
Effect of Levetiracetam on Oxidant–Antioxidant Activity during Long-Term Temporal Lobe Epilepsy in Rats

 , , ,
, , ,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
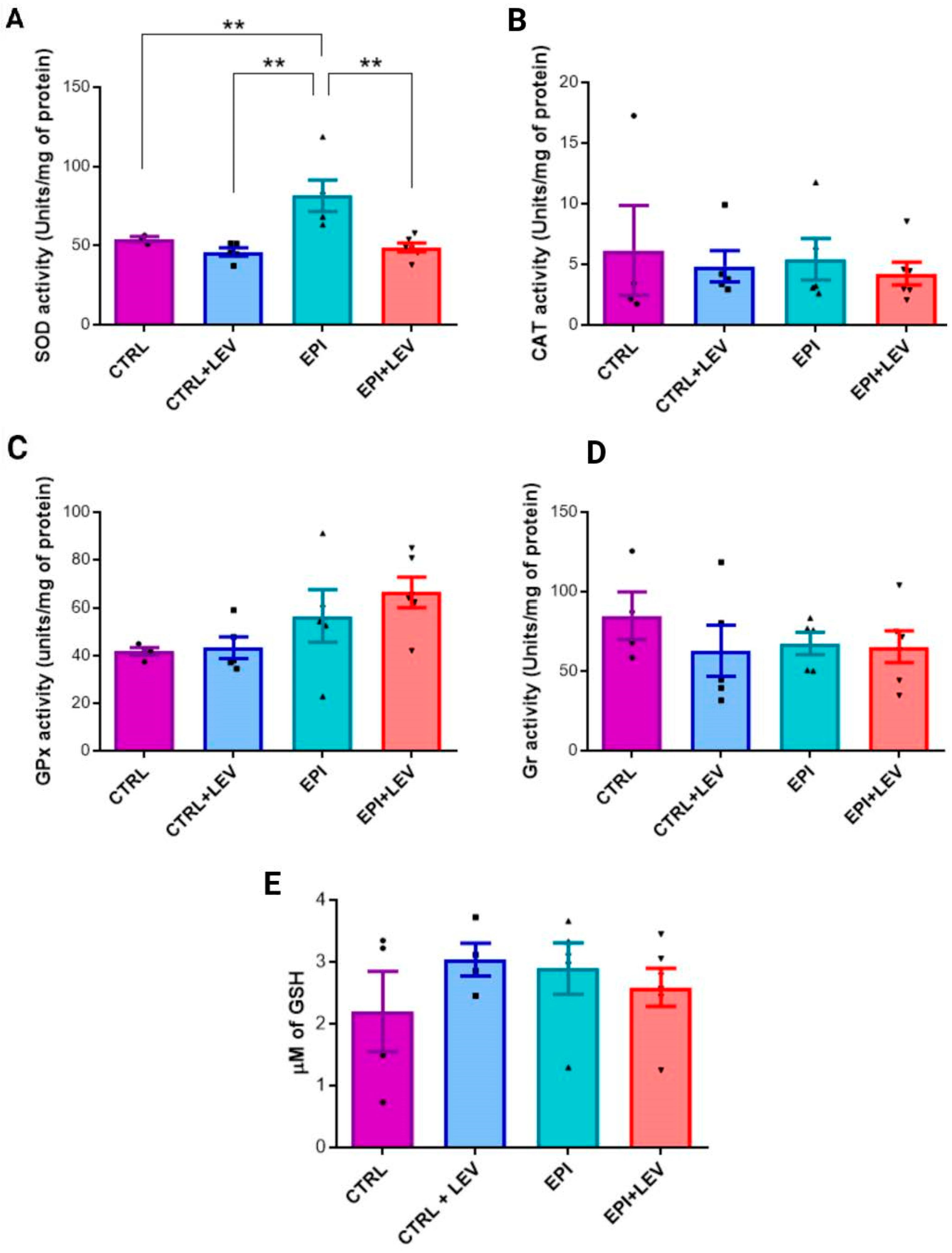
2.1. Antioxidant Markers
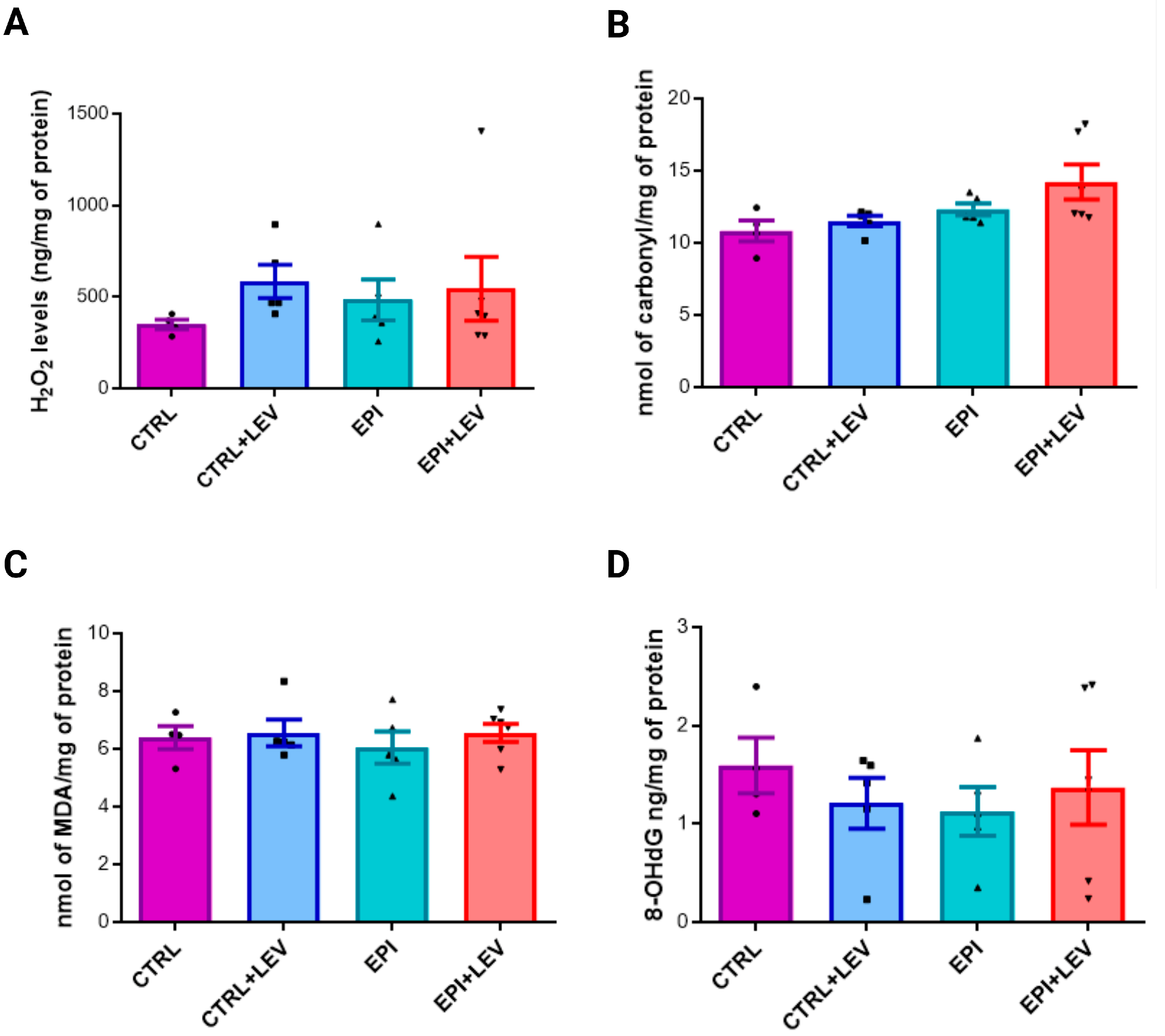
2.2. Oxidant Markers
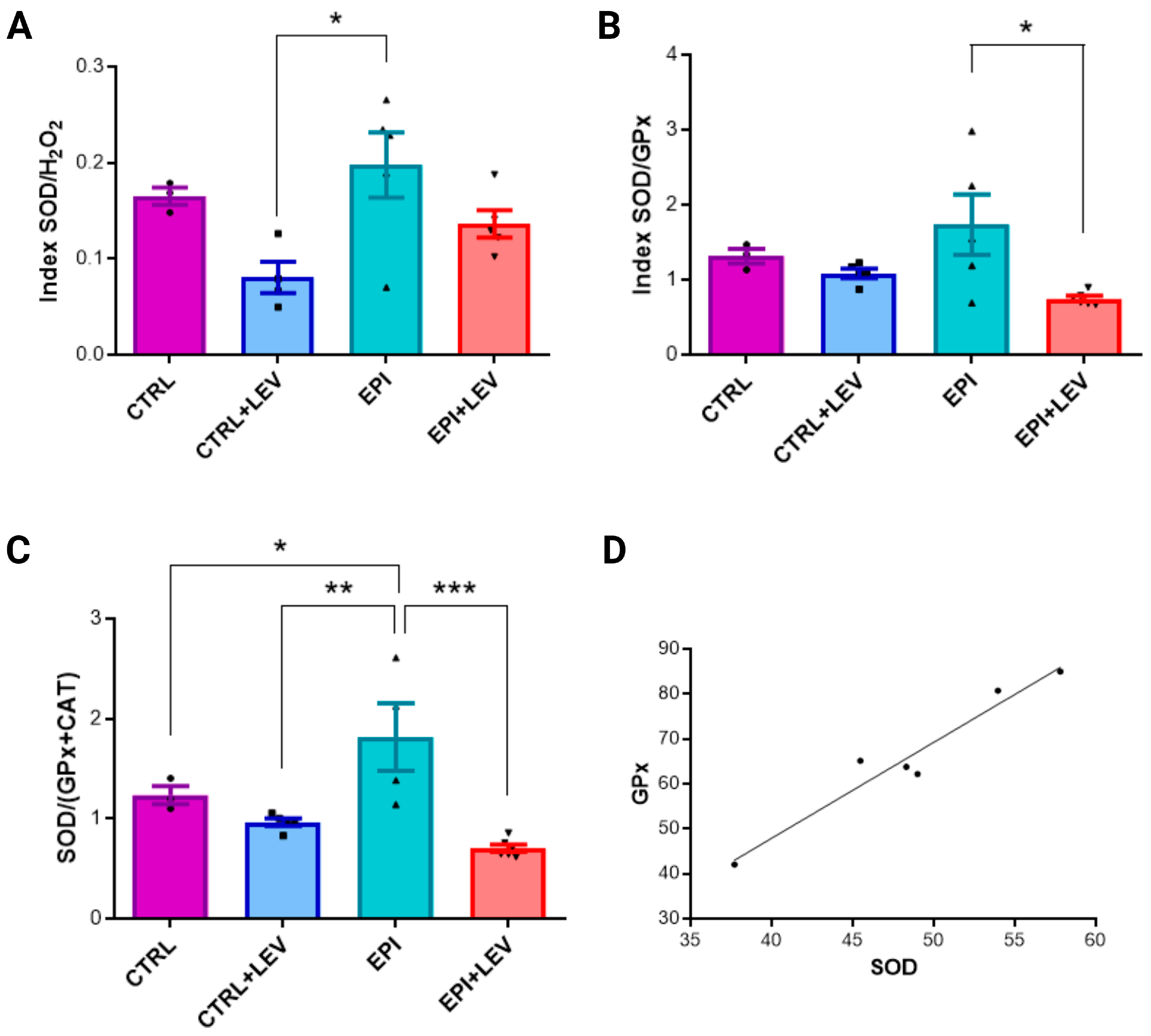
2.3. Ratio and Correlation Analysis of Biochemical Markers
2.4. Levetiracetam Blood Levels
3. Discussion
4. Materials and Methods
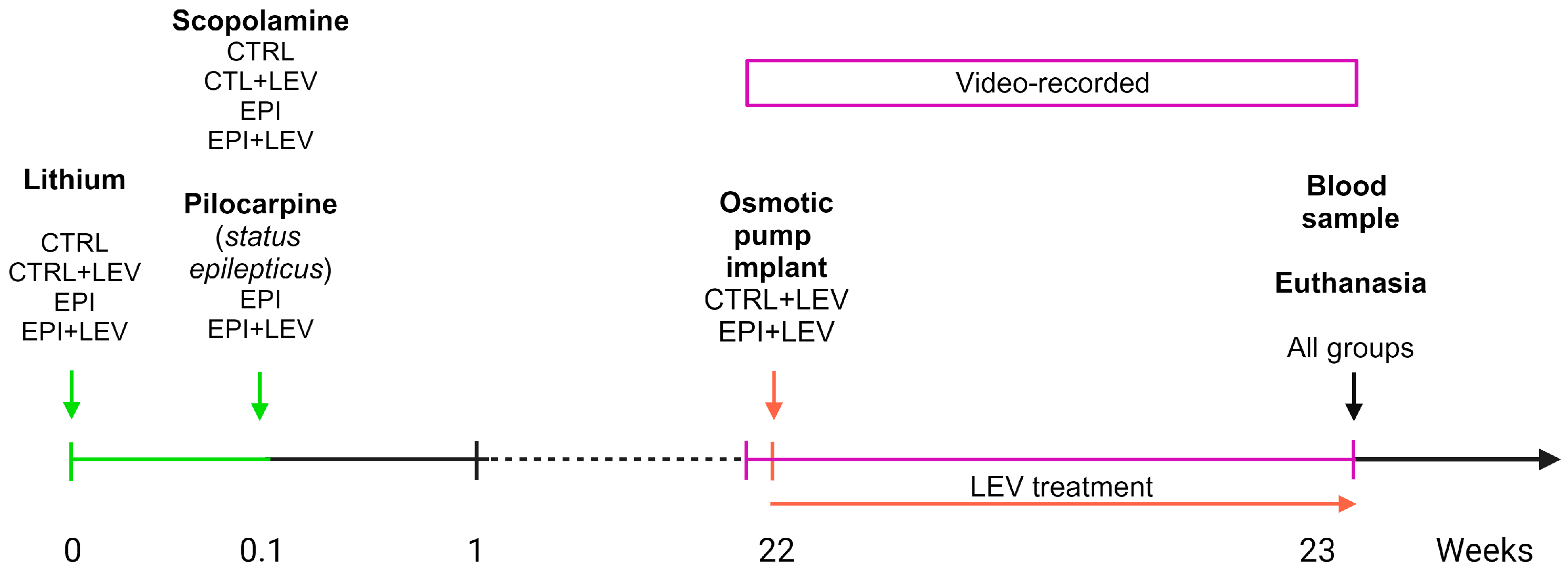
4.1. Animals
4.2. Experimental Design
4.3. Animal Model of Temporal Lobe Epilepsy
4.4. Monitoring of Spontaneous Behavioral Seizures
4.5. Levetiracetam Treatment
4.6. Tissue Sample Collection and Processing
4.7. Total Protein Measurement
4.8. Determination of Antioxidant Markers
4.8.1. SOD Activity Assay
4.8.2. CAT Activity Assay
4.8.3. GPx Activity Assay
4.8.4. GR Activity Assay
4.8.5. GSH Level Determination
4.9. Determinatuon of Oxidant Markers
4.9.1. Determination of H2O2 Levels
4.9.2. Protein Carbonylation Determination
4.9.3. MDA Level Determination
4.9.4. 8-OHdG Level Determination
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J., Jr.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Henning, O.; Heuser, K.; Larsen, V.S.; Kyte, E.B.; Kostov, H.; Marthinsen, P.B.; Egge, A.; Alfstad, K.A.; Nakken, K.O. Temporal lobe epilepsy. Temporallappsepilepsi. Tidsskr. Nor. Laegeforen. 2023, 143, 1–11. [Google Scholar] [CrossRef]
- Jarero-Basulto, J.J.; Gasca-Martínez, Y.; Rivera-Cervantes, M.C.; Ureña-Guerrero, M.E.; Feria-Velasco, A.I.; Beas-Zarate, C. Interactions Between Epilepsy and Plasticity. Pharmaceuticals 2018, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Geronzi, U.; Lotti, F.; Grosso, S. Oxidative stress in epilepsy. Expert Rev. Neurother. 2018, 18, 427–434. [Google Scholar] [CrossRef]
- Aguiar, C.C.; Almeida, A.B.; Araújo, P.V.; Cavalcante de Abreu, R.N.D.; Camelo-Chaves, E.M.; Cardoso do Vale, O.; Macêdo, D.S.; Woods, D.J.; de França Fonteles, M.M.; Mendes-Vasconcelos, S.M. Oxidative stress and epilepsy: Literature review. Oxid. Med. Cell. Longev. 2012, 2012, 795259. [Google Scholar] [CrossRef] [PubMed]
- Yilgor, A.; Demir, C. Determination of oxidative stress level and some antioxidant activities in refractory epilepsy patients. Sci. Rep. 2024, 14, 6688. [Google Scholar] [CrossRef]
- Glazyrin, Y.E.; Veprintsev, D.V.; Timechko, E.E.; Minic, Z.; Zamay, T.N.; Dmitrenko, D.V.; Berezovski, M.V.; Kichkailo, A.S. Comparative Proteomic Profiling of Blood Plasma Revealed Marker Proteins Involved in Temporal Lobe Epilepsy. Int. J. Mol. Sci. 2024, 25, 7935. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Guo, Y.; Wang, W. Bioinformatics Analysis Reveals Multiple Functional Changes in Astrocytes in Temporal Lobe Epilepsy. Brain Res. 2024, 1831, 148820. [Google Scholar] [CrossRef]
- Farooq, T.; Javaid, S.; Ashraf, W.; Rasool, M.F.; Anjum, S.M.M.; Sabir, A.; Ahmad, T.; Alqarni, S.A.; Alqahtani, F.; Imran, I. Neuroprotective Effect of Brivaracetam and Perampanel Combination on Electrographic Seizures and Behavior Anomalies in Pentylenetetrazole-Kindled Mice. ACS Omega 2024, 9, 26004–26019. [Google Scholar] [CrossRef]
- Ignacio-Mejía, I.; Contreras-García, I.J.; Mendoza-Torreblanca, J.G.; Omar Noel Medina-Campos, O.N.; José Pedraza-Chaverri, J.; García-Cruz, M.E.; Romo-Mancillas, A.; Gómez-Manzo, S.; Bandala, C.; Sánchez-Mendoza, M.E.; et al. Evaluation of the Antioxidant Activity of Levetiracetam in a Temporal Lobe Epilepsy Model. Biomedicines 2023, 11, 848. [Google Scholar] [CrossRef]
- Pichardo, L.A.; Contreras, I.J.; Zamudio, S.R.; Mixcoha, E.; Mendoza, J.G. Synaptic Vesicle Protein 2A as a novel pharmacological target with broad potential for new antiepileptic drugs. In Antiepileptic Drug Discovery: Novel Approaches, Methods in Pharmacology and Toxicology, 1st ed.; Talevi, A., Rocha, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 53–65. [Google Scholar] [CrossRef]
- Lynch, B.A.; Lambeng, N.; Nocka, K.; Kensel-Hammes, P.; Bajjalieh, S.M.; Matagne, A.; Fuks, B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl. Acad. Sci. USA 2004, 101, 9861–9866. [Google Scholar] [CrossRef]
- Janz, R.; Goda, Y.; Geppert, M.; Missler, M.; Südhof, T.C. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron 1999, 24, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Nowack, A.; Yao, J.; Custer, K.L.; Bajjalieh, S.M. SV2 regulates neurotransmitter release via multiple mechanisms. Am. J. Physiol. Cell Physiol. 2010, 299, C960–C967. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Nowack, A.; Kensel-Hammes, P.; Gardner, R.G.; Bajjalieh, S.M. Cotrafficking of SV2 and synaptotagmin at the synapse. J. Neurosci. 2010, 30, 5569–5578. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Bajjalieh, S.M. SV2 modulates the size of the readily releasable pool of secretory vesicles. Nat. Cell Biol. 2001, 3, 691–698. [Google Scholar] [CrossRef]
- Surges, R.; Volynski, K.E.; Walker, M.C. Is levetiracetam different from other antiepileptic drugs? Levetiracetam and its cellular mechanism of action in epilepsy. Ther. Adv. Neurol. Disord. 2008, 1, 13–24. [Google Scholar] [CrossRef]
- Niespodziany, I.; Klitgaard, H.; Margineanu, D.G. Levetiracetam inhibits the high-voltage-activated Ca(2+) current in pyramidal neurones of rat hippocampal slices. Neurosci. Lett. 2001, 306, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Bonsi, P.; Martella, G.; de Persis, C.; Costa, C.; Pisani, F.; Bernardi, G.; Calabresi, P. Intracellular calcium increase in epileptiform activity: Modulation by levetiracetam and lamotrigine. Epilepsia 2004, 45, 719–728. [Google Scholar] [CrossRef]
- Lukyanetz, E.A.; Shkryl, V.M.; Kostyuk, P.G. Selective blockade of N-type calcium channels by levetiracetam. Epilepsia 2002, 43, 9–18. [Google Scholar] [CrossRef]
- Yan, H.D.; Ishihara, K.; Seki, T.; Hanaya, R.; Kurisu, K.; Arita, K.; Serikawa, T.; Sasa, M. Inhibitory effects of levetiracetam on the high-voltage-activated L-type Ca2+ channels in hippocampal CA3 neurons of spontaneously epileptic rat (SER). Brain Res. Bull. 2013, 90, 142–148. [Google Scholar] [CrossRef]
- Madeja, M.; Margineanu, D.G.; Gorji, A.; Siep, E.; Boerrigter, P.; Klitgaard, H.; Speckmann, E.J. Reduction of voltage-operated potassium currents by levetiracetam: A novel antiepileptic mechanism of action? Neuropharmacology 2003, 45, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Nagarkatti, N.; Deshpande, L.S.; DeLorenzo, R.J. Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci. Lett. 2008, 436, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Angehagen, M.; Margineanu, D.G.; Ben-Menachem, E.; Rönnbäck, L.; Hansson, E.; Klitgaard, H. Levetiracetam reduces caffeine-induced Ca2+ transients and epileptiform potentials in hippocampal neurons. Neuroreport 2003, 14, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, I.J.; Bandala, C.; Ignacio-Mejía, I.; Pichardo-Macías, L.A.; Mendoza-Torreblanca, J.G.; García-Cruz, M.E.; Gómez-Manzo, S.; Cárdenas-Rodríguez, N. Effect of levetiracetam on DNA oxidation and glutathione content in a temporal lobe epilepsy model. Acta Neurobiol. Exp. 2024, 84, 51–58. [Google Scholar] [CrossRef]
- Ueda, Y.; Doi, T.; Takaki, M.; Nagatomo, K.; Nakajima, A.; Willmore, L.J. Levetiracetam Enhances Endogenous Antioxidant in the Hippocampus of Rats: In Vivo Evaluation by Brain Microdialysis Combined with ESR Spectroscopy. Brain Res. 2009, 1266, 1–7. [Google Scholar] [CrossRef]
- Pichardo-Macías, L.A.; Ramírez-Mendiola, B.A.; Contreras-García, I.J.; Zamudio-Hernández, S.R.; Chávez-Pacheco, J.-L.; Sánchez-Huerta, K.B.; Mendoza-Torreblanca, J.G. Effect of levetiracetam on extracellular amino acid levels in the dorsal hippocampus of rats with temporal lobe epilepsy. Epilepsy Res. 2018, 140, 111–119. [Google Scholar] [CrossRef]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. BioMed Res. Int. 2015, 2015, 745613. [Google Scholar] [CrossRef]
- Łukawski, K.; Czuczwar, S.J. Oxidative Stress and Neurodegeneration in Animal Models of Seizures and Epilepsy. Antioxidants 2023, 12, 1049. [Google Scholar] [CrossRef] [PubMed]
- Borowicz-Reutt, K.K.; Czuczwar, S.J. Role of oxidative stress in epileptogenesis and potential implications for therapy. Pharmacol. Rep. 2020, 72, 1218–1226. [Google Scholar] [CrossRef]
- Marques, F.V.B.S.; Kawamura, L.R.D.S.M.; Quintanilha, M.V.T.; Mota, I.F.L.; Vasconcelos, A.S.; da Silva, J.F.F.; Mortari, M.R. Pharmacological Perspectives and Mechanisms Involved in Epileptogenesis. Beni-Suef Univ. J. Basic Appl. Sci. 2022, 11, 97. [Google Scholar] [CrossRef]
- Madireddy, S.; Madireddy, S. Therapeutic Strategies to Ameliorate Neuronal Damage in Epilepsy by Regulating Oxidative Stress, Mitochondrial Dysfunction, and Neuroinflammation. Brain Sci. 2023, 13, 784. [Google Scholar] [CrossRef] [PubMed]
- Fabisiak, T.; Patel, M. Crosstalk Between Neuroinflammation and Oxidative Stress in Epilepsy. Front. Cell Dev. Biol. 2022, 10, 976953. [Google Scholar] [CrossRef]
- Méndez-Armenta, M.; Nava-Ruíz, C.; Juárez-Rebollar, D.; Rodríguez-Martínez, E.; Gómez, P.Y. Oxidative stress associated with neuronal apoptosis in experimental models of epilepsy. Oxid. Med. Cell. Longev. 2014, 2014, 293689. [Google Scholar] [CrossRef]
- Pardo-Peña, K.; Yañez-Hernández, A.; Medina-Ceja, L.; Morales-Villagrán, A. Ellagic Acid and Allopurinol Decrease H2O2 Concentrations, Epileptiform Activity and Astrogliosis after Status Epilepticus in the Hippocampus of Adult Rats. Exp. Brain Res. 2022, 240, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Malkov, A.; Ivanov, A.I.; Latyshkova, A.; Bregestovski, P.; Zilberter, M.; Zilberter, Y. Activation of Nicotinamide Adenine Dinucleotide Phosphate Oxidase is the Primary Trigger of Epileptic Seizures in Rodent Models. Ann. Neurol. 2019, 85, 907–920. [Google Scholar] [CrossRef]
- Ponomareva, D.; Ivanov, A.; Bregestovski, P. Analysis of the Effects of Pentose Phosphate Pathway Inhibition on the Generation of Reactive Oxygen Species and Epileptiform Activity in Hippocampal Slices. Int. J. Mol. Sci. 2024, 25, 1934. [Google Scholar] [CrossRef]
- Beltrán-Sarmiento, E.; Arregoitia-Sarabia, C.K.; Floriano-Sánchez, E.; Sandoval-Pacheco, R.; Galván-Hernández, D.E.; Coballase-Urrutia, E.; Carmona-Aparicio, L.; Ramos-Reyna, E.; Rodríguez-Silverio, J.; Cárdenas-Rodríguez, N. Effects of Valproate Monotherapy on the Oxidant-Antioxidant Status in Mexican Epileptic Children: A Longitudinal Study. Oxid. Med. Cell. Longev. 2018, 2018, 7954371. [Google Scholar] [CrossRef] [PubMed]
- Freitas, R.M.; Vasconcelos, S.M.; Souza, F.C.; Viana, G.S.; Fonteles, M.M. Oxidative stress in the hippocampus after pilocarpine-induced status epilepticus in Wistar rats. FEBS J. 2005, 272, 1307–1312. [Google Scholar] [CrossRef]
- Dal-Pizzol, F.; Klamt, F.; Vianna, M.M.; Schröder, N.; Quevedo, J.; Benfato, M.S.; Moreira, J.C.; Walz, R. Lipid peroxidation in hippocampus early and late after status epilepticus induced by pilocarpine or kainic acid in Wistar rats. Neurosci. Lett. 2000, 291, 179–182. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef]
- Liang, L.P.; Ho, Y.S.; Patel, M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Li, Q.Y.; Chang, L.Y.; Crapo, J.; Liang, L.P. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J. Neurochem. 2005, 92, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, A.; Li, L.; Huang, Y.; Xue, P.; Hao, A. Oxidative stress mediates hippocampal neuron death in rats after lithium-pilocarpine-induced status epilepticus. Seizure 2010, 19, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.A.; Almeida, J.P.; Freitas, R.M.; Nascimento, V.S.; Aguiar, L.M.V.; Júnior, H.V.N.; Fonseca, F.N.; Viana, G.S.B.; Sousa, F.C.F.; Fonteles, M.M.G. Effects of levetiracetam in lipid peroxidation level, nitrite-nitrate formation and antioxidant enzymatic activity in mice brain after pilocarpine-induced seizures. Cell. Mol. Neurobiol. 2007, 27, 395–406. [Google Scholar] [CrossRef]
- Dircio-Bautista, M.; Colín-González, A.L.; Aguilera, G.; Maya-López, M.; Villeda-Hernández, J.; Galván-Arzate, S.; García, E.; Túnez, I.; Santamaría, A. The Antiepileptic Drug Levetiracetam Protects Against Quinolinic Acid-Induced Toxicity in the Rat Striatum. Neurotox. Res. 2018, 33, 837–845. [Google Scholar] [CrossRef]
- Osuntokun, O.S.; Abdulwahab, U.F.; Akanji, N.O.; Adedokun, K.I.; Adekomi, A.D.; Olayiwola, G. Anticonvulsant and neuroprotective effects of carbamazepine-levetiracetam adjunctive treatment in convulsive status epilepticus rat model: Inhibition of cholinergic transmission. Neurosci. Lett. 2021, 762, 136167. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, B.A.; Shaikh, I.A.; Khateeb, M.M.; Habeeb, S.M. Omega 3 polyunsaturated fatty acids enhance the protective effect of levetiracetam against seizures, cognitive impairment and hippocampal oxidative DNA damage in young kindled rats. Pharmacol. Biochem. Behav. 2015, 135, 105–113. [Google Scholar] [CrossRef]
- Gibbs, J.E.; Walker, M.C.; Cock, H.R. Levetiracetam: Antiepileptic properties and protective effects on mitochondrial dysfunction in experimental status epilepticus. Epilepsia 2006, 47, 469–478. [Google Scholar] [CrossRef]
- Imran, I.; Koch, K.; Schöfer, H.; Lau, H.; Klein, J. Effects of Three Anti-Seizure Drugs on Cholinergic and Metabolic Activity in Experimental Status Epilepticus. J. Pharm. Pharm. Sci. 2019, 22, 340–351. [Google Scholar] [CrossRef]
- Mazhar, F.; Malhi, S.M.; Simjee, S.U. Comparative studies on the effects of clinically used anticonvulsants on the oxidative stress biomarkers in pentylenetetrazole-induced kindling model of epileptogenesis in mice. J. Basic Clin. Physiol. Pharmacol. 2017, 28, 31–42. [Google Scholar] [CrossRef]
- de Souza, A.G.; Chaves-Filho, A.J.M.; Souza-Oliveira, J.V.; Alves-Albuquerque de Souza, D.; Lopes, I.S.; Albuquerque-Jales de Carvalho, M.; Alves de Lima, K.; Florenço-Sousa, F.C.; Mendes-Vasconcelos, S.M.; Macedo, D.; et al. Prevention of pentylenetetrazole-induced kindling and behavioral comorbidities in mice by levetiracetam combined with the GLP-1 agonist liraglutide: Involvement of brain antioxidant and BDNF upregulating properties. Biomed. Pharmacother. 2019, 109, 429–439. [Google Scholar] [CrossRef]
- Sarangi, S.C.; Kakkar, A.K.; Kumar, R.; Gupta, Y.K. Effect of lamotrigine, levetiracetam & topiramate on neurobehavioural parameters & oxidative stress in comparison with valproate in rats. Indian J. Med. Res. 2016, 144, 104–111. [Google Scholar] [CrossRef]
- Yan, B.C.; Shen, H.; Zhang, Y.; Zhu, X.; Wang, J.; Xu, P.; Jiang, D.; Xing Yu, X. The antiepileptic drug levetiracetam promotes neuroblast differentiation and expression of superoxide dismutase in the mouse hippocampal dentate gyrus via PI3K/Akt signalling. Neurosci. Lett. 2018, 662, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Rowley, S.; Patel, M. Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free Radic. Biol. Med. 2013, 62, 121–131. [Google Scholar] [CrossRef]
- Tejada, S.; Sureda, A.; Roca, C.; Gamundí, A.; Esteban, S. Antioxidant response and oxidative damage in brain cortex after high dose of pilocarpine. Brain Res. Bull. 2007, 71, 372–375. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, R.M.; do Nascimento, K.G.; Ferreira, P.M.; Jordán, J. Neurochemical changes on oxidative stress in rat hippocampus during acute phase of pilocarpine-induced seizures. Pharmacol. Biochem. Behav. 2010, 94, 341–345. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, X.; Jia, W.; Zhang, C.; Boczek, T.; Harding, M.; Liu, Y.; Li, M.; Zhang, S.; Lei, S.; et al. Circulating glutathione peroxidase and superoxide dismutase levels in patients with epilepsy: A meta-analysis. Seizure 2021, 91, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10 (Suppl. 1), 23–48. [Google Scholar] [CrossRef]
- Ryan, K.; Backos, D.S.; Reigan, P.; Patel, M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J. Neurosci. 2012, 32, 11250–11258. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Liang, L.P.; Hellier, J.L.; Staley, K.J.; Patel, M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol. Dis. 2008, 30, 130–138. [Google Scholar] [CrossRef]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef]
- Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules 2021, 26, 1844. [Google Scholar] [CrossRef] [PubMed]
- Balmus, I.M.; Lefter, R.; Ciobica, A.; Cojocaru, S.; Guenne, S.; Timofte, D.; Stanciu, C.; Trifan, A.; Hritcu, L. Preliminary Biochemical Description of Brain Oxidative Stress Status in Irritable Bowel Syndrome Contention-Stress Rat Model. Medicina 2019, 55, 776. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yang, Z. Ferroptosis and Its Role in Epilepsy. Front. Cell. Neurosci. 2021, 15, 696889. [Google Scholar] [CrossRef]
- de Farias, A.C.S.; de Pieri-Pickler, K.; Bernardo, H.T.; Baldin, S.L.; Dondossola, E.R.; Rico, E.P. Melatonin Pretreatment Protects Against Status epilepticus, Glutamate Transport, and Oxidative Stress Induced by Kainic Acid in Zebrafish. Mol. Neurobiol. 2022, 59, 266–275. [Google Scholar] [CrossRef]
- de Haan, J.B.; Cristiano, F.; Iannello, R.C.; Kola, I. Cu/Zn-superoxide dismutase and glutathione peroxidase during aging. Biochem. Mol. Biol. Int. 1995, 35, 1281–1297. [Google Scholar] [PubMed]
- Bizoń, A.; Chojdak-Łukasiewicz, J.; Budrewicz, S.; Pokryszko-Dragan, A.; Piwowar, A. Exploring the Relationship between Antioxidant Enzymes, Oxidative Stress Markers, and Clinical Profile in Relapsing-Remitting Multiple Sclerosis. Antioxidants 2023, 12, 1638. [Google Scholar] [CrossRef]
- Garaiová, I.; Muchová, J.; Šustrová, M.; Blažíček, P.; Sivoňová, M.; Kvasnička, P.; Ďuračková, Z. The relationship between antioxidant systems and some markers of oxidative stress in persons with Down syndrome. Biologia 2004, 59, 787–794. [Google Scholar]
- Gupta, Y.K.; Gupta, M. Post traumatic epilepsy: A review of scientific evidence. Indian J. Physiol. Pharmacol. 2006, 50, 7–16. [Google Scholar]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Guan, Q.W.; Chen, F.H.; Xia, Q.X.; Yin, X.X.; Zhou, H.H.; Mao, X.Y. Antioxidants Targeting Mitochondrial Oxidative Stress: Promising Neuroprotectants for Epilepsy. Oxid. Med. Cell. Longev. 2020, 2020, 6687185. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, I.J.; Cárdenas-Rodríguez, N.; Romo-Mancillas, A.; Bandala, C.; Sergio, R.; Zamudio, S.R.; Gómez-Manzo, S.; Hernández-Ochoa, B.; Mendoza-Torreblanca, J.G.; Pichardo-Macías, L.A. Levetiracetam Mechanisms of Action: From Molecules to Systems. Pharmaceuticals 2022, 15, 475. [Google Scholar] [CrossRef] [PubMed]
- Ristić, A.J.; Savić, D.; Sokić, D.; Bogdanović Pristov, J.; Nestorov, J.; Baščarević, V.; Raičević, S.; Savić, S.; Spasojević, I. Hippocampal antioxidative system in mesial temporal lobe epilepsy. Epilepsia 2015, 56, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blümcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Contreras-García, I.J.; Pichardo-Macías, L.A.; Santana-Gómez, C.E.; Sánchez-Huerta, K.; Ramírez-Hernández, R.; Gómez-González, B.; Rocha, L.; Mendoza=Torreblanca, J.G. Differential expression of synaptic vesicle protein 2A after status epilepticus and during epilepsy in a lithium-pilocarpine model. Epilepsy Behav. 2018, 88, 283–294. [Google Scholar] [CrossRef]
- Glien, M.; Brandt, C.; Potschka, H.; Löscher, W. Effects of the novel antiepileptic drug levetiracetam on spontaneous recurrent seizures in the rat pilocarpine model of temporal lobe epilepsy. Epilepsia 2002, 43, 350–357. [Google Scholar] [CrossRef]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef]
- Contreras-García, I.J.; Gómez-Lira, G.; Phillips-Farfán, B.V.; Pichardo-Macías, L.A.; García-Cruz, M.E.; Chávez-Pacheco, J.J.; Mendoza-Torreblanca, J.G. Synaptic Vesicle Protein 2A Expression in Glutamatergic Terminals Is Associated with the Response to Levetiracetam Treatment. Brain Sci. 2021, 11, 531. [Google Scholar] [CrossRef]
- Oláh, E.; Bacsói, G.; Fekete, J.; Sharma, V.K. Determination of ng/mL levetiracetam using ultra-high-performance liquid chromatography-photodiode absorbance. J. Chromatogr. Sci. 2012, 50, 253–258. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Reznick, A.Z.; Packer, L. Oxidative damage to proteins: Spectrophotometric method for carbonyl assay. Methods Enzymol. 1994, 233, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Pedraza-Chaverri, J.; Yam-Canul, P.; Chirino, Y.I.; Sánchez-González, D.J.; Martínez-Martínez, C.M.; Cruz, C.; Medina-Campos, O.N. Protective effects of garlic powder against potassium dichromate-induced oxidative stress and nephrotoxicity. Food Chem. Toxicol. 2008, 46, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Gérard-Monnier, D.; Erdelmeier, I.; Régnard, K.; Moze-Henry, N.; Yadan, J.C.; Chaudière, J. Reactions of 1-methyl-2-phenylindole with malondialdehyde and 4-hydroxyalkenals. Analytical applications to a colorimetric assay of lipid peroxidation. Chem. Res. Toxicol. 1998, 11, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Colín-Barenque, L.; Bizarro-Nevares, P.; González Villalva, A.; Pedraza-Chaverri, J.; Medina-Campos, O.N.; Jimenez-Martínez, R.; Rodríguez-Rangel, D.S.; Reséndi, S.; Fortoul, T.I. Neuroprotective effect of carnosine in the olfactory bulb after vanadium inhalation in a mouse model. Int. J. Exp. Pathol. 2018, 99, 180–188. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ignacio-Mejía, I.; Contreras-García, I.J.; Pichardo-Macías, L.A.; García-Cruz, M.E.; Ramírez Mendiola, B.A.; Bandala, C.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Cárdenas-Rodríguez, N.; Mendoza-Torreblanca, J.G. Effect of Levetiracetam on Oxidant–Antioxidant Activity during Long-Term Temporal Lobe Epilepsy in Rats. Int. J. Mol. Sci. 2024, 25, 9313. https://doi.org/10.3390/ijms25179313
Ignacio-Mejía I, Contreras-García IJ, Pichardo-Macías LA, García-Cruz ME, Ramírez Mendiola BA, Bandala C, Medina-Campos ON, Pedraza-Chaverri J, Cárdenas-Rodríguez N, Mendoza-Torreblanca JG. Effect of Levetiracetam on Oxidant–Antioxidant Activity during Long-Term Temporal Lobe Epilepsy in Rats. International Journal of Molecular Sciences. 2024; 25(17):9313. https://doi.org/10.3390/ijms25179313
Chicago/Turabian StyleIgnacio-Mejía, Iván, Itzel Jatziri Contreras-García, Luz Adriana Pichardo-Macías, Mercedes Edna García-Cruz, Blanca Alcira Ramírez Mendiola, Cindy Bandala, Omar Noel Medina-Campos, José Pedraza-Chaverri, Noemí Cárdenas-Rodríguez, and Julieta Griselda Mendoza-Torreblanca. 2024. "Effect of Levetiracetam on Oxidant–Antioxidant Activity during Long-Term Temporal Lobe Epilepsy in Rats" International Journal of Molecular Sciences 25, no. 17: 9313. https://doi.org/10.3390/ijms25179313
APA StyleIgnacio-Mejía, I., Contreras-García, I. J., Pichardo-Macías, L. A., García-Cruz, M. E., Ramírez Mendiola, B. A., Bandala, C., Medina-Campos, O. N., Pedraza-Chaverri, J., Cárdenas-Rodríguez, N., & Mendoza-Torreblanca, J. G. (2024). Effect of Levetiracetam on Oxidant–Antioxidant Activity during Long-Term Temporal Lobe Epilepsy in Rats. International Journal of Molecular Sciences, 25(17), 9313. https://doi.org/10.3390/ijms25179313