
A Bioinformatics Investigation of Hub Genes Involved in Treg Migration and Its Synergistic Effects, Using Immune Checkpoint Inhibitors for Immunotherapies


Abstract
:1. Introduction
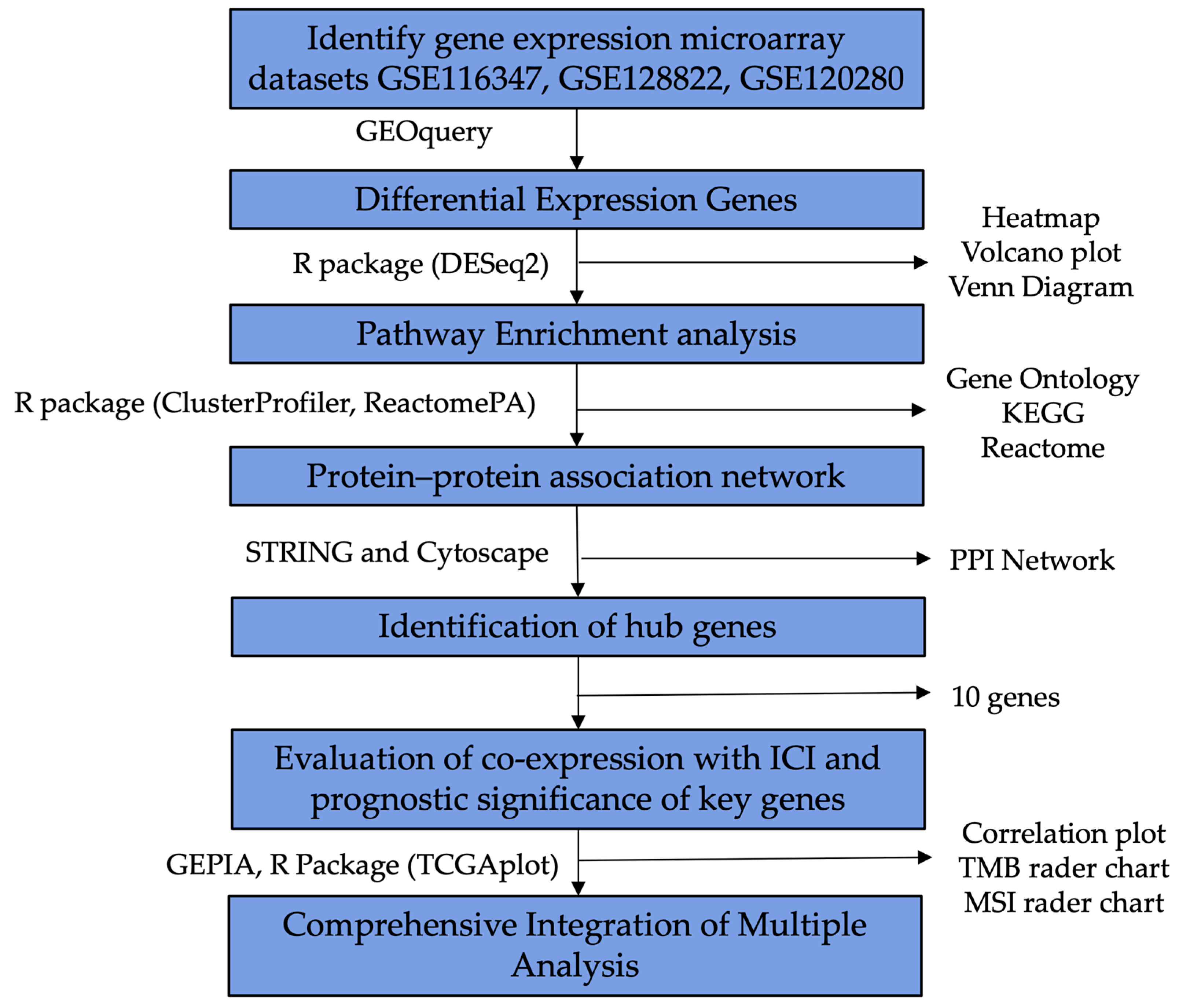
2. Results
2.1. GEO Dataset Selection
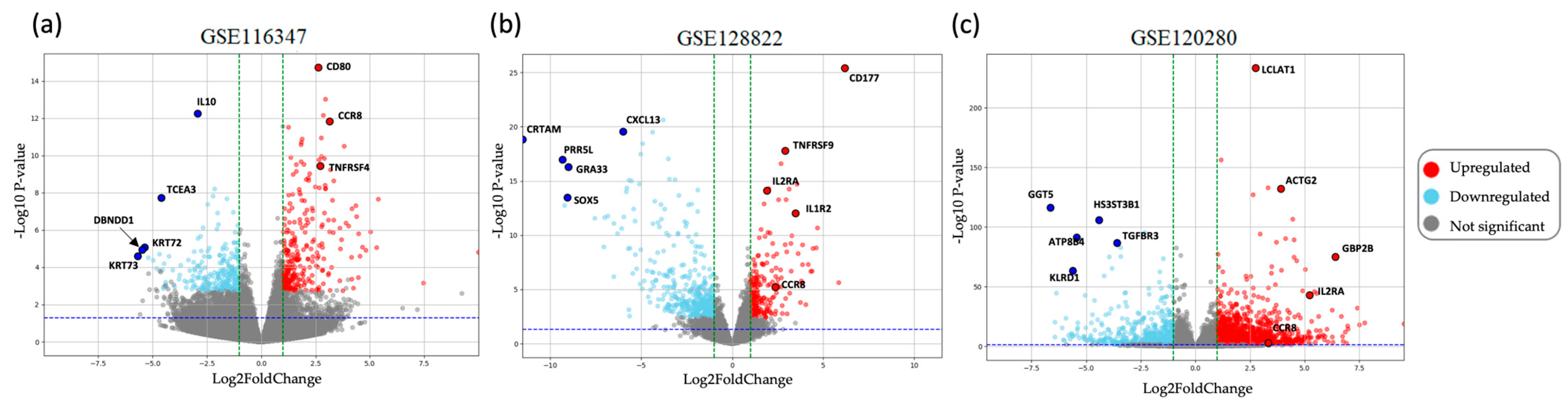
2.2. Identification of DEGs
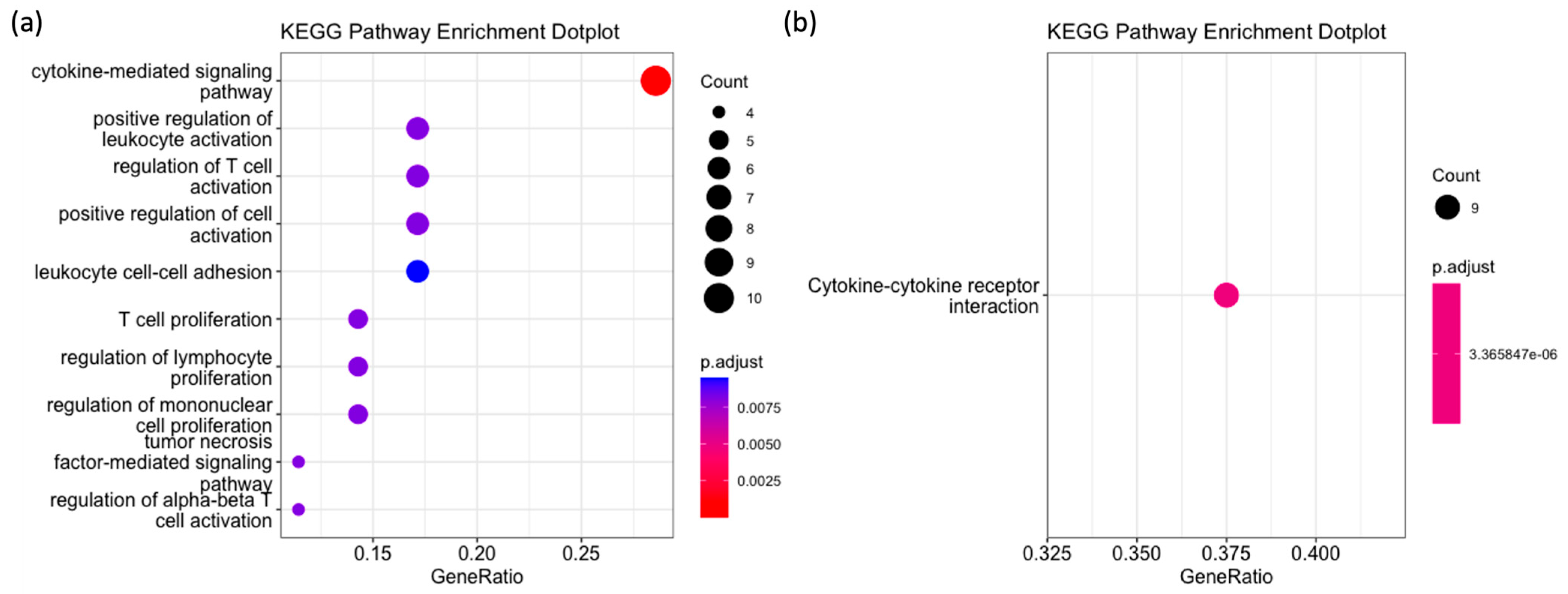
2.3. GO, KEGG and Reactome Pathway Enrichment Analysis
2.4. Hub Gene Identification from the PPI Network
2.5. Synergistic Effects of CCR8 and Immune Checkpoint Inhibitors
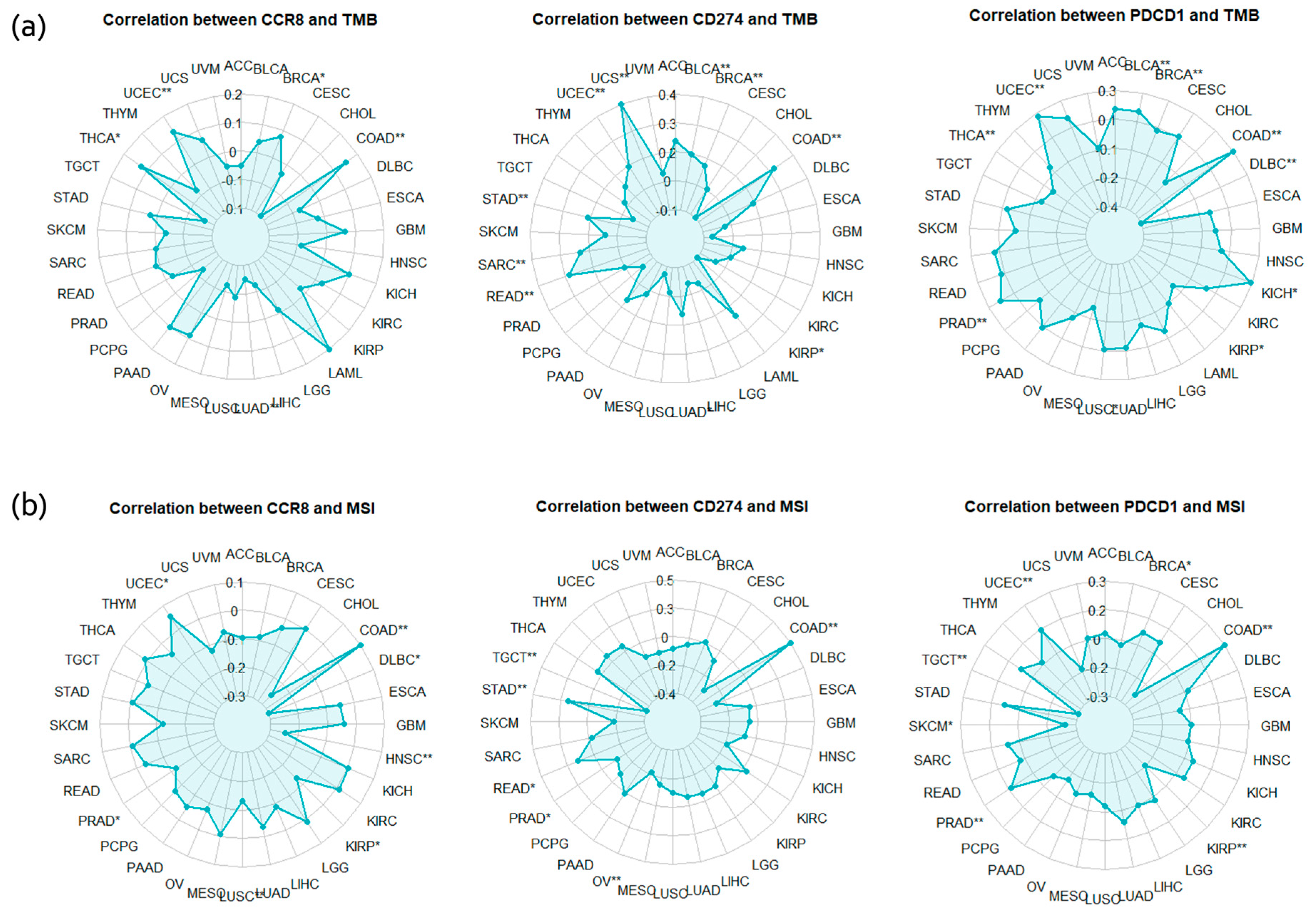
2.5.1. Correlation Analysis between CCR8 Expression and Immune Checkpoint Proteins
2.5.2. TMB, MSI Correlation Analysis
2.6. Comprehensive Integration of Multiple Analysis
3. Discussion
4. Materials and Methods
4.1. GEO Dataset Collection
4.2. Identification of DEGs in GEO Datasets
4.3. GO, KEGG and Reactome Pathway Enrichment Analysis
4.4. Hub Gene Identification from the PPI Network
4.5. Synergistic Effects of CCR8 and Immune Checkpoint Inhibitors
4.6. Comprehensive Integration of Multiple Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dees, S.; Ganesan, R.; Singh, S.; Grewal, I.S. Regulatory T cell targeting in cancer: Emerging strategies in immunotherapy. Eur. J. Immunol. 2021, 51, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Tiemessen, M.M.; Jagger, A.L.; Evans, H.G.; van Herwijnen, M.J.; John, S.; Taams, L.S. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. USA 2007, 104, 19446–19451. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Xu, Y.; Lin, C. Heterogeneity and subtypes of CD4+ regulatory T cells: Implications for tumor therapy. Front. Immunol. 2023, 14, 1291796. [Google Scholar] [CrossRef]
- Hori, S. FOXP3 as a master regulator of Treg cells. Nat. Rev. Immunol. 2021, 21, 618–619. [Google Scholar] [CrossRef] [PubMed]
- Barsheshet, Y.; Wildbaum, G.; Levy, E.; Vitenshtein, A.; Akinseye, C.; Griggs, J.; Lira, S.A.; Karin, N. CCR8+FOXp3+ Treg cells as master drivers of immune regulation. Proc. Natl. Acad. Sci. USA 2017, 114, 6086–6091. [Google Scholar] [CrossRef]
- Shan, F.; Somasundaram, A.; Bruno, T.C.; Workman, C.J.; Vignali, D.A.A. Therapeutic targeting of regulatory T cells in cancer. Trends Cancer 2022, 8, 944–961. [Google Scholar] [CrossRef]
- McRitchie, B.R.; Akkaya, B. Exhaust the exhausters: Targeting regulatory T cells in the tumor microenvironment. Front. Immunol. 2022, 13, 940052. [Google Scholar] [CrossRef]
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Yang, J.; Bae, H. Drug conjugates for targeting regulatory T cells in the tumor microenvironment: Guided missiles for cancer treatment. Exp. Mol. Med. 2023, 55, 1996–2004. [Google Scholar] [CrossRef]
- Nishikawa, H.; Koyama, S. Mechanisms of regulatory T cell infiltration in tumors: Implications for innovative immune precision therapies. J. Immunother. Cancer 2021, 9, e002591. [Google Scholar] [CrossRef]
- Kim, Y.H.; Bagot, M.; Pinter-Brown, L.; Rook, A.H.; Porcu, P.; Horwitz, S.M.; Whittaker, S.; Tokura, Y.; Vermeer, M.; Zinzani, P.L.; et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): An international, open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2018, 19, 1192–1204. [Google Scholar] [CrossRef]
- Chiang, Y.; Lu, L.F.; Tsai, C.L.; Tsai, Y.C.; Wang, C.C.; Hsueh, F.J.; Huang, C.Y.; Chen, C.H.; Pu, Y.S.; Cheng, J.C. C-C chemokine receptor 4 (CCR4)—Positive regulatory T cells interact with tumor-associated macrophages to facilitate metastatic potential after radiation. Eur. J. Cancer 2024, 198, 113521. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Pinter-Brown, L.C.; Foss, F.M.; Sokol, L.; Jorgensen, J.L.; Challagundla, P.; Dwyer, K.M.; Zhang, X.; Kurman, M.R.; Ballerini, R.; et al. Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood 2015, 125, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Evans, M.; Wang, C. Mogamulizumab for the treatment of cutaneous T-cell lymphoma: Recent advances and clinical potential. Ther. Adv. Hematol. 2016, 7, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Geskin, L.J.; Scarisbrick, J.; Bagot, M.; Fisher, D.C.; Elmets, C.; Duvic, M.; Beylot-Barry, M.; Kim, E.J.; Moriya, J.; Leoni, M.; et al. Efficacy of mogamulizumab in previously treated patients with less advanced mycosis fungoides: Results from the MAVORIC study. J. Clin. Oncol. 2019, 37, e19031. [Google Scholar] [CrossRef]
- Ogura, M.; Ishida, T.; Hatake, K.; Taniwaki, M.; Ando, K.; Tobinai, K.; Fujimoto, K.; Yamamoto, K.; Miyamoto, T.; Uike, N.; et al. Multicenter phase II study of mogamulizumab (KW-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T-cell lymphoma and cutaneous T-cell lymphoma. J. Clin. Oncol. 2014, 32, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Kufareva, I.; Salanga, C.L.; Handel, T.M. Chemokine and chemokine receptor structure and interactions: Implications for therapeutic strategies. Immunol. Cell Biol. 2015, 93, 372–383. [Google Scholar] [CrossRef]
- Liu, T.; Li, X.; You, S.; Bhuyan, S.S.; Dong, L. Effectiveness of AMD3100 in treatment of leukemia and solid tumors: From original discovery to use in current clinical practice. Exp. Hematol. Oncol. 2015, 5, 19. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, Z.; Gao, J.L.; Wan, W.; Ganesan, S.; McDermott, D.H.; Murphy, P.M. CXCR4 antagonist AMD3100 redistributes leukocytes from primary immune organs to secondary immune organs, lung, and blood in mice. Eur. J. Immunol. 2015, 45, 1855–1867. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Chen, R.; Wang, X.; Hu, K.; Huang, L.; Lu, M.; Hu, Q. CCL19 and CCR7 Expression, Signaling Pathways, and Adjuvant Functions in Viral Infection and Prevention. Front. Cell Dev. Biol. 2019, 7, 212. [Google Scholar] [CrossRef]
- Beyer, M.; Schultze, J.L. Regulatory T cells in cancer. Blood 2006, 108, 804–811. [Google Scholar] [CrossRef]
- Qiu, Y.; Ke, S.; Chen, J.; Qin, Z.; Zhang, W.; Yuan, Y.; Meng, D.; Zhao, G.; Wu, K.; Li, B.; et al. FOXP3+ regulatory T cells and the immune escape in solid tumours. Front. Immunol. 2022, 13, 982986. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ke, X.-Y. The Four types of Tregs in malignant lymphomas. J. Hematol. Oncol. 2011, 4, 50. [Google Scholar] [CrossRef]
- Kohli, K.; Pillarisetty, V.G.; Kim, T.S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 2022, 29, 10–21. [Google Scholar] [CrossRef]
- Cinier, J.; Hubert, M.; Besson, L.; Di Roio, A.; Rodriguez, C.; Lombardi, V.; Caux, C.; Ménétrier-Caux, C. Recruitment and Expansion of Tregs Cells in the Tumor Environment-How to Target Them? Cancers 2021, 13, 1850. [Google Scholar] [CrossRef]
- Liu, S.; Tao, Z.; Lou, J.; Li, R.; Fu, X.; Xu, J.; Wang, T.; Zhang, L.; Shang, W.; Mao, Y.; et al. CD4+CCR8+ Tregs in ovarian cancer: A potential effector Tregs for immune regulation. J. Transl. Med. 2023, 21, 803. [Google Scholar] [CrossRef]
- Zhu, W.; Wu, C.; Hu, S.; Liu, S.; Zhao, S.; Zhang, D.; Qiu, G.; Cheng, X.; Huang, J. Chemokine- and chemokine receptor-based signature predicts immunotherapy response in female colorectal adenocarcinoma patients. Sci. Rep. 2023, 13, 21358. [Google Scholar] [CrossRef]
- Jacquelot, N.; Duong, C.P.M.; Belz, G.T.; Zitvogel, L. Targeting Chemokines and Chemokine Receptors in Melanoma and Other Cancers. Front. Immunol. 2018, 9, 2480. [Google Scholar] [CrossRef]
- Mollica Poeta, V.; Massara, M.; Capucetti, A.; Bonecchi, R. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front. Immunol. 2019, 10, 379. [Google Scholar] [CrossRef] [PubMed]
- Rocamora-Reverte, L.; Melzer, F.L.; Wurzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2020, 11, 616949. [Google Scholar] [CrossRef] [PubMed]
- Kennedy-Batalla, R.; Acevedo, D.; Luo, Y.; Esteve-Sole, A.; Vlagea, A.; Correa-Rocha, R.; Seoane-Reula, M.E.; Alsina, L. Treg in inborn errors of immunity: Gaps, knowns and future perspectives. Front. Immunol. 2023, 14, 1278759. [Google Scholar] [CrossRef]
- Pereira, J.A.; Lanzar, Z.; Clark, J.T.; Hart, A.P.; Douglas, B.B.; Shallberg, L.; O’Dea, K.; Christian, D.A.; Hunter, C.A. PD-1 and CTLA-4 exert additive control of effector regulatory T cells at homeostasis. Front. Immunol. 2023, 14, 997376. [Google Scholar] [CrossRef]
- Li, X.; Zheng, Y. Regulatory T cell identity: Formation and maintenance. Trends Immunol. 2015, 36, 344–353. [Google Scholar] [CrossRef]
- Traxinger, B.R.; Richert-Spuhler, L.E.; Lund, J.M. Mucosal tissue regulatory T cells are integral in balancing immunity and tolerance at portals of antigen entry. Mucosal Immunol. 2022, 15, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Qiu, P.; Yao, Q.; Chen, J.; Lin, J. Integrated Bioinformatics Analysis for the Screening of Hub Genes and Therapeutic Drugs in Androgen Receptor-Positive TNBC. Dis. Markers 2022, 2022, 4964793. [Google Scholar] [CrossRef]
- Yan, Y.M.; Jin, M.Z.; Li, S.H.; Wu, Y.; Wang, Q.; Hu, F.F.; Shen, C.; Yin, W.H. Hub genes, diagnostic model, and predicted drugs in systemic sclerosis by integrated bioinformatics analysis. Front. Genet. 2023, 14, 1202561. [Google Scholar] [CrossRef]
- Darang, E.; Pezeshkian, Z.; Mirhoseini, S.Z.; Ghovvati, S. Bioinformatics and pathway enrichment analysis identified hub genes and potential biomarker for gastric cancer prognosis. Front. Oncol. 2023, 13, 1187521. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Huang, X.; Shao, K.; Li, G.; Wang, J.; Yang, H.; Hou, Y. Integrated bioinformatics analysis to identify 15 hub genes in breast cancer. Oncol. Lett. 2019, 18, 1023–1034. [Google Scholar] [CrossRef]
- Mo, X.C.; Zhang, Z.T.; Song, M.J.; Zhou, Z.Q.; Zeng, J.X.; Du, Y.F.; Sun, F.Z.; Yang, J.Y.; He, J.Y.; Huang, Y.; et al. Screening and identification of hub genes in bladder cancer by bioinformatics analysis and KIF11 is a potential prognostic biomarker. Oncol. Lett. 2021, 21, 205. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, M.K.; Mohammed, M.; Mwambi, H.; Omolo, B. Identification of Hub Genes Associated with Breast Cancer Using Integrated Gene Expression Data with Protein-Protein Interaction Network. Appl. Sci. 2023, 13, 2403. [Google Scholar] [CrossRef]
- Xue, J.M.; Liu, Y.; Wan, L.H.; Zhu, Y.X. Comprehensive Analysis of Differential Gene Expression to Identify Common Gene Signatures in Multiple Cancers. Med. Sci. Monit. 2020, 26, e919953. [Google Scholar] [CrossRef]
- Kuhn, D.J.; Dou, Q.P. The role of interleukin-2 receptor alpha in cancer. Front. Biosci. 2005, 10, 1462–1474. [Google Scholar] [CrossRef]
- Pan, Z.; Bao, L.; Lu, X.; Hu, X.; Li, L.; Chen, J.; Jin, T.; Zhang, Y.; Tan, Z.; Huang, P.; et al. IL2RA+VSIG4+ tumor-associated macrophage is a key subpopulation of the immunosuppressive microenvironment in anaplastic thyroid cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166591. [Google Scholar] [CrossRef]
- Hinterbrandner, M.; Rubino, V.; Stoll, C.; Forster, S.; Schnuriger, N.; Radpour, R.; Baerlocher, G.M.; Ochsenbein, A.F.; Riether, C. Tnfrsf4-expressing regulatory T cells promote immune escape of chronic myeloid leukemia stem cells. JCI Insight 2021, 6, e151797. [Google Scholar] [CrossRef]
- Ronchetti, S.; Ricci, E.; Petrillo, M.G.; Cari, L.; Migliorati, G.; Nocentini, G.; Riccardi, C. Glucocorticoid-induced tumour necrosis factor receptor-related protein: A key marker of functional regulatory T cells. J. Immunol. Res. 2015, 2015, 171520. [Google Scholar] [CrossRef]
- Soskic, B.; Jeffery, L.E.; Kennedy, A.; Gardner, D.H.; Hou, T.Z.; Halliday, N.; Williams, C.; Janman, D.; Rowshanravan, B.; Hirschfield, G.M.; et al. CD80 on Human T Cells Is Associated With FoxP3 Expression and Supports Treg Homeostasis. Front. Immunol. 2020, 11, 577655. [Google Scholar] [CrossRef]
- Jung, M.K.; Lee, J.S.; Kwak, J.E.; Shin, E.C. Tumor Necrosis Factor and Regulatory T Cells. Yonsei Med. J. 2019, 60, 126–131. [Google Scholar] [CrossRef]
- Chang, J.H.; Hu, H.; Jin, J.; Puebla-Osorio, N.; Xiao, Y.; Gilbert, B.E.; Brink, R.; Ullrich, S.E.; Sun, S.C. TRAF3 regulates the effector function of regulatory T cells and humoral immune responses. J. Exp. Med. 2014, 211, 137–151. [Google Scholar] [CrossRef]
- Nikolouli, E.; Elfaki, Y.; Herppich, S.; Schelmbauer, C.; Delacher, M.; Falk, C.; Mufazalov, I.A.; Waisman, A.; Feuerer, M.; Huehn, J. Recirculating IL-1R2+ Tregs fine-tune intrathymic Treg development under inflammatory conditions. Cell Mol. Immunol. 2021, 18, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yu, S.; Fitzgerald, D.C.; Elbehi, M.; Ciric, B.; Rostami, A.M.; Zhang, G.X. IL-12Rβ2 promotes the development of CD4+CD25+ regulatory T cells. J. Immunol. 2008, 181, 3870–3876. [Google Scholar] [CrossRef] [PubMed]
- Haruna, M.; Ueyama, A.; Yamamoto, Y.; Hirata, M.; Goto, K.; Yoshida, H.; Higuchi, N.; Yoshida, T.; Kidani, Y.; Nakamura, Y.; et al. The impact of CCR8+ regulatory T cells on cytotoxic T cell function in human lung cancer. Sci. Rep. 2022, 12, 5377. [Google Scholar] [CrossRef] [PubMed]
- Khatun, A.; Wu, X.; Qi, F.; Gai, K.; Kharel, A.; Kudek, M.R.; Fraser, L.; Ceicko, A.; Kasmani, M.Y.; Majnik, A.; et al. BATF is Required for Treg Homeostasis and Stability to Prevent Autoimmune Pathology. Adv. Sci. (Weinh) 2023, 10, e2206692. [Google Scholar] [CrossRef]
- Andersson, J.; Tran, D.Q.; Pesu, M.; Davidson, T.S.; Ramsey, H.; O’Shea, J.J.; Shevach, E.M. CD4+FoxP3+ regulatory T cells confer infectious tolerance in a TGF-β-dependent manner. J. Exp. Med. 2008, 205, 1975–1981. [Google Scholar] [CrossRef]
- Guo, C.; Dai, X.; Du, Y.; Xiong, X.; Gui, X. Preclinical development of a novel CCR8/CTLA-4 bispecific antibody for cancer treatment by disrupting CTLA-4 signaling on CD8 T cells and specifically depleting tumor-resident Tregs. Cancer Immunol. Immunother. 2024, 73, 210. [Google Scholar] [CrossRef]
- Chen, Q.; Shen, M.; Yan, M.; Han, X.; Mu, S.; Li, Y.; Li, L.; Wang, Y.; Li, S.; Li, T.; et al. Targeting tumor-infiltrating CCR8+ regulatory T cells induces antitumor immunity through functional restoration of CD4+ Tconvs and CD8+ T cells in colorectal cancer. J. Transl. Med. 2024, 22, 709. [Google Scholar] [CrossRef]
- Li, Y.; Tang, H.; Huang, Z.; Qin, H.; Cen, Q.; Meng, F.; Huang, L.; Lin, L.; Pu, J.; Yang, D. Bioinformatics analysis and identification of genes and pathways involved in patients with Wilms tumor. Transl. Cancer Res. 2022, 11, 2843–2857. [Google Scholar] [CrossRef]
- Matsuoka, T.; Yashiro, M. Bioinformatics Analysis and Validation of Potential Markers Associated with Prediction and Prognosis of Gastric Cancer. Int. J. Mol. Sci. 2024, 25, 5880. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Martinez, A.; Gong, C.; Wang, H.; Sove, R.J.; Mi, H.; Kimko, H.; Popel, A.S. Simulations of tumor growth and response to immunotherapy by coupling a spatial agent-based model with a whole-patient quantitative systems pharmacology model. PLoS Comput. Biol. 2022, 18, e1010254. [Google Scholar] [CrossRef] [PubMed]
- Macklin, P. Key challenges facing data-driven multicellular systems biology. Gigascience 2019, 8, giz127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Spath, S.S.; Marjani, S.L.; Zhang, W.; Pan, X. Characterization of cancer genomic heterogeneity by next-generation sequencing advances precision medicine in cancer treatment. Precis. Clin. Med. 2018, 1, 29–48. [Google Scholar] [CrossRef]
- Liston, A.; Humblet-Baron, S.; Duffy, D.; Goris, A. Human immune diversity: From evolution to modernity. Nat. Immunol. 2021, 22, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y. Bioinformatics. Current limitations and insights for the future. Plant Physiol. 2005, 138, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, J.; Chen, J. The integration of differentially expressed genes based on multiple microarray datasets for prediction of the prognosis in oral squamous cell carcinoma. Bioengineered 2021, 12, 3309–3321. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Harris, M.A.; Clark, J.; Ireland, A.; Lomax, J.; Ashburner, M.; Foulger, R.; Eilbeck, K.; Lewis, S.; Marshall, B.; Mungall, C.; et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kim, N.; Kim, M.H.; Pyo, J.; Lee, S.M.; Jang, J.S.; Lee, D.W.; Kim, K.W. CCR8 as a Therapeutic Novel Target: Omics-Integrated Comprehensive Analysis for Systematically Prioritizing Indications. Biomedicines 2023, 11, 2910. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Wang, X. TCGAplot: An R package for integrative pan-cancer analysis and visualization of TCGA multi-omics data. BMC Bioinform. 2023, 24, 483. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession No. | Platform | Total Samples | Case (Treg Group) | Control (Non-Treg Group) | Species | Disease/Condition | Pubmed ID |
|---|---|---|---|---|---|---|---|
| GSE128822 | GPL18573 | 10 | 5 | 5 | Homo sapiens | Lung adenocarcinoma | 32125291, 34552178 |
| GSE116347 | GPL18573 | 18 | 12 | 6 | Homo sapiens | Colorectal carcinoma | 30348759 |
| GSE120280 | GPL19057 | 12 | 6 | 6 | Mus musculus | Lung cancer | 32878747 |
| Pathway ID | Description | p-Value | Count |
|---|---|---|---|
| R-HSA-5669034 | TNFs bind their physiological receptors | 4.00 × 10−6 | 4 |
| R-HSA-5668541 | TNFR2 non-canonical NF-kB pathway | 4.00 × 10−5 | 5 |
| R-HSA-449147 | Signaling by Interleukins | 5.80 × 10−5 | 9 |
| R-HSA-8877330 | RUNX1 and FOXP3 control the development of regulatory T lymphocytes (Tregs) | 6.27 × 10−4 | 2 |
| R-HSA-8984722 | Interleukin-35 Signaling | 9.15 × 10−4 | 2 |
| Degree Centrality | Betweenness Centrality | Closeness Centrality | Clustering Coefficient | |
|---|---|---|---|---|
| IL2RA | 11 | 0.29 | 0.76 | 0.47 |
| TRAF1 | 8 | 0.13 | 0.62 | 0.57 |
| IL1R2 | 4 | 0.13 | 0.53 | 0.33 |
| BATF | 4 | 0.13 | 0.52 | 0.50 |
| IL12RB2 | 4 | 0.13 | 0.50 | 0.53 |
| CD80 | 9 | 0.12 | 0.64 | 0.69 |
| TNFRSF4 | 9 | 0.06 | 0.64 | 0.69 |
| TNFRSF18 | 9 | 0.06 | 0.64 | 0.69 |
| TRAF3 | 6 | 0.03 | 0.50 | 0.73 |
| CCR8 | 7 | 0.03 | 0.57 | 0.81 |
| Classification | Gene Symbol | Function |
|---|---|---|
| Immune Regulation | IL2RA | IL2RA (Interleukin-2 Receptor Alpha) is crucial for the development and function of Treg cells [46]. It plays a role in the high-affinity IL-2 receptor, which is important for Treg cell proliferation and survival [47]. |
| TNFRSF4 | TNFRSF4 (Tumor Necrosis Factor Receptor Superfamily, Member 4), also known as OX40, is involved in the activation, survival, and migration of Treg cells. It enhances Treg cell proliferation and function [48]. | |
| TNFRSF18 | TNFRSF18 (Tumor Necrosis Factor Receptor Superfamily, Member 18), also known as GITR, is important for Treg cell function and immune regulation. It contributes to Treg cell-mediated suppression and enhances their survival [49]. | |
| Cytokine Signaling | CD80 | CD80 (Cluster of Differentiation 80) is involved in costimulatory signaling essential for T cell activation. It provides necessary second signals for T cell activation and survival through interaction with CD28 and CTLA-4 [50]. |
| TRAF1 | TRAF1 (TNF Receptor-Associated Factor 1) plays roles in the downstream signaling of TNFRSF members, contributing to immune-response regulation and inflammation [51]. | |
| TRAF3 | TRAF3 (TNF Receptor-Associated Factor 3) is involved in various signaling pathways, including those triggered by TNF receptors, contributing to immune-response regulation and apoptosis [52]. | |
| IL1R2 | IL1R2 (Interleukin 1 Receptor Type 2) acts as a decoy receptor for IL-1, modulating immune and inflammatory responses by sequestering IL-1 and preventing it from interacting with the IL1 signaling receptor [53]. | |
| IL12RB2 | IL12RB2 (Interleukin 12 Receptor Beta 2) is involved in cytokine receptor activity and plays a role in the differentiation of Th1 cells, impacting immune cell signaling and response to infections [54]. | |
| Chemokine Signaling and Treg Migration | CCR8 | CCR8 (C-C Chemokine Receptor Type 8) has a significant role in chemokine signaling, crucial for the migration and positioning of Treg cells within the tumor microenvironment [55]. Its high clustering coefficient (0.81) highlights its role in chemokine-mediated processes that guide Treg cells to their functional sites (Table 3). |
| Transcription Factors | BATF | BATF (Basic Leucine Zipper ATF-Like Transcription Factor) is involved in T cell differentiation [56]. It functions as a transcription factor that regulates the expression of genes essential for the development and function of various T cell subsets, including Tregs. |
| Correlation with CCR8 Expression | Treg Infiltration Correlation | TMB | MSI | Clinical Status of ICIs | Final Score | |||
|---|---|---|---|---|---|---|---|---|
| PDCD1 | CD274 | CTLA4 | ||||||
| BRCA | 3 | 3 | 3 | 3 | 9 | 6 | 3 | 30 |
| HNSC | 2 | 2 | 2 | 3 | 6 | 6 | 3 | 24 |
| COAD | 2 | 2 | 2 | 2 | 6 | 6 | 3 | 23 |
| STAD | 2 | 2 | 2 | 3 | 6 | 5 | 3 | 23 |
| THCA | 2 | 0 | 2 | 3 | 4 | 3 | 0 | 14 |
| READ | 0 | 0 | 1 | 1 | 6 | 5 | 0 | 13 |
| Criteria | Score | Significance Level | Description |
|---|---|---|---|
| Correlation (p-Value) | |||
| p-value < 1 × 10−50 | 3 | Highly significant correlation | Calculated the correlation coefficients and p-values for the relationships between CCR8 and PD-1, PD-L1, and CTLA-4 |
| 1 × 10−50 ≤ p-value < 1 × 10−30 | 2 | Very significant correlation | |
| 1 × 10−30 ≤ p-value < 1 × 10−10 | 1 | Significant correlation | |
| p-value ≥ 1 × 10−10 | 0 | Not significant | |
| TMB & MSI Correlation | |||
| Correlation > 0.3 | 3 | Strong positive correlation | Correlations between TMB, MSI, and hub gene to measure genetic instability and mutational burden within the cancer types |
| 0.2 < Correlation ≤ 0.3 | 2 | Moderate positive correlation | |
| 0.1 < Correlation ≤ 0.2 | 1 | Weak positive correlation | |
| Correlation ≤ 0.1 | 0 | No correlation | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, N.; Na, S.; Pyo, J.; Jang, J.; Lee, S.-M.; Kim, K. A Bioinformatics Investigation of Hub Genes Involved in Treg Migration and Its Synergistic Effects, Using Immune Checkpoint Inhibitors for Immunotherapies. Int. J. Mol. Sci. 2024, 25, 9341. https://doi.org/10.3390/ijms25179341
Kim N, Na S, Pyo J, Jang J, Lee S-M, Kim K. A Bioinformatics Investigation of Hub Genes Involved in Treg Migration and Its Synergistic Effects, Using Immune Checkpoint Inhibitors for Immunotherapies. International Journal of Molecular Sciences. 2024; 25(17):9341. https://doi.org/10.3390/ijms25179341
Chicago/Turabian StyleKim, Nari, Seoungwon Na, Junhee Pyo, Jisung Jang, Soo-Min Lee, and Kyungwon Kim. 2024. "A Bioinformatics Investigation of Hub Genes Involved in Treg Migration and Its Synergistic Effects, Using Immune Checkpoint Inhibitors for Immunotherapies" International Journal of Molecular Sciences 25, no. 17: 9341. https://doi.org/10.3390/ijms25179341