley, N.J.; Hewitt, R.J.; Woods, L.M.; Larsen, L.; Harrison, J.C.; Rennison, D.; Brimble, M.A.; Sammut, I.A.; Larsen, D.S. Norborn-2-en-7-ones as physiologically-triggered carbon monoxide-releasing prodrugs. Chem. Sci. 2017, 8, 5454. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Clark, J.E.; Foresti, R.; Sarathchandra, P.; Mann, B.E.; Green, C.J. Carbon monoxide-releasing molecules: Characterization of biochemical and vascular activities. Circ. Res. 2002, 90, e17–e24. [Google Scholar] [CrossRef]
- Motterlini, R.; Sawle, P.; Bains, S.; Hammad, J.; Alberto, R.; Foresti, R.; Green, C.J. CORM-A1: A new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005, 19, 284–286. [Google Scholar] [CrossRef]
- Clark, J.E.; Naughton, P.; Shurey, S.; Green, C.J.; Johnson, T.R.; Mann, B.E.; Foresti, R.; Motterlini, R. Cardioprotective actions by a water-soluble carbon monoxide–releasing molecule. Circ. Res. 2003, 93, e2–e8. [Google Scholar] [CrossRef] [PubMed]
- Portal, L.; Morin, D.; Motterlini, R.; Ghaleh, B.; Pons, S. The CO-releasing molecule CORM-3 protects adult cardiomyocytes against hypoxia-reoxygenation by modulating pH restoration. Eur. J. Pharmacol. 2019, 862, 172636. [Google Scholar] [CrossRef]
- Soni, H.; Patel, P.; Rath, A.C.; Jain, M.; Mehta, A.A. Cardioprotective effect with carbon monoxide releasing molecule-2 (CORM-2) in isolated perfused rat heart: Role of coronary endothelium and underlying mechanism. Vasc. Pharmacol. 2010, 53, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Winburn, I.C.; Gunatunga, K.; McKernan, R.D.; Walker, R.J.; Sammut, I.A.; Harrison, J.C. Cell damage following carbon monoxide releasing molecule exposure: Implications for therapeutic applications. Basic Clin. Pharmacol. Toxicol. 2012, 111, 31–41. [Google Scholar] [CrossRef]
- Desmard, M.; Davidge, K.S.; Bouvet, O.; Morin, D.; Roux, D.; Foresti, R.; Ricard, J.D.; Denamur, E.; Poole, R.K.; Montravers, P. A carbon monoxide-releasing molecule (CORM-3) exerts bactericidal activity against Pseudomonas aeruginosa and improves survival in an animal model of bacteraemia. FASEB J. 2009, 23, 1023–1031. [Google Scholar] [CrossRef]
- Bauer, N.; Yang, X.; Yuan, Z.; Wang, B. Reassessing CORM-A1: Redox chemistry and idiosyncratic CO-releasing characteristics of the widely used carbon monoxide donor. Chem. Sci. 2023, 14, 3215–3228. [Google Scholar] [CrossRef]
- Bauer, N.; Yuan, Z.; Yang, X.; Wang, B. Plight of CORMs: The unreliability of four commercially available CO-releasing molecules, CORM-2, CORM-3, CORM-A1, and CORM-401, in studying CO biology. Biochem. Pharmacol. 2023, 214, 115642. [Google Scholar] [CrossRef]
- Bell, N.T.; Payne, C.M.; Sammut, I.A.; Larsen, D.S. Mechanistic Studies of Carbon Monoxide Release from Norborn-2-en-7-one CORMs. Asian J. Org. Chem. 2022, 11, e202200350. [Google Scholar] [CrossRef]
- Payne, F.M.; Nie, S.; Diffee, G.M.; Wilkins, G.T.; Larsen, D.S.; Harrison, J.C.; Baldi, J.C.; Sammut, I.A. The carbon monoxide prodrug oCOm-21 increases Ca2+ sensitivity of the cardiac myofilament. Physiol. Rep. 2024, 12, e15974. [Google Scholar] [CrossRef] [PubMed]
- Kharitonov, V.G.; Sharma, V.S.; Pilz, R.B.; Magde, D.; Koesling, D. Basis of guanylate cyclase activation by carbon monoxide. Proc. Natl. Acad. Sci. USA 1995, 92, 2568–2571. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Lu, H.T.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Alam, J.; Fu, X.-Y.; Lee, P.J. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J. Biol. Chem. 2005, 280, 8714–8721. [Google Scholar] [CrossRef]
- Sammut, I.A.; Foresti, R.; Clark, J.E.; Exon, D.J.; Vesely, M.J.; Sarathchandra, P.; Green, C.J.; Motterlini, R. Carbon monoxide is a major contributor to the regulation of vascular tone in aortas expressing high levels of haeme oxygenase-1. Br. J. Pharmacol. 1998, 125, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Brouard, S.; Berberat, P.O.; Tobiasch, E.; Seldon, M.P.; Bach, F.H.; Soares, M.P. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-κB to protect endothelial cells from tumor necrosis factor-α-mediated apoptosis. J. Biol. Chem. 2002, 277, 17950–17961. [Google Scholar] [CrossRef]
- Petrache, I.; Otterbein, L.E.; Alam, J.; Wiegand, G.W.; Choi, A.M. Heme oxygenase-1 inhibits TNF-α-induced apoptosis in cultured fibroblasts. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L312–L319. [Google Scholar] [CrossRef]
- Morse, D.; Pischke, S.E.; Zhou, Z.; Davis, R.J.; Flavell, R.A.; Loop, T.; Otterbein, S.L.; Otterbein, L.E.; Choi, A.M. Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 2003, 278, 36993–36998. [Google Scholar] [CrossRef]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef]
- Qin, S.; Du, R.; Yin, S.; Liu, X.; Xu, G.; Cao, W. Nrf2 is essential for the anti-inflammatory effect of carbon monoxide in LPS-induced inflammation. Inflamm. Res. 2015, 64, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, W.J.; Gadeberg, H.; Harrison, A.; Allen, N.D.; Riccardi, D.; Kemp, P.J. Carbon monoxide is a rapid modulator of recombinant and native P2X2 ligand-gated ion channels. Br. J. Pharmacol. 2009, 158, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, J.; Zhang, W.; Zhang, J.; Yang, J.; Li, K.; He, Y. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: A novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 2013, 45, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, M.; Katsnelson, M.A.; Dubyak, G.R.; Pearlman, E. Neutrophil P2X 7 receptors mediate NLRP3 inflammasome-dependent IL-1β secretion in response to ATP. Nat. Commun. 2016, 7, 10555. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, W.J.; Kemp, P.J. The carbon monoxide donor, CORM-2, is an antagonist of ATP-gated, human P2X4 receptors. Purinergic Signal. 2011, 7, 57–64. [Google Scholar] [CrossRef]
- Jung, S.-S.; Moon, J.-S.; Xu, J.-F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.; Nakahira, K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067. [Google Scholar] [CrossRef]
- Jiang, L.; Fei, D.; Gong, R.; Yang, W.; Yu, W.; Pan, S.; Zhao, M.; Zhao, M. CORM-2 inhibits TXNIP/NLRP3 inflammasome pathway in LPS-induced acute lung injury. Inflamm. Res. 2016, 65, 905–915. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Chen, D.; Dixon, B.J.; Doycheva, D.M.; Li, B.; Zhang, Y.; Hu, Q.; He, Y.; Guo, Z.; Nowrangi, D.; Flores, J. IRE1α inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic–ischemic brain injury in rats. J. Neuroinflamm. 2018, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.; Shin, D.-Y.; Zheng, M.; Joe, Y.; Pae, H.-O.; Ryter, S.W.; Chung, H.-T. Carbon monoxide, a reaction product of heme oxygenase-1, suppresses the expression of C-reactive protein by endoplasmic reticulum stress through modulation of the unfolded protein response. Mol. Immunol. 2011, 48, 1793–1799. [Google Scholar] [CrossRef]
- Kim, K.M.; Pae, H.-O.; Zheng, M.; Park, R.; Kim, Y.-M.; Chung, H.-T. Carbon Monoxide Induces Heme Oxygenase-1 via Activation of Protein Kinase R–Like Endoplasmic Reticulum Kinase and Inhibits Endothelial Cell Apoptosis Triggered by Endoplasmic Reticulum Stress. Circ. Res. 2007, 101, 919–927. [Google Scholar] [CrossRef]
- Zheng, G.; Zhan, Y.; Wang, H.; Luo, Z.; Zheng, F.; Zhou, Y.; Wu, Y.; Wang, S.; Wu, Y.; Xiang, G. Carbon monoxide releasing molecule-3 alleviates neuron death after spinal cord injury via inflammasome regulation. EBioMedicine 2019, 40, 643–654. [Google Scholar] [CrossRef]
- Zhang, W.; Tao, A.; Lan, T.; Cepinskas, G.; Kao, R.; Martin, C.M.; Rui, T. Carbon monoxide releasing molecule-3 improves myocardial function in mice with sepsis by inhibiting NLRP3 inflammasome activation in cardiac fibroblasts. Basic Res. Cardiol. 2017, 112, 16. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Ferrige, A.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.; Ashton, D.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef]
- Balligand, J.-L.; Ungureanu-Longrois, D.; Simmons, W.W.; Pimental, D.; Malinski, T.A.; Kapturczak, M.; Taha, Z.; Lowenstein, C.J.; Davidoff, A.J.; Kelly, R.A. Cytokine-inducible nitric oxide synthase (iNOS) expression in cardiac myocytes. Characterization and regulation of iNOS expression and detection of iNOS activity in single cardiac myocytes in vitro. J. Biol. Chem. 1994, 269, 27580–27588. [Google Scholar] [CrossRef]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef]
- Xu, K.Y.; Huso, D.L.; Dawson, T.M.; Bredt, D.S.; Becker, L.C. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1999, 96, 657–662. [Google Scholar] [CrossRef]
- Kelm, M.; Schrader, J. Control of coronary vascular tone by nitric oxide. Circ. Res. 1990, 66, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Suzuki, M.; Granger, D. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. USA 1991, 88, 4651–4655. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.; Palmer, R.; Moncada, S. Endogenous nitric oxide inhibits human platelet adhesion to vascular endothelium. Lancet 1987, 330, 1057–1058. [Google Scholar] [CrossRef]
- Stamler, J.S.; Loh, E.; Roddy, M.-A.; Currie, K.E.; Creager, M.A. Nitric oxide regulates basal systemic and pulmonary vascular resistance in healthy humans. Circulation 1994, 89, 2035–2040. [Google Scholar] [CrossRef]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Ishide, T.; Nauli, S.M.; Maher, T.J.; Ally, A. Cardiovascular responses and neurotransmitter changes following blockade of nNOS within the ventrolateral medulla during static muscle contraction. Brain Res. 2003, 977, 80–89. [Google Scholar] [CrossRef]
- Kurihara, N.; Alfie, M.E.; Sigmon, D.H.; Rhaleb, N.-E.; Shesely, E.G.; Carretero, O.A. Role of nNOS in blood pressure regulation in eNOS null mutant mice. Hypertension 1998, 32, 856–861. [Google Scholar] [CrossRef]
- Wang, Q.; Pelligrino, D.A.; Baughman, V.L.; Koenig, H.M.; Albrecht, R.F. The role of neuronal nitric oxide synthase in regulation of cerebral blood flow in normocapnia and hypercapnia in rats. J. Cereb. Blood Flow. Metab. 1995, 15, 774–778. [Google Scholar] [CrossRef]
- Griscavage, J.M.; Wilk, S.; Ignarro, L.J. Inhibitors of the proteasome pathway interfere with induction of nitric oxide synthase in macrophages by blocking activation of transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 3308–3312. [Google Scholar] [CrossRef]
- Connelly, L.; Palacios-Callender, M.; Ameixa, C.; Moncada, S.; Hobbs, A. Biphasic regulation of NF-κB activity underlies the pro-and anti-inflammatory actions of nitric oxide. J. Immunol. 2001, 166, 3873–3881. [Google Scholar] [CrossRef]
- Sato, M.; Hida, N.; Umezawa, Y. Imaging the nanomolar range of nitric oxide with an amplifier-coupled fluorescent indicator in living cells. Proc. Natl. Acad. Sci. USA 2005, 102, 14515–14520. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.-M.; Su, M.-J.; Chen, J.-K. Resveratrol protects myocardial ischemia–reperfusion injury through both NO-dependent and NO-independent mechanisms. Free Radic. Biol. Med. 2004, 36, 774–781. [Google Scholar] [CrossRef]
- Czapski, G.; Goldstein, S. The role of the reactions of· NO with superoxide and oxygen in biological systems: A kinetic approach. Free Radic. Biol. Med. 1995, 19, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, Z.; Li, L.-L. Advanced nitric oxide donors: Chemical structure of NO drugs, NO nanomedicines and biomedical applications. Nanoscale 2021, 13, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.K.; Dellinger, R.P.; Lundin, S.; Payen, D.; Vallet, B.; Gerlach, H.; Park, K.J.; Mehta, S.; Slutsky, A.S.; Friedrich, J.O. Inhaled nitric oxide does not reduce mortality in patients with acute respiratory distress syndrome regardless of severity: Systematic review and meta-analysis. Crit. Care Med. 2014, 42, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Pisarenko, O.; Studneva, I. Modulating the Bioactivity of Nitric Oxide as a Therapeutic Strategy in Cardiac Surgery. J. Surg. Res. 2021, 257, 178–188. [Google Scholar] [CrossRef]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite oxidation of sulfhydryls.: The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem. 1991, 266, 4244–4250. [Google Scholar] [CrossRef]
- Ishida, H.; Ichimori, K.; Hirota, Y.; Fukahori, M.; Nakazawa, H. Peroxynitrite-induced cardiac myocyte injury. Free Radic. Biol. Med. 1996, 20, 343–350. [Google Scholar] [CrossRef]
- Kaur, H.; Halliwell, B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation Nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett. 1994, 350, 9–12. [Google Scholar] [CrossRef]
- Li, D.-Y.; Tao, L.; Liu, H.; Christopher, T.; Lopez, B.; Ma, X. Role of ERK1/2 in the anti-apoptotic and cardioprotective effects of nitric oxide after myocardial ischemia and reperfusion. Apoptosis 2006, 11, 923–930. [Google Scholar] [CrossRef]
- Elliott, S.N.; McKnight, W.; Cirino, G.; Wallace, J.L. A nitric oxide-releasing nonsteroidal anti-inflammatory drug accelerates gastric ulcer healing in rats. Gastroenterology 1995, 109, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sayed, N.; Beuve, A.; Van Den Akker, F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007, 26, 578–588. [Google Scholar] [CrossRef]
- Martin, E.; Berka, V.; Bogatenkova, E.; Murad, F.; Tsai, A.-L. Ligand selectivity of soluble guanylyl cyclase: Effect of the hydrogen-bonding tyrosine in the distal heme pocket on binding of oxygen, nitric oxide, and carbon monoxide. J. Biol. Chem. 2006, 281, 27836–27845. [Google Scholar] [CrossRef]
- Snyder, C.M.; Shroff, E.H.; Liu, J.; Chandel, N.S. Nitric oxide induces cell death by regulating anti-apoptotic BCL-2 family members. PLoS ONE 2009, 4, e7059. [Google Scholar] [CrossRef]
- Grabowski, P.; Wright, P.; Van’t Hof, R.; Helfrich, M.; Ohshima, H.; Ralston, S. Immunolocalization of inducible nitric oxide synthase in synovium and cartilage in rheumatoid arthritis and osteoarthritis. Br. J. Rheumatol. 1997, 36, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Hinder, F.; Stubbe, H.D.; VAN AKEN, H.; Waurick, R.; Booke, M.; Meyer, J. Role of nitric oxide in sepsis-associated pulmonary edema. Am. J. Respir. Crit. Care Med. 1999, 159, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, D.; Stamler, J.; Bachwich, D.; Karmeli, F.; Ackerman, Z.; Podolsky, D. Enhanced colonic nitric oxide generation and nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Gut 1995, 36, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Tateya, S.; Cheng, A.M.; Rizzo-DeLeon, N.; Wang, N.F.; Handa, P.; Wilson, C.L.; Clowes, A.W.; Sweet, I.R.; Bomsztyk, K. M2 macrophage polarization mediates anti-inflammatory effects of endothelial nitric oxide signaling. Diabetes 2015, 64, 2836–2846. [Google Scholar] [CrossRef]
- Niedbala, W.; Cai, B.; Liu, H.; Pitman, N.; Chang, L.; Liew, F.Y. Nitric oxide induces CD4+ CD25+ Foxp3− regulatory T cells from CD4+ CD25− T cells via p53, IL-2, and OX40. Proc. Natl. Acad. Sci. USA 2007, 104, 15478–15483. [Google Scholar] [CrossRef]
- Niedbala, W.; Alves-Filho, J.C.; Fukada, S.Y.; Vieira, S.M.; Mitani, A.; Sonego, F.; Mirchandani, A.; Nascimento, D.C.; Cunha, F.Q.; Liew, F.Y. Regulation of type 17 helper T-cell function by nitric oxide during inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 9220–9225. [Google Scholar] [CrossRef]
- Hernandez-Cuellar, E.; Tsuchiya, K.; Hara, H.; Fang, R.; Sakai, S.; Kawamura, I.; Akira, S.; Mitsuyama, M. Cutting edge: Nitric oxide inhibits the NLRP3 inflammasome. J. Immunol. 2012, 189, 5113–5117. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Jaffrey, S.R.; Erdjument-Bromage, H.; Ferris, C.D.; Tempst, P.; Snyder, S.H. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001, 3, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef]
- Dimmeler, S.; Haendeler, J.; Nehls, M.; Zeiher, A.M. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1β–converting enzyme (ice)-like and cysteine protease protein (cpp)-32–like proteases. J. Exp. Med. 1997, 185, 601–608. [Google Scholar] [CrossRef]
- Mishra, B.B.; Rathinam, V.A.; Martens, G.W.; Martinot, A.J.; Kornfeld, H.; Fitzgerald, K.A.; Sassetti, C.M. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome–dependent processing of IL-1β. Nat. Immunol. 2013, 14, 52–60. [Google Scholar] [CrossRef]
- Asanuma, H.; Kitakaze, M.; Funaya, H.; Takashima, S.; Minamino, T.; Node, K.; Sakata, Y.; Asakura, M.; Sanada, S.; Shinozaki, Y. Nifedipine limits infarct size via NO-dependent mechanisms in dogs. Basic Res. Cardiol. 2001, 96, 497–505. [Google Scholar] [CrossRef]
- Kamenshchikov, N.O.; Mandel, I.A.; Podoksenov, Y.K.; Svirko, Y.S.; Lomivorotov, V.V.; Mikheev, S.L.; Kozlov, B.N.; Shipulin, V.M.; Nenakhova, A.A.; Anfinogenova, Y.J. Nitric oxide provides myocardial protection when added to the cardiopulmonary bypass circuit during cardiac surgery: Randomized trial. J. Thorac. Cardiovasc. Surg. 2019, 157, 2328–2336.e1. [Google Scholar] [CrossRef]
- Sun, J.; Aponte, A.M.; Menazza, S.; Gucek, M.; Steenbergen, C.; Murphy, E. Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovasc. Res. 2016, 110, 96–106. [Google Scholar] [CrossRef]
- Gianetti, J.; Del Sarto, P.; Bevilacqua, S.; Vassalle, C.; De Filippis, R.; Kacila, M.; Farneti, P.A.; Clerico, A.; Glauber, M.; Biagini, A. Supplemental nitric oxide and its effect on myocardial injury and function in patients undergoing cardiac surgery with extracorporeal circulation. J. Thorac. Cardiovasc. Surg. 2004, 127, 44–50. [Google Scholar] [CrossRef]
- Goodwin, L.R.; Francom, D.; Dieken, F.P.; Taylor, J.D.; Warenycia, M.W.; Reiffenstein, R.; Dowling, G. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: Postmortem studies and two case reports. J. Anal. Toxicol. 1989, 13, 105–109. [Google Scholar] [CrossRef]
- Ishigami, M.; Hiraki, K.; Umemura, K.; Ogasawara, Y.; Ishii, K.; Kimura, H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid. Redox Signal. 2009, 11, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef]
- Tripatara, P.; Patel, N.S.; Brancaleone, V.; Renshaw, D.; Rocha, J.; Sepodes, B.; Mota-Filipe, H.; Perretti, M.; Thiemermann, C. Characterisation of cystathionine gamma-lyase/hydrogen sulphide pathway in ischaemia/reperfusion injury of the mouse kidney: An in vivo study. Eur. J. Pharmacol. 2009, 606, 205–209. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical biology of H2S signaling through persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar] [CrossRef]
- Chou, C.-H.; Ogden, J.M.; Pohl, H.R.; Scinicariello, F.; Ingerman, L.; Barber, L.; Citra, M.J. Toxicological Profile for Hydrogen Sulfide and Carbonyl Sulfide; Agency for Toxic Substances and Disease Registry: Washington, DC, USA, 2016. [Google Scholar]
- Hill, B.; Woon, T.; Nicholls, P.; Peterson, J.; Greenwood, C.; Thomson, A. Interactions of sulphide and other ligands with cytochrome c oxidase. An electron-paramagnetic-resonance study. Biochem. J. 1984, 224, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Wu, H.; Wong, M.W.; Huang, D. Diallyl trisulfide is a fast H2S donor, but diallyl disulfide is a slow one: The reaction pathways and intermediates of glutathione with polysulfides. Org. Lett. 2015, 17, 4196–4199. [Google Scholar] [CrossRef]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L.; Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef]
- Papapetropoulos, A.; Whiteman, M.; Cirino, G. Pharmacological tools for hydrogen sulphide research: A brief, introductory guide for beginners. Br. J. Pharmacol. 2015, 172, 1633–1637. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Li, Z.; Pattillo, C.B.; Gojon, G., Sr.; Gojon, G., Jr.; Giordano, T.; Krum, H. A novel hydrogen sulfide prodrug, SG 1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc. Ther. 2015, 33, 216–226. [Google Scholar] [CrossRef]
- Nguyen, K.; Chau, V.Q.; Mauro, A.G.; Durrant, D.; Toldo, S.; Abbate, A.; Das, A.; Salloum, F.N. Hydrogen sulfide therapy suppresses cofilin-2 and attenuates ischemic heart failure in a mouse model of myocardial infarction. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, A.; Collino, M.; Yasin, M.; Benetti, E.; Gallicchio, M.; Mazzon, E.; Cuzzocrea, S.; Fantozzi, R.; Thiemermann, C. Anti-apoptotic and anti-inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock 2009, 31, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Karwi, Q.G.; Bice, J.S.; Baxter, G.F. Pre-and postconditioning the heart with hydrogen sulfide (H2S) against ischemia/reperfusion injury in vivo: A systematic review and meta-analysis. Basic. Res. Cardiol. 2018, 113, 6. [Google Scholar] [CrossRef]
- Ertugrul, I.A.; van Suylen, V.; Damman, K.; de Koning, M.-S.L.; van Goor, H.; Erasmus, M.E. Donor Heart Preservation with Hydrogen Sulfide: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2021, 22, 5737. [Google Scholar] [CrossRef]
- Johansen, D.; Ytrehus, K.; Baxter, G.F. Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia–reperfusion injury. Basic Res. Cardiol. 2006, 101, 53–60. [Google Scholar] [CrossRef]
- Meng, G.; Zhu, J.; Xiao, Y.; Huang, Z.; Zhang, Y.; Tang, X.; Xie, L.; Chen, Y.; Shao, Y.; Ferro, A. Hydrogen sulfide donor GYY4137 protects against myocardial fibrosis. Oxid. Med. Cell. Longev. 2015, 2015, 691070. [Google Scholar] [CrossRef]
- Meng, G.; Xiao, Y.; Ma, Y.; Tang, X.; Xie, L.; Liu, J.; Gu, Y.; Yu, Y.; Park, C.M.; Xian, M. Hydrogen sulfide regulates Krüppel-like factor 5 transcription activity via specificity protein 1 S-sulfhydration at Cys664 to prevent myocardial hypertrophy. J. Am. Heart Assoc. 2016, 5, e004160. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Feng, H.; Li, S.; Meng, G.; Liu, S.; Tang, X.; Ma, Y.; Han, Y.; Xiao, Y.; Gu, Y. SIRT3 mediates the antioxidant effect of hydrogen sulfide in endothelial cells. Antioxid. Redox Signal. 2016, 24, 329–343. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef]
- Lee, S.W.; Cheng, Y.; Moore, P.K.; Bian, J.-S. Hydrogen sulphide regulates intracellular pH in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2007, 358, 1142–1147. [Google Scholar] [CrossRef]
- Lim, J.J.; Liu, Y.-H.; Khin, E.S.W.; Bian, J.-S. Vasoconstrictive effect of hydrogen sulfide involves downregulation of cAMP in vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2008, 295, C1261–C1270. [Google Scholar] [CrossRef]
- Yong, Q.C.; Pan, T.-T.; Hu, L.-F.; Bian, J.-S. Negative regulation of β-adrenergic function by hydrogen sulphide in the rat hearts. J. Mol. Cell. Cardiol. 2008, 44, 701–710. [Google Scholar] [CrossRef]
- Sun, Y.-G.; Cao, Y.-X.; Wang, W.-W.; Ma, S.-F.; Yao, T.; Zhu, Y.-C. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc. Res. 2008, 79, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.M.; Kim, D.H.; Jandu, S.; Bergman, Y.; Tan, S.; Wang, H.; Pandey, D.R.; Abraham, T.P.; Shoukas, A.A.; Berkowitz, D.E. MPST but not CSE is the primary regulator of hydrogen sulfide production and function in the coronary artery. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H71–H79. [Google Scholar] [CrossRef]
- White, B.J.; Smith, P.A.; Dunn, W.R. Hydrogen sulphide–mediated vasodilatation involves the release of neurotransmitters from sensory nerves in pressurized mesenteric small arteries isolated from rats. Br. J. Pharmacol. 2013, 168, 785–793. [Google Scholar] [CrossRef]
- Ariyaratnam, P.; Loubani, M.; Morice, A.H. Hydrogen sulphide vasodilates human pulmonary arteries: A possible role in pulmonary hypertension? Microvasc. Res. 2013, 90, 135–137. [Google Scholar] [CrossRef]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, A.; McDonald, M.; Thiemermann, C. The production of hydrogen sulfide limits myocardial ischemia and reperfusion injury and contributes to the cardioprotective effects of preconditioning with endotoxin, but not ischemia in the rat. Shock 2006, 26, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef]
- Yang, G.; Sun, X.; Wang, R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004, 18, 1782–1784. [Google Scholar] [CrossRef]
- Yusof, M.; Kamada, K.; Kalogeris, T.; Gaskin, F.S.; Korthuis, R.J. Hydrogen sulfide triggers late-phase preconditioning in postischemic small intestine by an NO-and p38 MAPK-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H868–H876. [Google Scholar] [CrossRef]
- Lohninger, L.; Tomasova, L.; Praschberger, M.; Hintersteininger, M.; Erker, T.; Gmeiner, B.M.; Laggner, H. Hydrogen sulphide induces HIF-1α and Nrf2 in THP-1 macrophages. Biochimie 2015, 112, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liang, F.; Masood, W.S.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s-sulfhydration, MAPK dependent anti-apoptosis and NF-κB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liu, G.J.; Liu, N.; Zhang, G.; Zhang, J.X.; Li, L.F. Effect of hydrogen sulfide on inflammatory cytokines in acute myocardial ischemia injury in rats. Exp. Ther. Med. 2015, 9, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef]
- Xie, L.; Gu, Y.; Wen, M.; Zhao, S.; Wang, W.; Ma, Y.; Meng, G.; Han, Y.; Wang, Y.; Liu, G. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes 2016, 65, 3171–3184. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef]
- Zhang, D.; Macinkovic, I.; Devarie-Baez, N.O.; Pan, J.; Park, C.M.; Carroll, K.S.; Filipovic, M.R.; Xian, M. Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. 2014, 53, 575–581. [Google Scholar] [CrossRef]
- Lin, Z.; Altaf, N.; Li, C.; Chen, M.; Pan, L.; Wang, D.; Xie, L.; Zheng, Y.; Fu, H.; Han, Y. Hydrogen sulfide attenuates oxidative stress-induced NLRP3 inflammasome activation via S-sulfhydrating c-Jun at Cys269 in macrophages. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2890–2900. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Gan, W.; Ren, J.; Li, T.; Lv, S.; Li, C.; Liu, Z.; Yang, M. The SGK1 inhibitor EMD638683, prevents Angiotensin II–induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1–10. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pan, P.; Yang, Y.; Ge, H.; Chen, W.; Qu, J.; Shi, J.; Cui, G.; Liu, X.; Feng, H. Endogenous hydrogen sulphide attenuates NLRP3 inflammasome-mediated neuroinflammation by suppressing the P2X7 receptor after intracerebral haemorrhage in rats. J. Neuroinflamm. 2017, 14, 163. [Google Scholar] [CrossRef]
- Tian, D.; Dong, J.; Jin, S.; Teng, X.; Wu, Y. Endogenous hydrogen sulfide-mediated MAPK inhibition preserves endothelial function through TXNIP signaling. Free Radic. Biol. Med. 2017, 110, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Mehmood, S.; Liu, X.; Ma, S.; Yang, R. Hydrogen sulfide mitigates myocardial inflammation by inhibiting nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome activation in diabetic rats. Exp. Biol. Med. 2020, 245, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhuang, X.; Xie, C.; Hu, X.; Dong, X.; Guo, Y.; Li, S.; Liao, X. Exogenous hydrogen sulfide attenuates high glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome activation by suppressing TLR4/NF-κB pathway in H9c2 cells. Cell. Physiol. Biochem. 2016, 40, 1578–1590. [Google Scholar] [CrossRef]
- Mudaliar, H.; Pollock, C.; Ma, J.; Wu, H.; Chadban, S.; Panchapakesan, U. The role of TLR2 and 4-mediated inflammatory pathways in endothelial cells exposed to high glucose. PLoS ONE 2014, 9, e108844. [Google Scholar] [CrossRef]
- Qiu, Z.; Lei, S.; Zhao, B.; Wu, Y.; Su, W.; Liu, M.; Meng, Q.; Zhou, B.; Leng, Y.; Xia, Z.-y. NLRP3 inflammasome activation-mediated pyroptosis aggravates myocardial ischemia/reperfusion injury in diabetic rats. Oxid. Med. Cell. Longev. 2017, 2017, 9743280. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, W.; He, K.; Bai, L.; Miao, Y.; Liu, B.; Zhang, X.; Jin, S.; Wu, Y. Hydrogen sulfide alleviates lipopolysaccharide-induced myocardial injury through TLR4-NLRP3 pathway. Physiol. Res. 2023, 72, 15. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, P.; Sun, L. Exogenous hydrogen sulfide mitigates NLRP3 inflammasome-mediated inflammation through promoting autophagy via the AMPK-mTOR pathway. Biol. Open 2019, 8, bio043653. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Ding, Y.; Zhou, W.; Tao, L.; Lu, P.; Wang, Y.; Hu, R. Nuclear factor E2-related factor-2 negatively regulates NLRP3 inflammasome activity by inhibiting reactive oxygen species-induced NLRP3 priming. Antioxid. Redox Signal. 2017, 26, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Teng, X.; Jin, S.; Dong, J.; Guo, Q.; Tian, D.; Wu, Y. Hydrogen sulfide improves endothelial dysfunction by inhibiting the vicious cycle of NLRP3 inflammasome and oxidative stress in spontaneously hypertensive rats. J. Hypertens. 2019, 37, 1633–1643. [Google Scholar] [CrossRef]
- Su, Y.; Wang, Y.; Liu, M.; Chen, H. Hydrogen sulfide attenuates renal I/R-induced activation of the inflammatory response and apoptosis via regulating Nrf2-mediated NLRP3 signaling pathway inhibition. Mol. Med. Rep. 2021, 24, 518. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Jong, W.M.; Eerbeek, O.; Koeman, A.; Pulskens, W.P.; Butter, L.M.; Leemans, J.C.; Hollmann, M.W. Deletion of the innate immune NLRP3 receptor abolishes cardiac ischemic preconditioning and is associated with decreased Il-6/STAT3 signaling. PLoS ONE 2012, 7, e40643. [Google Scholar] [CrossRef] [PubMed]
- Sandanger, Ø.; Gao, E.; Ranheim, T.; Bliksøen, M.; Kaasbøll, O.; Alfsnes, K.; Nymo, S.H.; Rashidi, A.; Ohm, I.; Attramadal, H. NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem. Biophys. Res. Commun. 2016, 469, 1012–1020. [Google Scholar] [CrossRef]
- Hollmann, M.W.; Zuurbier, C.J. Nlrp3 plays no role in acute cardiac infarction due to low cardiac expression. Int. J. Cardiol. 2014, 177, 41–43. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payne, F.M.; Dabb, A.R.; Harrison, J.C.; Sammut, I.A. Inhibitors of NLRP3 Inflammasome Formation: A Cardioprotective Role for the Gasotransmitters Carbon Monoxide, Nitric Oxide, and Hydrogen Sulphide in Acute Myocardial Infarction. Int. J. Mol. Sci. 2024, 25, 9247. https://doi.org/10.3390/ijms25179247
Payne FM, Dabb AR, Harrison JC, Sammut IA. Inhibitors of NLRP3 Inflammasome Formation: A Cardioprotective Role for the Gasotransmitters Carbon Monoxide, Nitric Oxide, and Hydrogen Sulphide in Acute Myocardial Infarction. International Journal of Molecular Sciences. 2024; 25(17):9247. https://doi.org/10.3390/ijms25179247
Chicago/Turabian StylePayne, Fergus M., Alisha R. Dabb, Joanne C. Harrison, and Ivan A. Sammut. 2024. "Inhibitors of NLRP3 Inflammasome Formation: A Cardioprotective Role for the Gasotransmitters Carbon Monoxide, Nitric Oxide, and Hydrogen Sulphide in Acute Myocardial Infarction" International Journal of Molecular Sciences 25, no. 17: 9247. https://doi.org/10.3390/ijms25179247