
Crucial Interactions between Altered Plasma Trace Elements and Fatty Acids Unbalance Ratio to Management of Systemic Arterial Hypertension in Diabetic Patients: Focus on Endothelial Dysfunction


Abstract
:
1. Introduction
2. Results
2.1. Clinical Characterization According Cardiometabolic Syndrome of Cohort Study
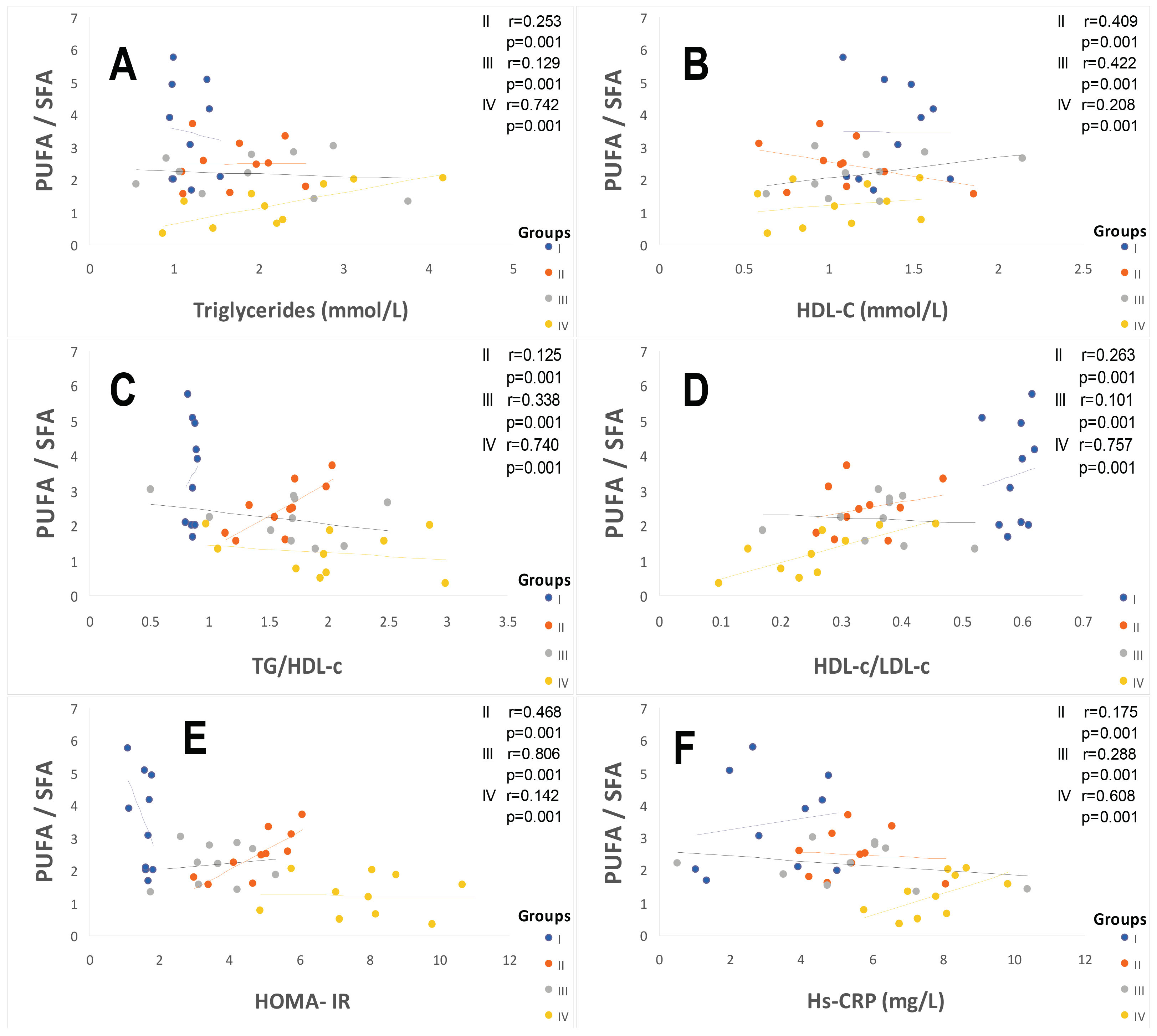
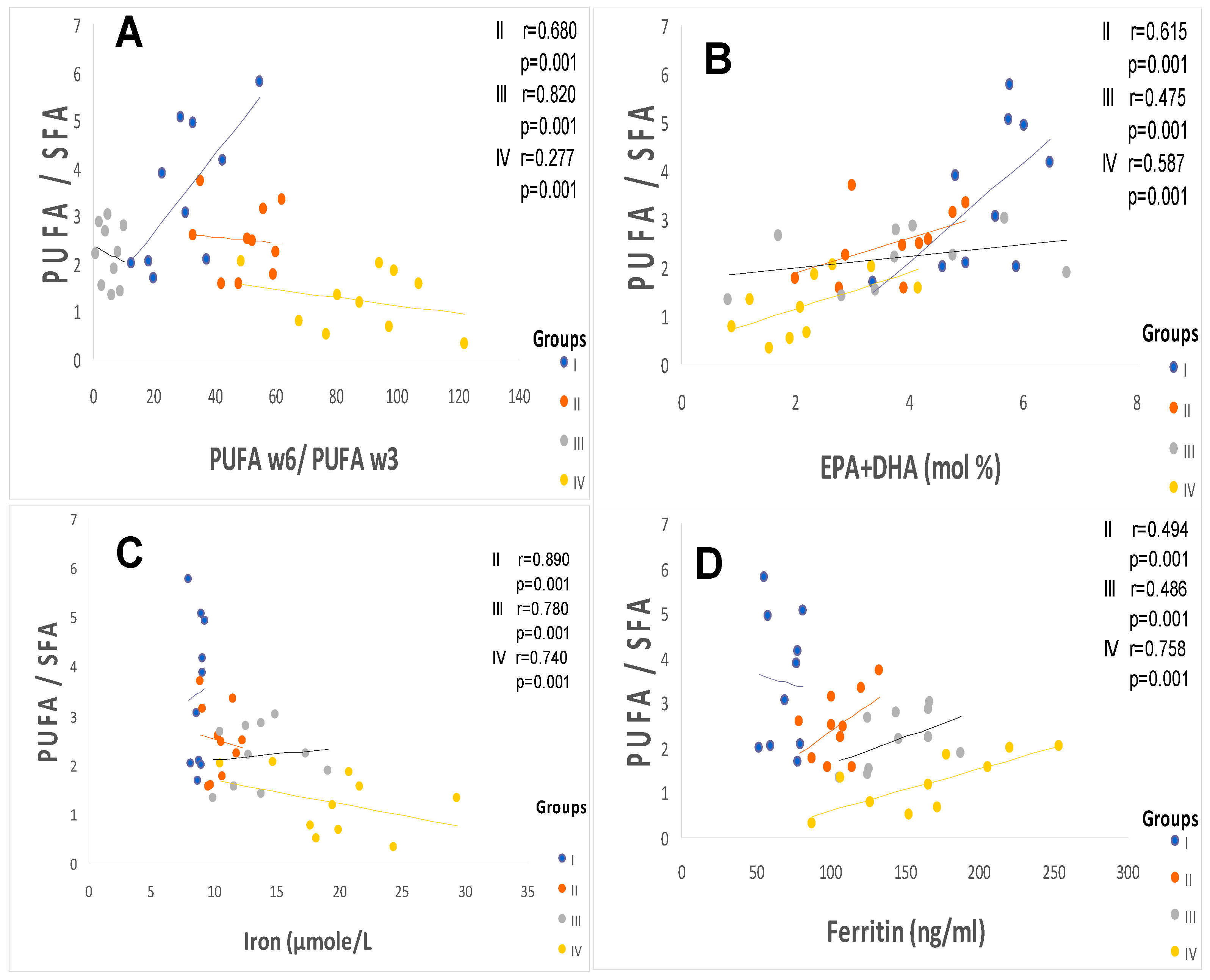
2.2. Plasma Fatty Acids Profile
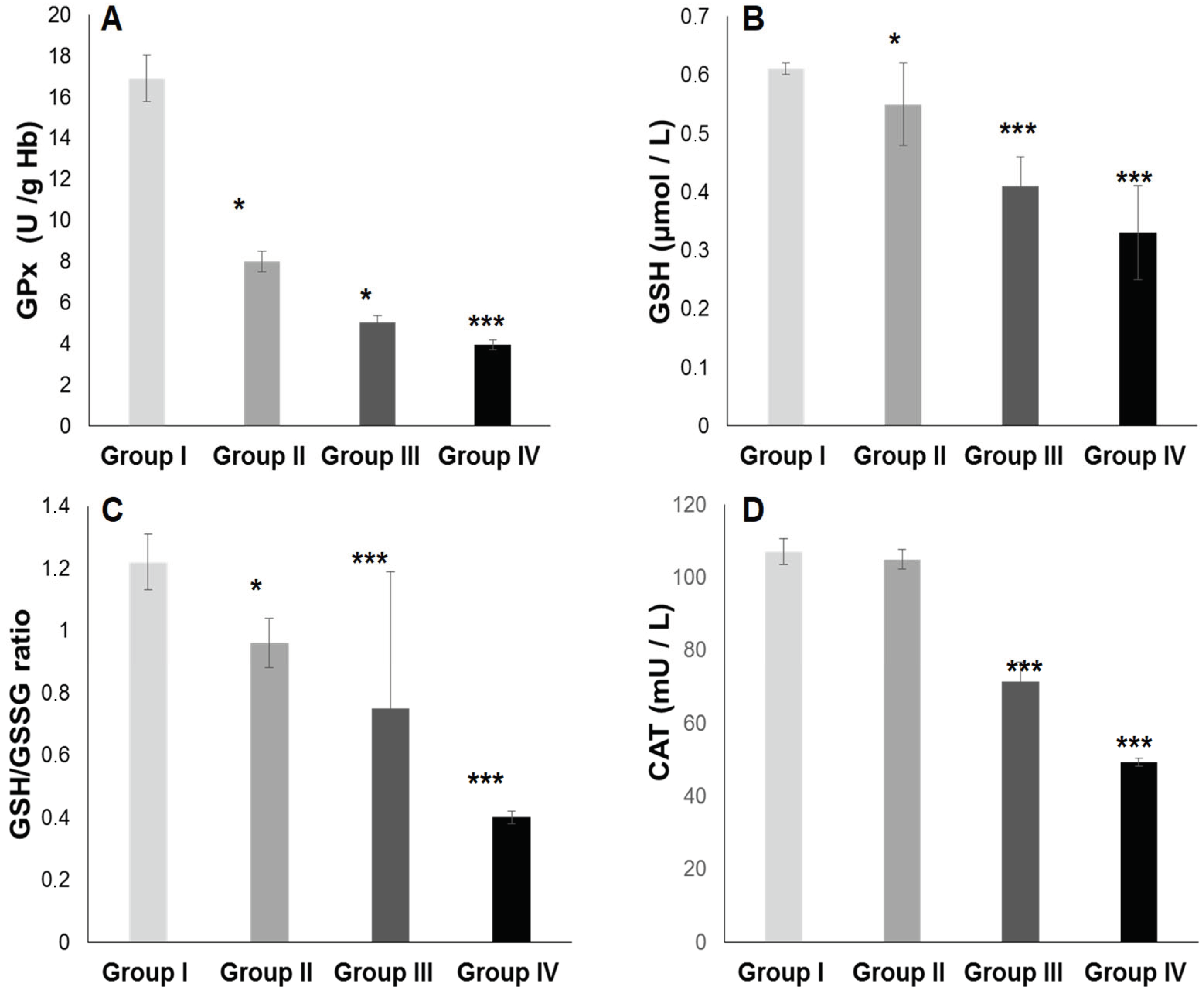
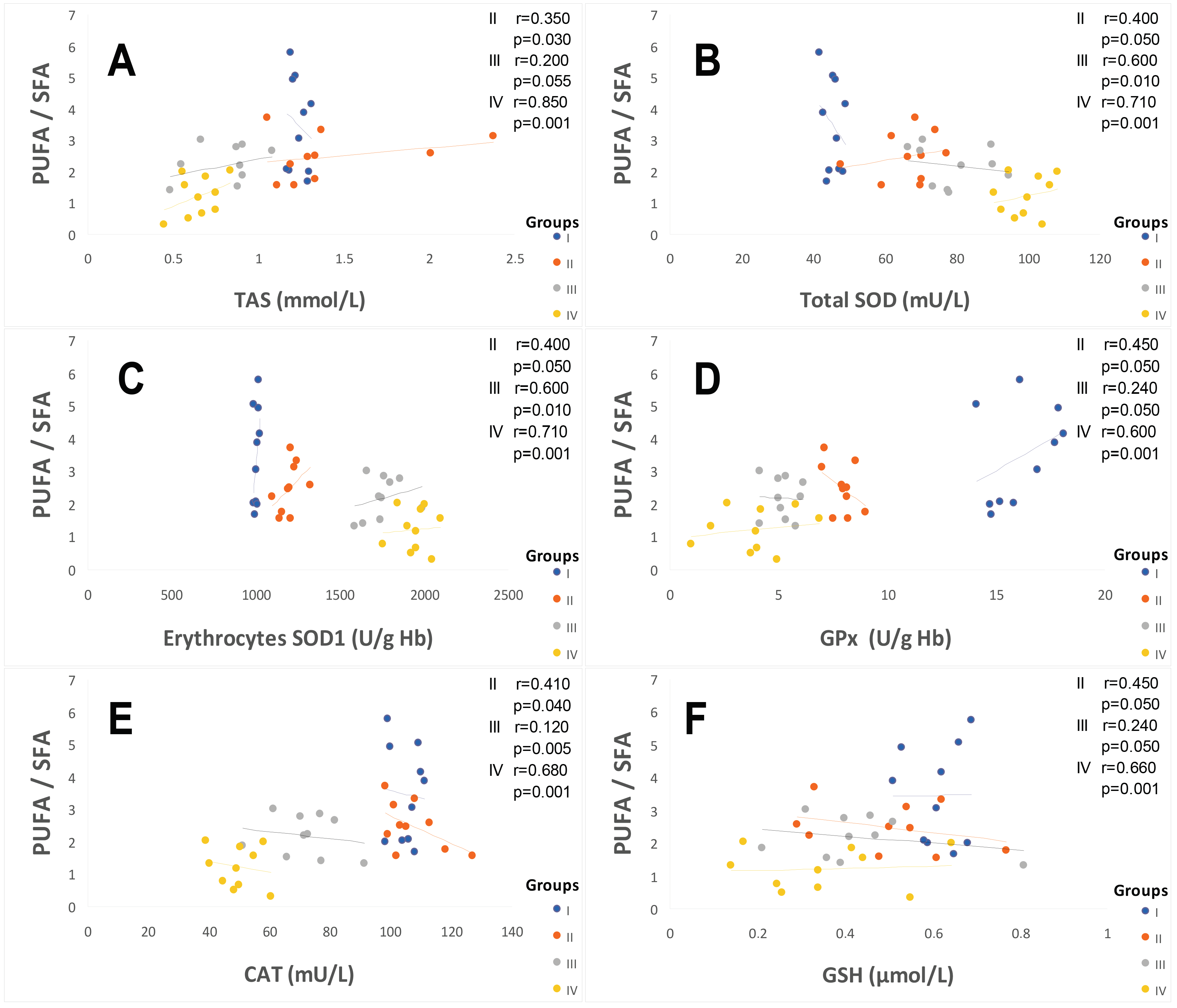
2.3. Oxidative Stress Status
2.3.1. Total Plasma Antioxidant Activity (TPAA) and Plasma Antioxidant Enzymatic Profile
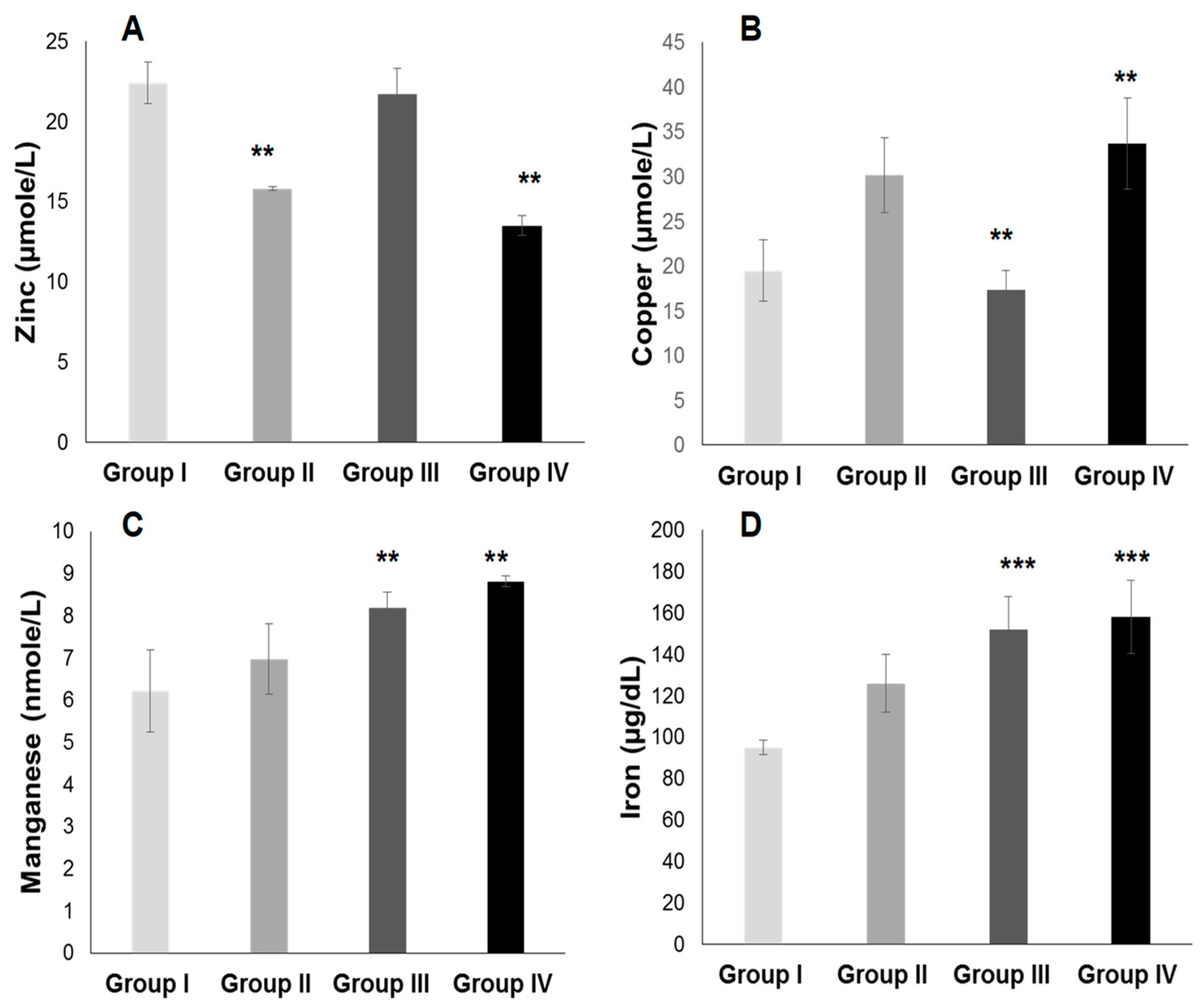
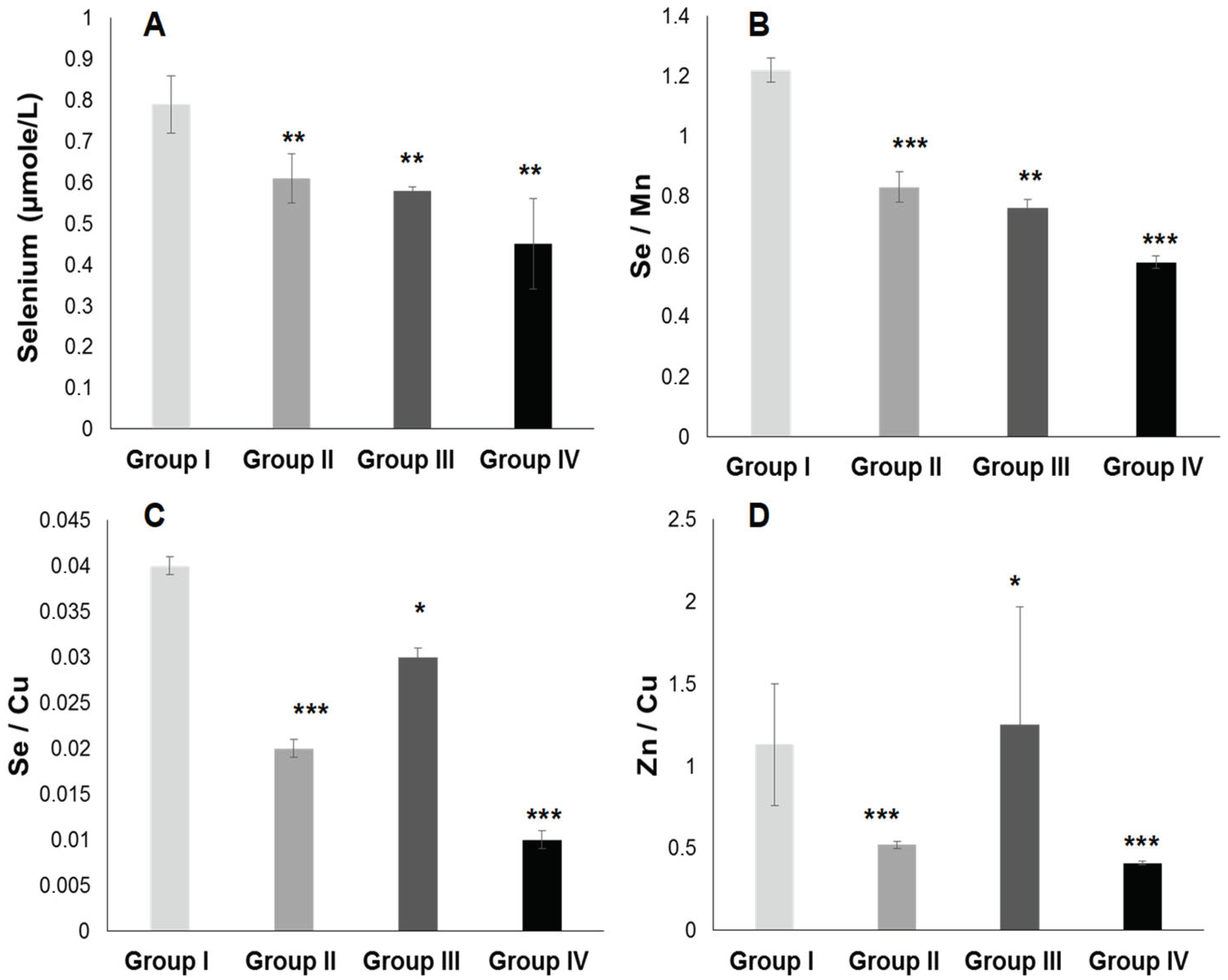
2.3.2. Plasma Antioxidant Trace Elements (PATE) Profile
3. Discussion
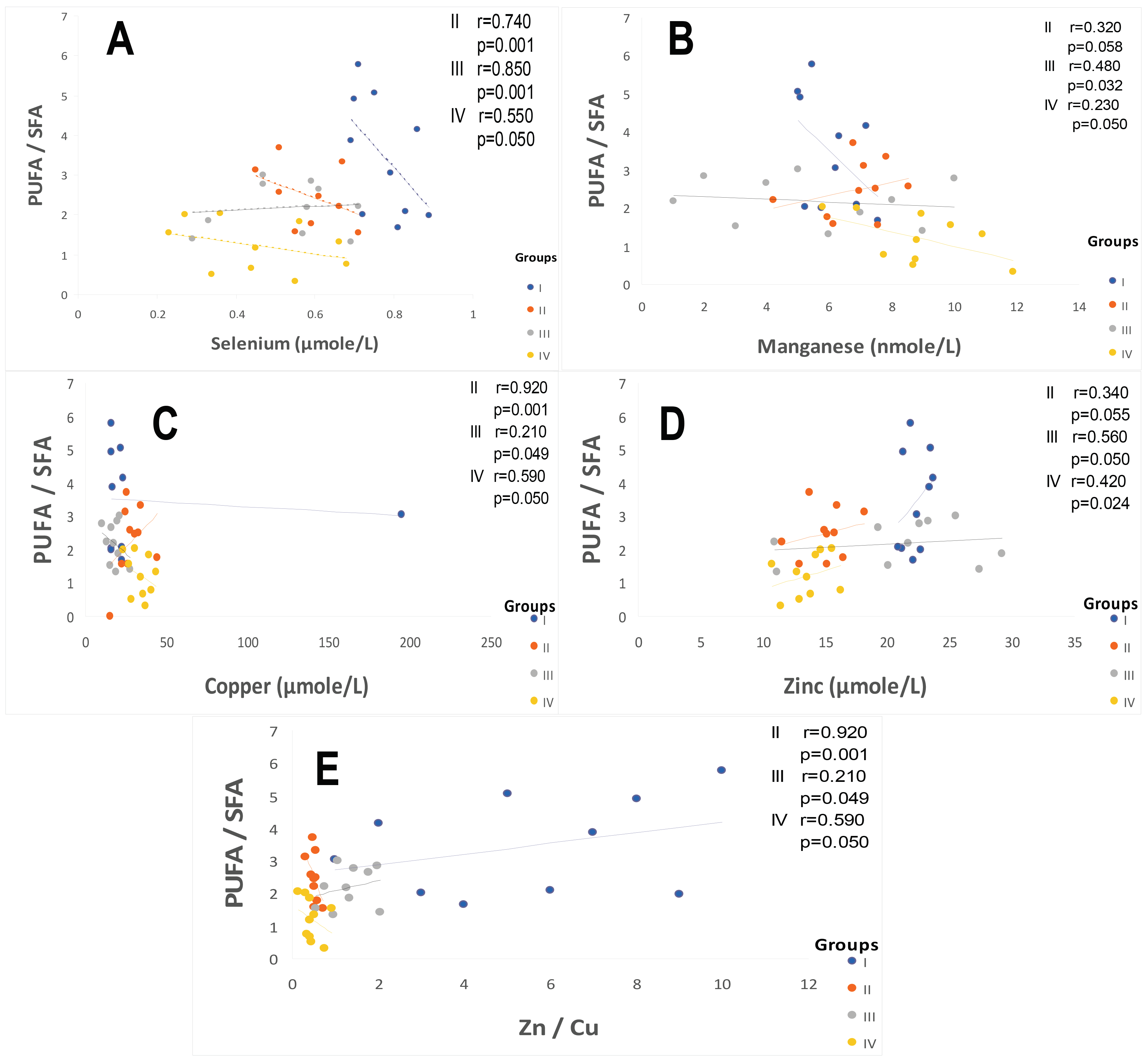
3.1. The First Major Point Is Linked to the ATE–Fatty Acids–Lipid Disorders
3.2. The Second Point Is Linked between ATE–Fatty Acids–Oxidative Stress (OxS)
3.3. The Third Crucial Point Is Related to the ATE–Fatty Acids–Atherothroboembolic Risk
3.4. The Specific Point Is Linked to Relationship between Insulin Resistance/Hypertension/Hyperinsulinism in Hypertensive Patient without Diabetes
3.5. The Particular Point Is Related to Relationship between Sex/Gender and Cardiometabolic Syndrome–Hypertension–Type 2 Diabetes Mellitus (T2DM)
3.6. The Important Point Is Linked to Relationship between Hs-CRP-Systemic Inflammation and Hypertension with or without Type 2 Diabetes Mellitus
3.7. The Prominent Point Is Related to Potential Mechanism and Clinical Implications of SOD Activity in Diabetic and Hypertensive Patients with or without Type 2 Diabetes Mellitus
4. Materials and Methods
4.1. Informed Consent Statement and Ethical Considerations
4.2. Participants and Clinical Protocol Design
4.3. Cardiometabolic Syndrome (CMS) Screening
4.4. Classification and Diagnosis Criteria of Hypertension
4.5. Classification and Diagnosis Criteria of Diabetes Mellitus
4.6. Plasma Samples and Biochemical Analysis
4.7. Plasma Fatty Acids Extraction and Assay
4.8. Trace Elements Determination and Assessment Methods
4.9. Plasma Oxidative Stress Biomarkers and Analytical Process
4.9.1. Total Blood Antioxidant Status (TAS), Plasma Thiobarbituric Acid Reactive Substances (TBARS), and Plasma Malondialdehyde (MDA) Levels Quantification
4.9.2. SOD, GPx, Catalase Activities, and Glutathione Levels Determination
4.10. Atherothromboembolic Risk Assessment
4.11. Statistical Analysis
5. Conclusions
6. Limitations of This Study
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: A pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 2021, 398, 957–980. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Buckley, J.; Ali, M.K.; Davies, J.; Flood, D.; Mehta, R.; Griffiths, B.; Lim, L.L.; Manne-Goehler, J.; Pearson-Stuttard, J.; et al. Improving health outcomes of people with diabetes: Target setting for the WHO Global Diabetes Compact. Lancet 2023, 401, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Relationships among insulin resistance, type 2 diabetes, essential hypertension, and cardiovascular disease: Similarities and differences. J. Clin. Hypertens. 2011, 13, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Chew, N.W.S.; Ng, C.H.; Tan, D.J.H.; Kong, G.; Lin, C.; Chin, Y.H.; Lim, W.H.; Huang, D.Q.; Quek, J.; Fu, C.E.; et al. The global burden of metabolic disease: Data from 2000 to 2019. Cell Metab. 2023, 35, 414–428.e3. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo-and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Yu, Y.; Lyons, T.J. A lethal tetrad in diabetes: Hyperglycemia, dyslipidemia, oxidative stress, and endothelial dysfunction. Am. J. Med. Sci. 2005, 330, 227–232. [Google Scholar] [CrossRef]
- Gouaref, I.; Bouazza, A.; AitAbderrhmane, S.; Koceir, E.A. Lipid Profile Modulates Cardiometabolic Risk Biomarkers Including Hypertension in People with Type-2 Diabetes: A Focus on Unbalanced Ratio of Plasma Polyunsaturated/Saturated Fatty Acids. Molecules 2020, 25, 4315. [Google Scholar] [CrossRef]
- Arashi, H.; Yamaguchi, J.; Kawada-Watanabe, E.; Koyanagi, R.; Sekiguchi, H.; Mori, F.; Haruta, S.; Ishii, Y.; Murasaki, S.; Suzuki, K.; et al. Polyunsaturated Fatty Acid Impact on Clinical Outcomes in Acute Coronary Syndrome Patients With Dyslipidemia: Subanalysis of HIJ-PROPE.R. J. Am. Heart Assoc. 2019, 8, e012953. [Google Scholar] [CrossRef]
- Nozue, T.; Michishita, I. Statin treatment alters serum n-3 to n-6 polyunsaturated fatty acids ratio in patients with dyslipidemia. Lipids Health Dis. 2015, 14, 67. [Google Scholar] [CrossRef]
- Kim, J.A.; Montagnani, M.; Chandrasekran, S.; Quon, M.J. Role of lipotoxicity in endothelial dysfunction. Heart Fail. Clin. 2012, 8, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Gou, R.; Gou, Y.; Qin, J.; Luo, T.; Gou, Q.; He, K.; Xiao, S.; Li, R.; Li, T.; Xiao, J.; et al. Association of dietary intake of saturated fatty acids with hypertension: 1999–2018 National Health and Nutrition Examination Survey. Front. Nutr. 2022, 9, 1006247. [Google Scholar] [CrossRef] [PubMed]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle: Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Rostoff, P.; Siniarski, A.; Mostowik, M.; Golebiowska-Wiatrak, R.; Nessler, J.; Undas, A.; Gajos, G. Relationship between polyunsaturated fatty acid composition in serum phospholipids, systemic low-grade inflammation, and glycemic control in patients with type 2 diabetes and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2018, 17, 29. [Google Scholar] [CrossRef]
- Wu, J.H.; Lemaitre, R.N.; King, I.B.; Song, X.; Psaty, B.M.; Siscovick, D.S.; Moza_arian, D. Circulating omega-6 polyunsaturated fatty acids and total and cause-specific mortality: The Cardiovascular Health Study. Circulation 2014, 130, 1245–1253. [Google Scholar] [CrossRef]
- Steffen, B.T.; Steen, L.M.; Zhou, X.; Ouyang, P.; Weir, N.L.; Tsai, M.Y. n-3 Fatty acids attenuate the risk of diabetes associated with elevated serum non esterified fatty acids: The multi-ethnic study of atherosclerosis. Diabetes Care 2015, 38, 575–580. [Google Scholar] [CrossRef]
- Colussi, G.; Catena, C.; Mos, L.; Sechi, L.A. The Metabolic Syndrome and the Membrane Content of Polyunsaturated Fatty Acids in Hypertensive Patients. Metab. Syndr. Relat. Disord. 2015, 13, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Salter, A. Saturated fatty acids and coronary heart disease risk: The debate goes on. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 97–102. [Google Scholar] [CrossRef]
- Xie, W.; Zhai, Z.; Yang, Y.; Kuang, T.; Wang, C. Free fatty acids inhibit TM-EPCR expression through JNK pathway: An implication for the development of the prothrombotic state in metabolic syndrome. J. Thromb. Thrombolysis 2012, 34, 468–474. [Google Scholar] [CrossRef]
- Joy, N.G.; Perkins, J.M.; Mikeladze, M.; Younk, L.; Tate, D.B.; Davis, S.N. Comparative effects of acute hypoglycemia and hyperglycemia on pro-atherothrombotic biomarkers and endothelial function in non-diabetic humans. J. Diabetes Complicat. 2016, 30, 1275–1281. [Google Scholar] [CrossRef]
- Felmeden, D.C.; Lip, G.Y. Endothelial function and its assessment. Expert Opin. Investig. Drugs 2005, 14, 1319–1336. [Google Scholar] [CrossRef]
- Millan-Orge, M.; Torres-Peña, J.D.; Arenas-Larriva, A.; Quintana-Navarro, G.M.; Peña-Orihuela, P.; Alcala-Diaz, J.F.; Luque, R.M.; Rodriguez-Cantalejo, F.; Katsiki, N.; Lopez-Miranda, J.; et al. Influence of dietary intervention on microvascular endothelial function in coronary patients and atherothrombotic risk of recurrence. Sci. Rep. 2021, 11, 20301. [Google Scholar] [CrossRef] [PubMed]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; Yazbi, A.E.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci 2022, 27, 105. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Romano, M.; Basarali, M.K.; Elzagallaai, A.; Karaman, M.; Demir, Z.; Demir, M.F.; Akcay, F.; Seyrek, M.; Haksever, N.; et al. The Effect of Corrected Inflammation, Oxidative Stress and Endothelial Dysfunction on Fmd Levels in Patients with Selected Chronic Diseases: A Quasi-Experimental Study. Sci. Rep. 2020, 10, 9018. [Google Scholar] [CrossRef]
- Kassab, A.; Ajmi, T.; Issaoui, M.; Chaeib, L.; Miled, A.; Hammami, M. Homocysteine enhances LDL fatty acid peroxidation, promoting microalbuminuria in type 2 diabetes. Ann. Clin. Biochem. 2008, 45, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Shreenivas, S.; Oparil, S. The role of endothelin-1 in human hypertension. Clin. Hemorheol. Microcirc. 2007, 37, 157–178. [Google Scholar]
- Rahmani, E.; Samimi, M.; Ebrahimi, F.A.; Foroozanfard, F.; Ahmadi, S.; Rahimi, M.; Jamilian, M.; Aghadavod, E.; Bahmani, F.; Taghizadeh, M.; et al. The effects of omega-3 fatty acids and vitamin E co-supplementation on gene expression of lipoprotein(a) and oxidized low-density lipoprotein, lipid profiles and biomarkers of oxidative stress in patients with polycystic ovary syndrome. Mol. Cell Endocrinol. 2017, 439, 247–255. [Google Scholar] [CrossRef]
- Amponsah-Offeh, M.; Diaba-Nuhoho, P.; Speier, S.; Morawietz, H. Oxidative Stress, Antioxidants and Hypertension. Antioxidants 2023, 12, 281. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can. J. Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef]
- Appel, L.J.; Frohlich, E.D.; Hall, J.E.; Pearson, T.A.; Sacco, R.L.; Seals, D.R.; Sacks, F.M.; Smith, S.C., Jr.; Vafiadis, D.K.; Van Horn, L.V. The importance of population-wide sodium reduction as a means to prevent cardiovascular disease and stroke: A call to action from the American Heart Association. Circulation 2011, 123, 1138–1143. [Google Scholar] [CrossRef]
- He, F.J.; MacGregor, G.A. Salt, blood pressure and the renin-angiotensin system. J. Renin Angiotensin Aldosterone Syst. 2003, 4, 11–16. [Google Scholar] [CrossRef]
- Perez, V.; Chang, E.T. Sodium-to-potassium ratio and blood pressure, hypertension, and related factors. Adv. Nutr. 2014, 5, 712–741. [Google Scholar] [CrossRef]
- Baqar, S.; Michalopoulos, A.; Jerums, G.; Ekinci, E.I. Dietary sodium and potassium intake in people with diabetes: Are guidelines being met? Nutr. Diabetes. 2020, 10, 23. [Google Scholar] [CrossRef]
- Mendis, S.; Davis, S.; Norrving, B. Organizational update: The world health organization global status report on noncommunicable diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke 2015, 46, e121–e122. [Google Scholar] [CrossRef]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R., 3rd; Simons-Morton, D.G.; et al. DASH-Sodium Collaborative Research Group. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef]
- Little, R.; Ellison, D.H. Modifying Dietary Sodium and Potassium Intake: An End to the ‘Salt Wars’? Hypertension 2024, 81, 415–425. [Google Scholar] [CrossRef]
- Cormick, G.; Ciapponi, A.; Cafferata, M.L.; Cormick, M.S.; Belizán, J.M. Calcium supplementation for prevention of primary hypertension. Cochrane Database Syst Rev. 2022, 1, CD010037. [Google Scholar] [PubMed]
- Gouaref, I.; Bellahsene, Z.; Zekri, S.; Alamir, B.; Koceir, E.A. The link between trace elements and metabolic syndrome/oxidative stress in essential hypertension with or without type 2 diabetes. Ann. Biol. Clin. 2016, 74, 233–243. [Google Scholar] [CrossRef]
- Loyke, H.F. Effects of Elements in Human Blood Pressure Control. Biol. Trace Elem.Res. 2002, 85, 193–209. [Google Scholar] [CrossRef]
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Banning, A.; Schnurr, K. Selenium-dependent enzymes in endothelial cell function. Antioxid. Redox Signal. 2003, 5, 205–215. [Google Scholar] [CrossRef]
- Sakhaei, F.; Keshvari, M.; Asgary, S.; Salehizadeh, L.; Rastqar, A.; Samsam-Shariat, S.Z. Enzymatic antioxidant system and endothelial function in patients with metabolic syndrome. ARYA Atheroscler. 2020, 16, 94–101. [Google Scholar]
- Bastola, M.M.; Locatis, C.; Maisiak, R.; Fontelo, P. Selenium, copper, zinc and hypertension: An analysis of the National Health and Nutrition Examination Survey (2011–2016). BMC Cardiovasc. Disord. 2020, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Stawarska, A.; Czerwonka, M.; Wyrębiak, R.; Wrzesień, R.; Bobrowska-Korczak, B. Zinc affects cholesterol oxidation products and fatty acids composition in rats’ serum. Nutrients 2021, 13, 1563. [Google Scholar] [CrossRef] [PubMed]
- Kitala, K.; Tanski, D.; Godlewski, J.; Krajewska-Włodarczyk, M.; Gromadziński, L.; Majewski, M. Copper and Zinc Particles as Regulators of Cardiovascular System Function-A Review. Nutrients 2023, 15, 3040. [Google Scholar] [CrossRef]
- Hernandez, M.C.; Rojas, P.; Carrasco, F.; Basfifer, K.; Codoce, J.; Inostroza, J.; Ruz, M. Zinc supplementation reduces free fatty acid concentration in patients with type 2 diabetes. Rev. Chil. Nutr. 2020, 47, 1000–1008. [Google Scholar]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 532–542. [Google Scholar] [CrossRef]
- Kuruppu, D.; Hendrie, H.C.; Yang, L.; Gao, S. Selenium levels and hypertension: A systematic review of the literature. Public Health Nutr. 2014, 17, 1342–1352. [Google Scholar] [CrossRef]
- Tubek, S. Zinc balance normalization: An important mechanism of angiotensin-converting enzyme inhibitors and other drugs decreasing the activity of the rennin-angiotensin-aldosterone system. Biol. Trace Elem. Res. 2007, 115, 223–226. [Google Scholar] [CrossRef]
- Tang, Z.; Gasperkova, D.; Xu, J.; Baillie, R.; Lee, J.; Clarke, S. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J. Nutr. 2000, 130, 2915–2921. [Google Scholar] [CrossRef]
- Huster, D.; Purnat, T.D.; Burkhead, J.L.; Ralle, M.; Fiehn, O.; Stuckert, F.; Olson, N.E.; Teupser, D.; Lutsenko, S. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 2007, 282, 8343–8355. [Google Scholar] [CrossRef]
- Morrell, A.; Tallino, S.; Yu, L.; Burkhead, J.L. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life 2017, 69, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Stelmańska, E. Regulation of extramitochondrial malic enzyme gene expression in lipogenic tissues. Postepy Hig. Med. Dosw. 2007, 61, 664–671. [Google Scholar]
- DiSilvestro, R.A.; Joseph, E.L.; Zhang, W.; Raimo, A.E.; Kim, Y.M. A randomized trial of copper supplementation effects on blood copper enzyme activities and parameters related to cardiovascular health. Metabolism 2012, 61, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Lutsenko, S.; Washington-Hughes, C.; Ralle, M.; Schmidt, K. Copper and the brain noradrenergic system. J. Biol. Inorg. Chem. 2019, 24, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Geethika, M.; Eswarappa, S.M.; Mugesh, G. Manganese-Based Nanozymes: Multienzyme Redox Activity and Effect on the Nitric Oxide Produced by Endothelial Nitric Oxide Synthase. Chemistry 2018, 24, 8393–8403. [Google Scholar] [CrossRef]
- Klimis-Zacas, D.; Kalea, A. Manganese: Modulator of vascular function, structure, and metabolism. Cell Biol. Toxicol. 2008, 24, S-S130. [Google Scholar]
- Farkas, C.S. Manganese and hepatic cholesterol. N. Engl. J. Med. 1980, 302, 585. [Google Scholar]
- Lu, L.; Wang, M.; Liao, X.; Zhang, L.; Luo, X. Manganese influences the expression of fatty acid synthase and malic enzyme in cultured primary chicken hepatocytes. Br. J. Nutr. 2017, 118, 881–888. [Google Scholar] [CrossRef]
- Zhuang, X.; Pang, X.; Zhang, W.; Wu, W.; Zhao, J.; Yang, H.; Qu, W. Effects of zinc and manganese on advanced glycation end products (AGEs) formation and AGEs-mediated endothelial cell dysfunction. Life Sci. 2012, 90, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Mondola, P.; Damiano, S.; Sasso, A.; Santillo, M. The Cu, Zn Superoxide Dismutase: Not Only a Dismutase Enzyme. Front. Physiol. 2016, 7, 594. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; Mazzon, E.; Dugo, L.; Di Paola, R.; Caputi, A.P.; Salvemini, D. Superoxide: A key player in hypertension. FASEB J. 2004, 18, 94–101. [Google Scholar] [CrossRef]
- Handy, D.E.; Loscalzo, J. The role of glutathione peroxidase-1 in health and disease. Free Radic Biol Med. 2022, 188, 146–161. [Google Scholar] [CrossRef]
- Chrissobolis, S.; Didion, S.P.; Kinzenbaw, D.A.; Schrader, L.I.; Dayal, S.; Lentz, S.R.; Faraci, F.M. Glutathione peroxidase-1 plays a major role in protecting against angiotensin II- induced vascular dysfunction. Hypertension 2008, 51, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Niu, X.; Ma, X.; Wei, J. Is selenium deficiency really the cause of Keshan disease? Environ. Geochem. Health 2011, 33, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Vivoli, G.; Bergomi, M.; Borella, P.; Rovesti, S. Trace elements in hypertension. In Trace Elements in Man and Animals, 10th ed.; Roussel, A.M., Anderson, R.A., Favier, A.E., Eds.; Kluwer Academic/PlenumPublishers: New York, NY, USA, 2000; p. 581. [Google Scholar]
- Tubek, S. Role of Trace Elements in Primary Arterial Hypertension: Is Mineral Water Style or Prophylaxis? Biol. Trae Elem. Res. 2006, 114, 1–4. [Google Scholar] [CrossRef]
- Cerf, M.E. Cardiac Glucolipotoxicity and Cardiovascular Outcomes. Medicina 2018, 54, 70. [Google Scholar] [CrossRef]
- Karupaiah, T.; Tan, C.H.; Chinna, K.; Sundram, K. The chain length of dietary saturated fatty acids affects human postprandial lipemia. J. Am. Coll. Nutr. 2011, 30, 511–521. [Google Scholar] [CrossRef]
- Khajeh, M.; Hassanizadeh, S.; Pourteymour Fard Tabrizi, F.; Hassanizadeh, R.; Vajdi, M.; Askari, G. Effect of Zinc Supplementation on Lipid Profile and Body Composition in Patients with Type 2 Diabetes Mellitus: A GRADE-Assessed Systematic Review and Dose-Response Meta-analysis. Biol. Trace Elem. Res. 2024. [Google Scholar] [CrossRef]
- Kröger, J.; Schulze, M.B. Recent insights into the relation of Δ5 desaturase and Δ6 desaturase activity to the development of type 2 diabetes. Curr. Opin. Lipidol. 2012, 23, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Knez, M.; Stangoulis, J.C.R.; Glibetic, M.; Tako, E. The Linoleic Acid: Dihomo-γ-Linolenic Acid Ratio (LA: DGLA)-An Emerging Biomarker of Zn Status. Nutrients 2017, 9, 825. [Google Scholar] [CrossRef] [PubMed]
- Takic, M.; Zekovic, M.; Terzic, B.; Stojsavljevic, A.; Mijuskovic, M.; Radjen, S.; Ristic-Medic, D. Zinc Deficiency, Plasma Fatty Acid Profile and Desaturase Activities in Hemodialysis Patients: Is Supplementation Necessary? Front. Nutr. 2021, 8, 700450. [Google Scholar] [CrossRef] [PubMed]
- Stiles, L.I.; Ferrao, K.; Mehta, K.J. Role of zinc in health and disease. Clin. Exp. Med. 2024, 24, 38. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Kothapalli, K.S.; Brenna, J.T. Desaturase and elongase-limiting endogenous long-chain polyunsaturated fatty acid biosynthesis. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 103–110. [Google Scholar] [CrossRef]
- Knez, M.; Boy, E. Existing knowledge on Zn status biomarkers (1963–2021) with a particular focus on FADS1 and FADS2 diagnostic performance and recommendations for further research. Front. Nutr. 2023, 9, 1057156. [Google Scholar] [CrossRef]
- Mansoori, A.; Ghiasi Hafezi, S.; Ansari, A.; Arab Yousefabadi, S.; Kolahi Ahari, R.; Darroudi, S.; Eshaghnezhad, M.; Ferns, G.; Ghayour-Mobarhan, M.; Esmaily, H.; et al. Serum Zinc and Copper Concentrations and Dyslipidemia as Risk Factors of Cardiovascular Disease in Adults: Data Mining Techniques. Biol. Trace Elem. Res. 2024. [Google Scholar] [CrossRef]
- Mirończuk, A.; Kapica-Topczewska, K.; Socha, K.; Soroczyńska, J.; Jamiołkowski, J.; Kułakowska, A.; Kochanowicz, J. Selenium, Copper, Zinc Concentrations and Cu/Zn, Cu/Se Molar Ratios in the Serum of Patients with Acute Ischemic Stroke in North eastern Poland—A New Insight into Stroke Pathophysiology. Nutrients 2021, 13, 2139. [Google Scholar] [CrossRef]
- Hu, X.F.; Sharin, T.; Chan, H.M. Dietary and blood selenium are inversely associated with the prevalence of stroke among Inuit in Canada. J. Trace Elem. Med. Biol. Organ. Soc. Miner. Trace Elem. (GMS) 2017, 44, 322–330. [Google Scholar] [CrossRef]
- Xie, C.; Xian, J.; Zeng, M.; Cai, Z.; Li, S.; Zhao, Y.; Shi, Z. Regional Difference in the Association between the Trajectory of Selenium Intake and Hypertension: A 20-Year Cohort Study. Nutrients 2021, 13, 1501. [Google Scholar] [CrossRef]
- Franco, C.; Sciatti, E.; Favero, G.; Bonomini, F.; Vizzardi, E.; Rezzani, R. Essential Hypertension and Oxidative Stress: Novel Future Perspectives. Int. J. Mol. Sci. 2022, 23, 14489. [Google Scholar] [CrossRef]
- Griendling, K.K.; Camargo, L.L.; Rios, F.J.; Alves-Lopes, R.; Montezano, A.C.; Touyz, R.M. Oxidative Stress and Hypertension. Circ. Res. 2021, 128, 993–1020. [Google Scholar] [CrossRef]
- Sinha, N.; Dabla, P.K. Oxidative stress and antioxidants in hypertension—A current review. Curr. Hypertens. Rev. 2015, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Fitsanakis, V.A.; Zhang, N.; Garcia, S.; Aschner, M. Manganese (Mn) and Iron (Fe): Interdependency of Transport and Regulation. Neurotox. Res. 2010, 18, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Jouihan, H.A.; Cobine, P.A.; Cooksey, R.C.; Hoagland, E.A.; Boudina, S.; Abel, E.D.; Winge, D.R.; McClain, D.A. Iron-mediated inhibition of mitochondrial manganese uptake mediates mitochondrial dysfunction in a mouse model of hemochromatosis. Mol. Med. 2008, 14, 98–108. [Google Scholar] [CrossRef]
- Chen, H.; Cui, Z.; Lu, W.; Wang, P.; Wang, J.; Zhou, Z.; Zhang, N.; Wang, Z.; Lin, T.; Song, Y.; et al. Association between serum manganese levels and diabetes in Chinese adults with hypertension. J. Clin. Hypertens 2022, 24, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Rauhala, P.; Chiueh, C.C. Effects of atypical antioxidative agents, S-nitroso glutathione and manganese, on brain lipid peroxidation induced by iron leaking from tissue disruption. Ann. N. Y. Acad. Sci. 2000, 899, 238–254. [Google Scholar] [CrossRef] [PubMed]
- Friederich, M.; Hansell, P.; Palm, F. Diabetes, oxidative stress, nitric oxide and mitochondria function. Curr. Diabetes Rev. 2009, 5, 120–144. [Google Scholar] [CrossRef]
- Malecki, E.A.; Greger, J.L. Manganese protects against heart mitochondrial lipid peroxidation in rats fed high levels of polyunsaturated fatty acids. J. Nutr. 1996, 126, 27–33. [Google Scholar] [CrossRef]
- Ekmekcioglu, C.; Prohaska, C.; Pomazal, K.; Steffan, I.; Schernthaner, G.; Marktl, X. Concentrations of seven trace elements in different hematological matrices in patients with type 2 diabetes as compared to healthy controls. Biol. Trace Elem. Res. 2001, 79, 205–219. [Google Scholar] [CrossRef]
- Leverve, X.M.; Guigas, B.; Detaille, D.; Batandier, C.; Koceir, E.A.; Chauvin, C.; Fontaine, E.; Wiernsperger, N.F. Mitochondrial metabolism and type-2 diabetes: A specific target of metformin. Diabetes Metab. 2003, 29, 6S88–6S94. [Google Scholar] [CrossRef]
- Koh, E.S.; Kim, S.J.; Yoon, H.E.; Chung, J.H.; Chung, S.; Park, C.W.; Chang, Y.S.; Shin, S.J. Association of blood manganese level with diabetes and renal dysfunction: A cross-sectional study of the Korean general population. BMC Endocr. Disord. 2014, 14, 24. [Google Scholar] [CrossRef]
- Wiernsperger, N.; Rapin, J.R. Trace elements in glucometabolic disorders: An update. Diabetol. Metab. Syndr. 2010, 2, 70. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Meltzer, H.M.; Folmer, M.; Wang, S.; Lie, O.; Maage, A.; Mundal, H.H.; Ydersbond, T.A. Supplementary selenium influences the response to fatty acid-induced oxidative stress in humans. Biol. Trace Elem. Res. 1997, 60, 51–68. [Google Scholar] [CrossRef]
- Wen, Y.; Zhang, L.; Li, S.; Wang, T.; Jiang, K.; Zhao, L.; Zhu, Y.; Zhao, W.; Lei, X.; Sharma, M.; et al. Effect of dietary selenium intake on CVD: A retrospective cohort study based on China Health and Nutrition Survey (CHNS) data. Public Health Nutr. 2024, 27, e122. [Google Scholar] [CrossRef]
- Mattmiller, S.A.; Carlson, B.A.; Sordillo, L.M. Regulation of inflammation by selenium and selenoproteins: Impact on eicosanoid biosynthesis. J. Nutr. Sci. 2013, 2, e28. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H. Interference of selenium and selenoproteins with the insulin-regulated carbohydrate and lipid metabolism. Free Radic. Biol. Med. 2013, 65, 1538–1547. [Google Scholar] [CrossRef]
- Brown, K.M.; Arthur, J.R. Selenium, selenoproteins and human health: A review. Public Health Nutr. 2001, 4, 593–599. [Google Scholar] [CrossRef]
- Hampel, G.; Watanabe, K.; Weksler, B.B.; Jaffe, E.A. Selenium deficiency inhibits prostacyclin release and enhances production of platelet activating factor by human endothelial cells. Biochem. Biophys. Acta 1989, 1006, 151–158. [Google Scholar] [CrossRef]
- Haberland, A.; Neubert, K.; Kruse, I.; Behne, D.; Schimk, I. Consequences of long-term selenium-deficient diet on the prostacyclin and thromboxane release from rat aorta. Biol. Trace Elem. Res. 2001, 81, 71–78. [Google Scholar] [CrossRef]
- Montazerifar, F.; Hashemi, M.; Karajibani, M.; Sanadgol, H.; Dikshit, M. Evaluation of lipid peroxidation and erythrocyte glutathione peroxidase and superoxide dismutase in hemodialysis patients. Saudi J. Kidney Dis. Transpl. 2012, 23, 274–279. [Google Scholar] [PubMed]
- Pedro-Botet, J.; Covas, M.I.; Martin, S.; Rubiés-Prat, J. Decreased endogenous antioxidant enzymatic status in essential hypertension. J. Hum. Hypertens. 2000, 14, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Jun, T.; Ke-yan, F.; Catalano, M. Increased superoxide anion production in humans: A possible mechanism for the pathogenesis of hypertension. J. Hum. Hypertens. 1996, 10, 305–309. [Google Scholar]
- Kasperczyk, S.; Kasperczyk, J.; Ostałowska, A.; Zalejska-Fiolka, J.; Wielkoszyński, T.; Swietochowska, E.; Birkner, E. The role of the antioxidant enzymes in erythrocytes in the development of arterial hypertension among humans exposed to lead. Biol. Trace Elem. Res. 2009, 130, 95–106. [Google Scholar] [CrossRef]
- Hennig, B.; Chow, K.C. Lipid peroxidation and endothelial cell injury: Implications in atherosclerosis. Free Radic Biol Med. 1988, 4, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.T.; Zhou, L.N.; Huang, C.J.; Hua, X.; Jian, R.; Su, B.H. Selenium inhibits high glucose—and high insulin-induced adhesion molecule expression in vascular endothelial cells. Arch. Med. Res. 2008, 39, 373–379. [Google Scholar] [CrossRef]
- Xun, P.; Liu, K.; Morris, J.S.; Daviglus, M.L.; He, K. Longitudinal association between toenail selenium levels and measures of subclinical atherosclerosis: The CARDIA trace element study. Atherosclerosis 2010, 210, 662–667. [Google Scholar] [CrossRef]
- Rodrigo, R.; Libuy, M.; Feliú, F.; Hasson, D. Oxidative stress-related biomarkers in essential hypertension and ischemia-reperfusion myocardial damage. Dis. Markers. 2013, 35, 773–790. [Google Scholar] [CrossRef]
- Meng, H.; Ruan, J.; Chen, Y.; Yan, Z.; Liu, J.; Wang, X.; Meng, X.; Wang, J.; Zhang, Q.; Li, X.; et al. Trace Elements Open a New Direction for the Diagnosis of Atherosclerosis. Rev. Cardiovasc. Med. 2023, 24, 23. [Google Scholar] [CrossRef]
- Meng, X.L.; Wang, Y.; Wang, H.L.; Nie, H.H.; Cheng, B.J.; Cao, H.J.; Li, X.D.; Wang, S.F.; Chen, G.M.; Tao, F.B.; et al. The association between essential trace element mixture and atherosclerotic cardiovascular disease risk among Chinese community-dwelling older adults. Environ. Sci. Pollut. Res. Int. 2022, 29, 90351–90363. [Google Scholar] [CrossRef]
- Wechselberger, C.; Messner, B.; Bernhard, D. The Role of Trace Elements in Cardiovascular Diseases. Toxics. 2023, 11, 956. [Google Scholar] [CrossRef] [PubMed]
- Trpkovic, A.; Resanovic, I.; Stanimirovic, J.; Radak, D.; Mousa, S.A.; Cenic-Milosevic, D.; Jevremovic, D.; Isenovic, E.R. Oxidized low-density lipoprotein as a biomarker of cardiovascular diseases. Crit. Rev. Clin. Lab. Sci. 2015, 52, 70–85. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. [Google Scholar] [CrossRef] [PubMed]
- Seppanen, C.M.; Cho, H.; Csallany, A.S. Comparison between High-PUFA and Low-PUFA Fats on Lipid Peroxidation and LDL Oxidation. Food Nutr. Sci. 2013, 04, 572–579. [Google Scholar] [CrossRef]
- Staprans, I.; Pan, X.M.; Rapp, J.H.; Feingold, K.R. The role of dietary oxidized cholesterol and oxidized fatty acids in the development of atherosclerosis. Mol. Nutr. Food Res. 2005, 49, 1075–1082. [Google Scholar] [CrossRef]
- Martinelli, N.; Consoli, L.; Olivieri, O. A ‘desaturase hypothesis’ for atherosclerosis: Janus-faced enzymes in omega-6 and omega-3 polyunsaturated fatty acid metabolism. J. Nutrigenet. Nutr. 2009, 2, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Chimhashu, T.; Malan, L.; Baumgartner, J.; van Jaarsveld, P.J.; Galetti, V.; Moretti, D.; Smuts, C.M.; Zimmermann, M.B. Sensitivity of fatty acid desaturation and elongation to plasma zinc concentration: A randomised controlled trial in beninese children. Br. J. Nutr. 2018, 119, 610–619. [Google Scholar] [CrossRef]
- Sherratt, S.C.R.; Juliano, R.A.; Mason, R.P. Eicosapentaenoic acid (EPA) has optimal chain length and degree of unsaturation to inhibit oxidation of small dense LDL and membrane cholesterol domains as compared to related fatty acids in vitro. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183254. [Google Scholar] [CrossRef]
- Erkan, L.G.; Guvenc, G.; Altinbas, B.; Niaz, N.; Yalcin, M. The effects of centrally injected arachidonic acid on respiratory system: Involvement of cyclooxygenase to thromboxane signaling pathway. Respir. Physiol. Neurobiol. 2016, 225, 1–7. [Google Scholar] [CrossRef]
- Meng, H.; McClendon, C.L.; Dai, Z.; Li, K.; Zhang, X.; He, S.; Shang, E.; Liu, Y.; Lai, L. Discovery of Novel 15-Lipoxygenase Activators To Shift the Human Arachidonic Acid Metabolic Network toward Inflammation Resolution. J. Med. Chem. 2016, 59, 4202–4209. [Google Scholar] [CrossRef] [PubMed]
- Tretjakovs, P.; Kalnins, U.; Dabina, I.; Erglis, A.; Dinne, I.; Jurka, A.; Latkovskis, G.; Zvaigzne, A.; Pirags, V. Nitric oxide production and arachidonic acid metabolism in platelet mem-branes of coronary heart disease patients with and without diabetes. Med. Princ. Pract. 2003, 12, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Nabli, N.; Slimene, M.; Bouslama, A.; Omezzine, A.; Laradi, S.; Garcia, I.; Drai, J.; Barnier, E.; Boughzala, E.; Hammami, M.; et al. Arachidonate to saturated fatty acid ratio of circulating sterides and phospholipids: Can it be risk marker of coronary stenosis? A Tunisian study. Ann. Biol. Clin. 2001, 59, 743–749. [Google Scholar]
- Al-Shabrawey, M.; Elmarakby, A.; Samra, Y.; Moustafa, M.; Looney, S.W.; Maddipati, K.R.; Tawfik, A. Hyperhomocysteinemia dysregulates plasma levels of polyunsaturated fatty acids-derived eicosanoids. Life Res. 2022, 5, 14. [Google Scholar] [CrossRef]
- Wang, M.H.; Hsiao, G.; Al-Shabrawey, M. Eicosanoids and Oxidative Stress in Diabetic Retinopathy. Antioxidants 2020, 9, 520. [Google Scholar] [CrossRef]
- Zhou, F.; Chen, S. Hyperhomocysteinemia and risk of incident cognitive outcomes: An updated dose-response meta-analysis of prospective cohort studies. Ageing Res. Rev. 2019, 51, 55–66. [Google Scholar] [CrossRef]
- de Souza, A.W.; Silva, N.P.; de Carvalho, J.F.; D’Almeida, V.; Noguti, M.A.; Sato, E.I. Impact of hypertension and hyperhomocysteinemia on arterial thrombosis in primary anti phospholipid syndrome. Lupus 2007, 16, 782–787. [Google Scholar] [CrossRef]
- Koubaa, N.; Nakbi, A.; Smaoui, M.; Abid, N.; Chaaba, R.; Abid, M.; Hammami, M. Hyperhomocysteinemia and elevated ox-LDL in Tunisian type 2 diabetic patients: Role of genetic and dietary factors. Clin. Biochem. 2007, 40, 1007–1014. [Google Scholar] [CrossRef]
- Toshima, S.; Hasegawa, A.; Kurabayashi, M.; Itabe, H.; Takano, T.; Sugano, J.; Shimamura, K.; Kimura, J.; Michishita, I.; Suzuki, T.; et al. Circulating oxidized low density lipoprotein levels. A biochemical risk marker for coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2243–2247. [Google Scholar] [CrossRef]
- Holvoet, P.; Mertens, A.; Verhamme, P.; Bogaerts, K.; Beyens, G.; Verhaeghe, R.; Collen, D.; Muls, E.; Van de Werf, F. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 844–848. [Google Scholar] [CrossRef]
- Millian, N.S.; Garrow, T.A. Human betaine-homocysteine methyltransferase is a zinc metalloenzyme. Arch. Biochem. Biophys. 1998, 356, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Kirsch, H.; Domergue, F.; Abbadi, A.; Sperling, P.; Bauer, J.; Cirpus, P.; Zank, T.K.; Moreau, H.; Roscoe, T.J.; et al. Novel fatty acid elongases and their use for the reconstitution of docosahexaenoic acid biosynthesis. J. Lipid. Res. 2004, 45, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Laganà, A.S. The Link between Homocysteine and Omega-3 Polyunsaturated Fatty Acid: Critical Appraisal and Future Directions. Biomolecules. 2020, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.M.; Sapp, P.A.; Kris-Etherton, P.; Petersen, K. Effects of saturated fatty acid consumption on lipoprotein(a): A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2024, S0002-9165(24)00591-4. [Google Scholar] [CrossRef]
- Law, H.G.; Meyers, F.J.; Berglund, L.; Enkhmaa, B. Lipoprotein(a) and diet-a challenge for a role of saturated fat in cardiovascular disease risk reduction? Am. J. Clin. Nutr. 2023, 118, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Ward, N.C.; Ying, Q.; Chan, D.C.; Pang, J.; Mori, T.A.; Schultz, C.J.; Dwivedi, G.; Francis, R.J.; Watts, G.F. Improved arterial inflammation with high dose omega-3 fatty acids in patients with elevated lipoprotein(a): Selective effect of eicosapentaenoic acid? J. Clin. Lipidol. 2023, 17, 694–699. [Google Scholar] [CrossRef]
- Nakajima, K.; Yamashita, T.; Kusuhara, M.; Yonemura, A.; Ito, T.; Higashi, K.; Ayaori, M.; Ohmori, R.; Nakamura, H.; Ohsuzu, F. The susceptibility of lipoprotein (a) to copper oxidation is correlated with the susceptibility of autologous low density lipoprotein to oxidation. Clin. Biochem. 2003, 36, 113–120. [Google Scholar] [CrossRef]
- Antonicelli, R.; Testa, R.; Bonfigli, A.R.; Sirolla, C.; Pieri, C.; Marra, M.; Marcovina, S.M. Relationship between lipoprotein (a) levels, oxidative stress, and blood pressure levels in patients with essential hypertension. Clin. Exp. Med. 2001, 1, 145–150. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, W.S.; Wang, X.; Xu, L.; Yang, X.C. Palmitic Acid Increases Endothelin-1 Expression in Vascular Endothelial Cells through the Induction of Endoplasmic Reticulum Stress and Protein Kinase C Signaling. Cardiology 2018, 140, 133–140. [Google Scholar] [CrossRef]
- Yakubu, M.A.; Leffler, C.W. Regulation of ET-1 biosynthesis in cerebral microvascular endothelial cells by vasoactive agents and PK. C. Am. J. Physiol. 1999, 276, C300–C305. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; Martin-Nizard, F.; Chinetti, G.; Trottein, F.; Fruchart, J.C.; Najib, J.; Duriez, P.; Staels, B. Peroxisome proliferator-activated receptor activators inhibit thrombin-induced endothelin-1 production in human vascular endothelial cells by inhibiting the activator protein-1 signaling pathway. Circ. Res. 1999, 85, 394–402. [Google Scholar] [CrossRef]
- Baier-Bitterlich, G.; Uberall, F.; Bauer, B.; Fresser, F.; Wachter, H.; Grunicke, H.; Utermann, G.; Altman, A.; Baier, G. Protein kinase C-θ isoenzyme selective stimulation of the transcription factor complex AP-1 in T lymphocytes. Mol. Cell. Biol. 1996, 16, 1842–1850. [Google Scholar] [CrossRef]
- Christensen, M.S.; Therkelsen, K.; Møller, J.M.; Dyerberg, J.; Schmidt, E.B. n-3 fatty acids do not decrease plasma endothelin levels in healthy individuals. Scand. J. Clin. Lab. Investig. 1997, 57, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, H.; Moridaira, K.; Wada, O. Zinc deficiency further increases the enhanced expression of endothelin-1 in glomeruli of the obstructed kidney. Kidney Int. 2000, 58, 575–586. [Google Scholar] [CrossRef]
- Morand, J.; Briançon-Marjollet, A.; Lemarie, E.; Gonthier, B.; Arnaud, J.; Korichneva, I.; Godin-Ribuot, D. Zinc deficiency promotes endothelin secretion and endothelial cell migration through nuclear hypoxia-inducible factor-1 translocation. Am. J. Physiol. Cell Physiol. 2019, 317, C270–C276. [Google Scholar] [CrossRef]
- Dive, V.; Chang, C.F.; Yiotakis, A.; Sturrock, E.D. Inhibition of zinc metallopeptidases in cardiovascular disease--from unity to trinity, or duality? Curr. Pharm. Des. 2009, 15, 3606–3621. [Google Scholar] [CrossRef]
- López-Ongil, S.; Senchak, V.; Saura, M.; Zaragoza, C.; Ames, M.; Ballermann, B.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; Lowenstein, C.J. Superoxide regulation of endothelin-converting enzyme. J. Biol. Chem. 2000, 275, 26423–26427. [Google Scholar] [CrossRef]
- Lankhorst, S.; Kappers, M.H.; van Esch, J.H.; Danser, A.H.; van den Meiracker, A.H. Hypertension during vascular endothelial growth factor inhibition: Focus on nitric oxide, endothelin-1, and oxidative stress. Antioxid. Redox Signal. 2014, 20, 135–145. [Google Scholar] [CrossRef]
- González-Manchón, C.; Martín-Requero, A.; Ayuso, M.S.; Parrilla, R. Role of endogenous fatty acids in the control of hepatic gluconeogenesis. Arch. Biochem. Biophys. 1992, 292, 95–101. [Google Scholar] [CrossRef]
- Sarafidis, P.A.; Bakris, G.L. Non-esterified fatty acids and blood pressure elevation: A mechanism for hypertension in subjects with obesity/insulin resistance? J. Hum. Hypertens. 2007, 21, 12–19. [Google Scholar] [CrossRef]
- da Silva, A.A.; do Carmo, J.M.; Li, X.; Wang, Z.; Mouton, A.J.; Hall, J.E. Role of Hyperinsulinemia and Insulin Resistance in Hypertension: Metabolic Syndrome Revisited. Can. J. Cardiol. 2020, 36, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.S.; Wang, A.; Yu, H. Link between insulin resistance and hypertension: What is the evidence from evolutionary biology? Diabetol. Metab. Syndr. 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Uzu, T.; Fujii, T.; Nishimura, M.; Nakamura, S.; Inenaga, T.; Kimura, G. Role of insulin resistance in the genesis of sodium sensitivity in essential hypertension. J. Hum. Hypertens. 1999, 13, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Salvetti, A.; Brogi, G.; Di Legge, V.; Bernini, G.P. The inter-relationship between insulin resistance and hypertension. Drugs 1993, 46, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Seghieri, C.; Policardo, L.; Francesconi, P.; Seghieri, G. Gender differences in the relationship between diabetes process of care indicators and cardiovascular outcomes. Eur. J. Public Health 2016, 26, 219–224. [Google Scholar] [CrossRef]
- Sattar, N. Gender aspects in type 2 diabetes mellitus and cardiometabolic risk. Best. Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 501–507. [Google Scholar] [CrossRef]
- Lofty, M.; Adeghate, J.; Kalasz, H.; Singh, J.; Adeghate, E. Chronic Complications of Diabetes Mellitus: A Mini Review. Curr. Diabetes Rev. 2017, 13, 3–10. [Google Scholar]
- Campesi, I.; Franconi, F.; Seghieri, G.; Meloni, M. Sex-gender-related therapeutic approaches for cardiovascular complications associated with diabetes. Pharmacol. Res. 2017, 119, 195–207. [Google Scholar] [CrossRef]
- Ostman, J.; Lönnberg, G.; Arnqvist, H.J.; Blohmé, G.; Bolinder, J.; Ekbom Schnell, A.; Eriksson, J.W.; Gudbjörnsdottir, S.; Sundkvist, G.; Nyström, L. Gender differences and temporal variation in the incidence of type 1 diabetes: Results of 8012 cases in the nationwide Diabetes Incidence Study in Sweden 1983–2002. J. Intern. Med. 2008, 263, 386–394. [Google Scholar] [CrossRef]
- Liang, X.; Xing, Z.; Lai, K.; Li, X.; Gui, S.; Li, Y. Sex differences in the association between metabolic score for insulin resistance and the reversion to normoglycemia in adults with prediabetes: A cohort study. Diabetol. Metab. Syndr. 2024, 16, 183. [Google Scholar] [CrossRef]
- Ramezankhani, A.; Azizi, F.; Hadaegh, F. Gender differences in changes in metabolic syndrome status and its components and risk of cardiovascular disease: A longitudinal cohort study. Cardiovasc. Diabetol. 2022, 21, 227. [Google Scholar] [CrossRef]
- Cooper, A.J.; Gupta, S.R.; Moustafa, A.F.; Chao, A.M. Sex/Gender Differences in Obesity Prevalence, Comorbidities, and Treatment. Curr. Obes. Rep. 2021, 10, 458–466. [Google Scholar] [CrossRef]
- Kautzky-Willer, A.; Leutner, M.; Harreiter, J. Sex differences in type 2 diabetes. Diabetologia 2023, 66, 986–1002. [Google Scholar] [CrossRef]
- Connelly, P.J.; Currie, G.; Delles, C. Sex Differences in the Prevalence, Outcomes and Management of Hypertension. Curr. Hypertens. Rep. 2022, 24, 185–192. [Google Scholar] [CrossRef]
- Gerdts, E.; Sudano, I.; Brouwers, S.; Borghi, C.; Bruno, R.M.; Ceconi, C.; Cornelissen, V.; Diévart, F.; Ferrini, M.; Kahan, T.; et al. Sex differences in arterial hypertension. Eur. Heart J. 2022, 43, 4777–4788. [Google Scholar] [CrossRef] [PubMed]
- Ciarambino, T.; Crispino, P.; Leto, G.; Mastrolorenzo, E.; Para, O.; Giordano, M. Influence of Gender in Diabetes Mellitus and Its Complication. Int. J. Mol. Sci. 2022, 23, 8850. [Google Scholar] [CrossRef]
- Hage, F.G. C-reactive protein and hypertension. J. Hum. Hypertens. 2014, 28, 410–415. [Google Scholar] [CrossRef]
- Virdis, A.; Ghiadoni, L.; Plantinga, Y.; Taddei, S.; Salvetti, A. C-reactive protein and hypertension: Is there a causal relationship? Curr. Pharm. Des. 2007, 13, 1693–1698. [Google Scholar] [CrossRef]
- Schein, A.S.O.; Bock, P.M.; Massierer, D.; Gus, M.; Schaan, B.D. C-reactive protein and blood pressure variability in type 2 hypertensive diabetic patients. Blood Press. Monit. 2019, 24, 52–58. [Google Scholar] [CrossRef]
- Sesso, H.D.; Buring, J.E.; Rifai, N.; Blake, G.J.; Gaziano, J.M.; Ridker, P.M. C-reactive protein and the risk of developing hypertension. JAMA 2003, 290, 2945–2951. [Google Scholar] [CrossRef]
- Schillaci, G.; Pirro, M. C-reactive protein in hypertension: Clinical significance and predictive value. Nutr. Metab. Cardiovasc. Dis. 2006, 16, 500–508. [Google Scholar] [CrossRef]
- Mouliou, D.S. C-Reactive Protein: Pathophysiology, Diagnosis, False Test Results and a Novel Diagnostic Algorithm for Clinicians. Diseases 2023, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Emerging Risk Factors Collaboration; Kaptoge, S.; Di Angelantonio, E.; Lowe, G.; Pepys, M.B.; Thompson, S.G.; Collins, R.; Danesh, J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: An individual participant meta-analysis. Lancet 2010, 375, 132–140. [Google Scholar]
- Gomez-Marcos, M.A.; Recio-Rodríguez, J.I.; Patino-Alonso, M.C.; Agudo-Conde, C.; Gomez-Sanchez, L.; Rodriguez-Sanchez, E.; Gomez-Sanchez, M.; Martinez-Vizcaino, V.; Garcia-Ortiz, L. Relationships between high-sensitive C-reactive protein and markers of arterial stiffness in hypertensive patients. Differences by sex. BMC Cardiovasc. Disord. 2012, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Chu, J.; Hu, W.; Sun, N.; Shen, Y. Joint effect of elevated-c-reactive protein level and hypertension on new-onset stroke: A nationwide prospective cohort study of CHARL.S. Front. Public Health 2022, 10, 919506. [Google Scholar] [CrossRef]
- Liu, H.H.; Cao, Y.X.; Sun, D.; Jin, J.L.; Zhang, H.W.; Guo, Y.L.; Zhu, C.G.; Wu, N.Q.; Gao, Y.; Dong, Q.T.; et al. High-sensitivity C-reactive protein and hypertension: Combined effects on coronary severity and cardiovascular outcomes. Hypertens. Res. 2019, 42, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhang, Z.; Zhang, H.; Hao, Y.; Zhang, J.; Liu, W.; Xu, G.; Liu, X. Prognostic Value of C-Reactive Protein and Homocysteine in Large-Artery Atherosclerotic Stroke: A Prospective Observational Study. J. Stroke Cerebrovasc. Dis. 2017, 26, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Naess, H.; Nyland, H.; Idicula, T.; Waje-Andreassen, U. C-reactive protein and homocysteine predict long-term mortality in young ischemic stroke patients. J. Stroke Cerebrovasc. Dis. 2013, 22, e435–e440. [Google Scholar] [CrossRef]
- Wang, C.H.; Li, S.H.; Weisel, R.D.; Fedak, P.W.; Dumont, A.S.; Szmitko, P.; Li, R.K.; Mickle, D.A.; Verma, S. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation 2003, 107, 1783–1790. [Google Scholar] [CrossRef]
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L.; Han, C.; Li, M.; Wang, S.; Wu, D. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-kappaB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Dow, C.A.; Templeton, D.L.; Lincenberg, G.M.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Elevations in C-reactive protein and endothelin-1 system activity in humans. Life Sci. 2016, 159, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Gemici, G.; Erdim, R.; Tokay, S.; Tezcan, H.; Fak, A.S.; Oktay, A. Interaction between C-reactive protein and endothelin-1 in coronary artery disease. Cardiology 2007, 107, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Giannopoulos, S.; Kosmidou, M.; Hatzitolios, A.I.; Savopoulos, C.G.; Ziakas, A.; Karamouzis, M. Measurements of endothelin-1, C-reactive protein and fibrinogen plasma levels in patients with acute ischemic stroke. Neurol. Res. 2008, 30, 727–730. [Google Scholar] [CrossRef]
- Kaur, G.; Abdelrahman, K.; Berman, A.N.; Biery, D.W.; Shiyovich, A.; Huck, D.; Garshick, M.; Blankstein, R.; Weber, B. Lipoprotein(a): Emerging insights and therapeutics. Am. J. Prev. Cardiol. 2024, 18, 100641. [Google Scholar] [CrossRef]
- Duarte Lau, F.; Giugliano, R.P. Lipoprotein(a) and its Significance in Cardiovascular Disease: A Review. JAMA Cardiol. 2022, 7, 760–769. [Google Scholar] [CrossRef]
- Alhomoud, I.S.; Talasaz, A.; Mehta, A.; Kelly, M.S.; Sisson, E.M.; Bucheit, J.D.; Brown, R.; Dixon, D.L. Role of lipoprotein(a) in atherosclerotic cardiovascular disease: A review of current and emerging therapies. Pharmacotherapy 2023, 43, 1051–1063. [Google Scholar] [CrossRef]
- Tangvarasittichai, S.; Pingmuanglaew, P.; Tangvarasittichai, O. Association of Elevated Serum Lipoprotein(a), Inflammation, Oxidative Stress and Chronic Kidney Disease with Hypertension in Non-diabetes Hypertensive Patients. Indian J. Clin. Biochem. 2016, 31, 446–451. [Google Scholar] [CrossRef]
- Takagi, H.; Umemoto, T. Atorvastatin decreases lipoprotein(a): A meta-analysis of randomized trials. Int. J. Cardiol. 2012, 154, 183–186. [Google Scholar] [CrossRef]
- Small, A.M.; Pournamdari, A.; Melloni, G.E.M.; Scirica, B.M.; Bhatt, D.L.; Raz, I.; Braunwald, E.; Giugliano, R.P.; Sabatine, M.S.; Peloso, G.M.; et al. Lipoprotein(a), C-Reactive Protein, and Cardiovascular Risk in Primary and Secondary Prevention Populations. JAMA Cardiol. 2024, 9, 385–391. [Google Scholar] [CrossRef]
- Kono, Y.; Fridovich, I. Superoxide radical inhibits catalase. J. Biol. Chem. 1982, 257, 5751–5754. [Google Scholar] [CrossRef]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef]
- Lin, S.J.; Shyue, S.K.; Hung, Y.Y.; Chen, Y.H.; Ku, H.H.; Chen, J.W.; Tam, K.B.; Chen, Y.L. Superoxide dismutase inhibits the expression of vascular cell adhesion molecule-1 and intracellular cell adhesion molecule-1 induced by tumor necrosis factor-alpha in human endothelial cells through the JNK/p38 pathways. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 334–340. [Google Scholar] [CrossRef]
- Wynants, M.; Vengethasamy, L.; Ronisz, A.; Meyns, B.; Delcroix, M.; Quarck, R. NF-kappaB pathway is involved in CRP-induced effects on pulmonary arterial endothelial cells in chronic thromboembolic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L934–L942. [Google Scholar] [CrossRef]
- Miller, F.J., Jr.; Gutterman, D.D.; Rios, C.D.; Heistad, D.D.; Davidson, B.L. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ. Res. 1998, 82, 1298–1305. [Google Scholar] [CrossRef]
- Zhang, L.; Tu, H.; Li, Y.L. Angiotensin II enhances hyperpolarization-activated currents in rat aortic baroreceptor neurons: Involvement of superoxide. Am. J. Physiol. Cell Physiol. 2010, 298, C98–C106. [Google Scholar] [CrossRef]
- Lob, H.E.; Vinh, A.; Li, L.; Blinder, Y.; Offermanns, S.; Harrison, D.G. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension 2011, 58, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef]
- Dinić, S.; Arambašić Jovanović, J.; Uskoković, A.; Mihailović, M.; Grdović, N.; Tolić, A.; Rajić, J.; Đorđević, M.; Vidaković, M. Oxidative stress-mediated beta cell death and dysfunction as a target for diabetes management. Front. Endocrinol. 2022, 13, 1006376. [Google Scholar] [CrossRef]
- Faraci, F.M.; Didion, S.P. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1367–1373. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, Y.; Zhang, G.; Yang, Z.; Xu, W.; Chen, Q. The Applications and Mechanisms of Superoxide Dismutase in Medicine, Food, and Cosmetics. Antioxidants 2023, 12, 1675. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 8. [Google Scholar]
- Expert Panel on Detection Evaluation Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National cholesterol education program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (adult treatment panel III). JAMA 2001, 285, 2486–2497. [Google Scholar] [CrossRef]
- Bonora, E.; Targher, G.; Alberiche, M.; Bonadonna, R.C.; Saggiani, F.; Zenere, M.B.; Monauni, T.; Muggeo, M. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: Studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 2000, 23, 57–63. [Google Scholar] [CrossRef]
- Deurenberg, P.; Westrate, J.A.; Seidell, J.C. Body mass index as a measure of body fatness: Age- and sex-specific prediction formulas. Br. J. Nutr. 1991, 65, 105–111. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Mee, F.; Atkins, N.; Thomas, M. Evaluation of three devices for self measurement of blood pressure according to the revised British Hypertension Society Protocol: The Omron HEM-705CP, Philips HP5332, and Nissei DS-175. Blood Press. Monit. 1996, 1, 55–61. [Google Scholar] [PubMed]
- Bangalore, S.; Gong, Y.; Cooper-DeHoff, R.M.; Pepine, C.J.; Messerli, F.H. 2014 Eighth Joint National Committee panel recommendation for blood pressure targets revisited: Results from the INVEST study. J. Am. Coll. Cardiol. 2014, 64, 784–793. [Google Scholar] [CrossRef]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; et al. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- Mancia, G.; Kreutz, R.; Brunström, M.; Burnier, M.; Grassi, G.; Januszewicz, A.; Muiesan, M.L.; Tsioufis, K.; Agabiti-Rosei, E.; Algharably, E.A.E.; et al. 2023 ESH Guidelines for the management of arterial hypertension The Task Force for the management of arterial hypertension of the European Society of Hypertension: Endorsed by the International Society of Hypertension (ISH) and the European Renal Association (ERA). J. Hypertens. 2023, 41, 1874–2071. [Google Scholar]
- Bergman, M.; Manco, M.; Satman, I.; Chan, J.; Schmidt, M.I.; Sesti, G.; Vanessa Fiorentino, T.; Abdul-Ghani, M.; Jagannathan, R.; Kumar Thyparambil Aravindakshan, P.; et al. International Diabetes Federation Position Statement on the 1-hour post-load plasma glucose for the diagnosis of intermediate hyperglycaemia and type 2 diabetes. J. Diabetes Res. Clin. Pract. 2024, 209, 111589. [Google Scholar] [CrossRef]
- Knopfholz, J.; Disserol, C.C.; Pierin, A.J.; Schirr, F.L.; Streisky, L.; Takito, L.L.; Massucheto Ledesma, P.; Faria-Neto, J.R.; Olandoski, M.; da Cunha, C.L.; et al. Validation of the Friedewald formula in patients with metabolic syndrome. Cholesterol 2014, 2014, 261878. [Google Scholar] [CrossRef] [PubMed]
- Itabe, H.; Ueda, M. Measurement of plasma oxidized low-density lipoprotein and its clinical implications. J. Atheroscler. Thromb. 2007, 14, 1–11. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Dole, V.P.; Meinertz, H. Micro-determination of long chain fatty acids in plasma and tissues. J. Biol. Chem. 1960, 235, 2595–2599. [Google Scholar] [CrossRef] [PubMed]
- Duncombe, W.G.; Rising, T.J. Quantitative extraction and determination of non esterified fatty acids in plasma. J. Lipid Res. 1973, 14, 258–261. [Google Scholar] [CrossRef]
- Arnaud, J.; Bellanger, J.; Bienvenu, F.; Chappuis, P.; Favier, A. Recommended method for assaying serum zinc with flame atomic absorption. Ann. Biol. Clin. 1986, 44, 77–87. [Google Scholar]
- Aihara, K.; Nishi, Y.; Hatano, S.; Kihara, M.; Yoshimitsu, K.; Takeichi, N.; Ito, T.; Ezaki, H.; Usui, T. Zinc, copper, manganese, and selenium metabolism in thyroid disease. Am. J. Clin. Nutr. 1984, 40, 26–35. [Google Scholar] [CrossRef]
- Beckett, J.M.; Hartley, T.F.; Ball, M.J. Evaluation of the Randox colorimetric serum copper and zinc assays against atomic absorption spectroscopy. Ann. Clin. Biochem. 2009, 46, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Koracevic, D.; Koracevic, G.; Djordjevic, V.; Andrejevic, S.; Cosic, V. Method for the measurement of antioxidant activity in human fluids. J. Clin. Pathol. 2001, 54, 356–361. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Ghani, M.A.; Barril, C.; Bedgood, D.R., Jr.; Prenzler, P.D. Measurement of antioxidant activity with the thiobarbituric acid reactive substances assay. Food Chem. 2017, 230, 195–207. [Google Scholar] [CrossRef]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P/G | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|
| (N = 100) | (N = 209) | (N = 107) | (N = 298) | |
| Age (year) | 46 ± 2 | 50 ± 4 | 53 ± 5 | 55 ± 3 |
| Sex-gender repartition (%) | 50 (M) 50 (F) | 47 (M) 53 (F) *** | 71 (M) 29 (F) *** | 35.5 (M) 64.5 (F) *** |
| Body Weight (Kg) | 69 ± 3 | 81 ± 5 | 79 ± 6 | 85 ± 2 |
| BMI (Kg/m2) | 23 ± 2 | 34 ± 4 *** | 31 ± 1 *** | 35 ± 3 *** |
| WC (cm) | 76 ± 3 (F) | 110 ± 5 (F) *** | 109 ± 4 (F) *** | 113 ± 5 (F) *** |
| 74 ± 2 (M) | 94 ± 2 (M) | 101 ± 5 (M) *** | 111 ± 3 (M) *** | |
| WC/WH ratio | 0.81 ± 0.02 (F) | 1.07 ± 0.01 (F) *** | 0.99 ± 0.02 (F) *** | 1.13 ± 0.03 (F) *** |
| 0.87 ± 0.02 (M) | 1.05 ± 0.03 (M) | 0.93 ± 0.01 (M) *** | 1.09 ± 0.04 (M) *** | |
| BF (%) | 13.0 ± 2.55 (F) | 47.8 ± 5.11 (F) *** | 33.7 ± 2.58 (F) *** | 61.9 ± 5.22 (F) *** |
| 9.11 ± 0.56 (M) | 43.4 ± 7.13 (M) *** | 31.8 ± 3.23 (M) *** | 49.9 ± 5.18 (M) *** |
| P/G | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|
| (N = 100) | (N =209) | (N = 107) | (N = 298) | |
| Glycemia (mmol/L) | 4.62 ± 0.22 | 7.99 ± 0.59 ** | 5.33 ± 0.81 *** | 9.63 ± 0.66 *** |
| Insulinemia (pmol/L) | 67 ± 1.44 | 149 ± 5.25 ** | 126 ± 3.19 *** | 174 ± 8.33 |
| HOMA-IR | 1.68 ± 0.05 | 4.91 ± 0.23 *** | 3.68 ± 0.54 *** | 7.97 ± 0.81 *** |
| HbA1C (%) | 5.11 ± 0.22 | 6.43 ± 0.52 ** | 5.64 ± 0.17 *** | 9.21 ± 0.64 *** |
| Triglycerides (mmol/L) | 1.19 ± 0.21 | 1.98 ± 0.32 *** | 1.87 ± 0.54 *** | 2.08 ± 0.62 *** |
| Total Cholesterol (mmol/L) | 3.25 ± 0.17 | 5.27 ± 0.63 *** | 5.89 ± 0.81 *** | 6.67 ± 0.22 *** |
| HDL-C (mmol/L) | 1.52 ± 0.22 (F) | 1.08 ± 0.31 (F) | 1.10 ± 0.47 (F) * | 1.04 ± 0.19 (F) * |
| 1.24 ± 0.1 (M) | 1.06 ± 0.2 (M) | 1.09 ± 0.1 (M) * | 0.98 ± 0.1 (M) * | |
| LDL-C (mmol/L) | 2.45 ± 0.5 | 3.48 ± 0.6 ** | 4.23 ± 0.5 *** | 4.69 ± 0.3 *** |
| AST (IU/L) | 20.2 ± 1.55 | 21.6 ± 4.07 | 23.2 ± 5.66 | 25.1 ± 3.27 |
| ALT (IU/L) | 20.1 ± 2.64 | 25.4 ± 3.07 | 28.2 ± 4.46 *** | 34.7 ± 8.09 *** |
| AST/ALT Ratio | 1.04 ± 0.06 | 0.85 ± 0.02 | 0.82 ± 0.01 *** | 0.72 ± 0.04 *** |
| GGT (IU/L) | 19.2 ± 7.11 | 28.3 ± 6.19 *** | 37.4 ± 5.51 *** | 62.1 ± 9.04 *** |
| Hs-CRP (mg/L) | 2.81 ± 1.79 | 5.66 ± 0.91 ** | 5.41 ± 0.66 *** | 7.82 ± 0.53 *** |
| Fibrinogen (mg/L) | 2.97 ± 0.41 | 3.38 ± 0.13 | 3.19 ± 0.11 | 3.42 ± 0.10 |
| Ferritin (ng/mL) | 69.4 ± 9.22 | 109 ± 11.9 *** | 146 ± 19.8 *** | 166 ± 12.7 *** |
| Creatinine (µmol/L) | 68 ± 3.41 | 71 ± 2.52 | 80 ± 4.11 *** | 81 ± 6.32 *** |
| Uric acid (µmol/L) | 284 ± 12 | 404 ± 16 | 321 ± 14 | 517 ± 18 *** |
| Microalbuminuria (mg/24 h) | 14.3 ± 2.74 | 27.8 ± 4.63 *** | 34.6 ± 5.27 *** | 47.9 ± 6.47 *** |
| P/G | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|
| (N = 100) | (N = 209) | (N = 107) | (N = 298) | |
| SBP (mm Hg) | 120 ± 7 | 128 ± 5 | 146 ± 3 *** | 158 ± 3 *** |
| DBP (mm Hg) | 63 ± 4 | 71 ± 3 | 91 ± 5 *** | 97 ± 6 *** |
| HDL-c/LDL-c | 0.58 ± 0.02 | 0.33 ± 0.01 ** | 0.37 ± 0.03 *** | 0.25 ± 0.02 *** |
| TG/HDL-c | 0.87 ± 0.02 | 1.68 ± 0.04 ** | 1.70 ± 0.01 *** | 1.97 ± 0.03 *** |
| ApoA1 (g/L) | 1.71 ± 0.01 | 1.37 ± 0.05 | 0.85 ± 0.01 *** | 0.80 ± 0.02 *** |
| ApoB100 (g/L) | 0.84 ± 0.02 | 0.93 ± 0.05 | 0.92 ± 0.01 | 0.99 ± 0.07 |
| ApoB100/ApoA1 | 0.49 ± 0.01 | 0.67 ± 0.01 | 1.08 ± 0.02 *** | 1.23 ± 0.06 *** |
| Lp(a) (g/L) | 0.21 ± 0.06 | 0.23 ± 0.02 ** | 0.34 ± 0.03 *** | 0.85 ± 0.01 *** |
| tHcy (µmol/L) | 10.3 ± 0.77 | 10.9 ± 0.06 | 15.7 ± 0.22 *** | 17.4 ± 0.11 *** |
| Ox-LDL (µmol/L) | 15.7 ± 1.08 | 33.6 ± 6.23 *** | 47.9 ± 5.56 *** | 55.3 ± 6.11 *** |
| ET-1 (pg/mL) | 51.5 ± 8.75 | 77.30 ± 9.03 ** | 99.3 ± 5.11 *** | 114 ± 7.22 *** |
| P/G | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|
| (N = 100) | (N = 209) | (N = 107) | (N = 298) | |
| NEFFA (µmol/L) | 540 ± 25 | 627 ± 13 ** | 594 ± 77 *** | 894 ± 89 *** |
| Total SFA (mol %) | 16.2 ± 4.63 | 19.4 ± 3.24 ** | 21.7 ± 4.89 *** | 34.3 ± 5.29 *** |
| Lauric acid | 0.37 ± 0.03 | 0.48 ± 0.06 * | 0.50 ± 0.03 ** | 0.61 ± 0.03 ** |
| Myristic acid | 0.40 ± 0.07 | 0.68 ± 0.02 * | 0.76 ± 0.04 *** | 0.98 ± 0.04 *** |
| Palmitic acid | 11.01 ± 1.02 | 15.1 ± 1.05 ** | 16.5 ± 1.01 *** | 25.1 ± 1.07 *** |
| Stearic acid | 4.44 ± 0.51 | 3.17 ± 0.11 | 3.99 ± 2.81 | 7.61 ± 1.78 ** |
| Total MUFA (mol %) | 17.2 ± 1.49 | 21.7 ± 1.98 | 23.9 ± 1.33 *** | 29.7 ± 1.71 *** |
| Total PUFA (mol %) | 49.6 ± 3.23 | 47.9 ± 2.88 | 47.6 ± 3.21 | 40.2 ± 3.55 ** |
| Linoleic acid(w6) | 40.2 ± 1.66 | 37.1 ± 1.99 * | 36.1 ± 2.55 * | 27.9 ± 1.50 *** |
| Linolenic acid (w3) | 1.31 ± 0.13 | 0.71 ± 0.20 | 0.73 ± 0.32 *** | 0.41 ± 0.13 *** |
| Arachidonic acid | 8.1 ± 1.44 | 10.1 ± 0.87 ** | 10.8 ± 1.94 *** | 11.9 ± 1.94 *** |
| PUFA/SFA | 3.06 ± 0.69 | 2.46 ± 0.88 | 2.19 ± 0.65 *** | 1.17 ± 0.67 *** |
| PUFA w6/PUFA w3 | 30.6 ± 12.2 | 52.2 ± 9.95 *** | 49.4 ± 7.96 *** | 87.5 ± 11.5 *** |
| EPA (mol %) | 2.98 ± 0.33 | 1.81 ± 0.22 *** | 1.75 ± 0.21 *** | 1.22 ± 0.17 *** |
| DHA (mol %) | 2.55 ± 0.61 | 2.08 ± 0.89 ns | 1.99 ± 0.11 *** | 0.88 ± 0.03 *** |
| EPA+DHA (mol %) | 5.53 ± 0.94 | 3.89 ± 1.11 * | 3.74 ± 0.32 *** | 2.10 ± 0.20 *** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gouaref, I.; Otmane, A.; Makrelouf, M.; Abderrhmane, S.A.; Haddam, A.E.M.; Koceir, E.-A. Crucial Interactions between Altered Plasma Trace Elements and Fatty Acids Unbalance Ratio to Management of Systemic Arterial Hypertension in Diabetic Patients: Focus on Endothelial Dysfunction. Int. J. Mol. Sci. 2024, 25, 9288. https://doi.org/10.3390/ijms25179288
Gouaref I, Otmane A, Makrelouf M, Abderrhmane SA, Haddam AEM, Koceir E-A. Crucial Interactions between Altered Plasma Trace Elements and Fatty Acids Unbalance Ratio to Management of Systemic Arterial Hypertension in Diabetic Patients: Focus on Endothelial Dysfunction. International Journal of Molecular Sciences. 2024; 25(17):9288. https://doi.org/10.3390/ijms25179288
Chicago/Turabian StyleGouaref, Ines, Amel Otmane, Mohamed Makrelouf, Samir Ait Abderrhmane, Ali El Mahdi Haddam, and Elhadj-Ahmed Koceir. 2024. "Crucial Interactions between Altered Plasma Trace Elements and Fatty Acids Unbalance Ratio to Management of Systemic Arterial Hypertension in Diabetic Patients: Focus on Endothelial Dysfunction" International Journal of Molecular Sciences 25, no. 17: 9288. https://doi.org/10.3390/ijms25179288