
A Multi-Omics Study of Epigenetic Changes in Type II Alveolar Cells of A/J Mice Exposed to Environmental Tobacco Smoke

 , , , , , , and
, , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Animal Studies
2.2. Histopathological Examination of Lung Tissues
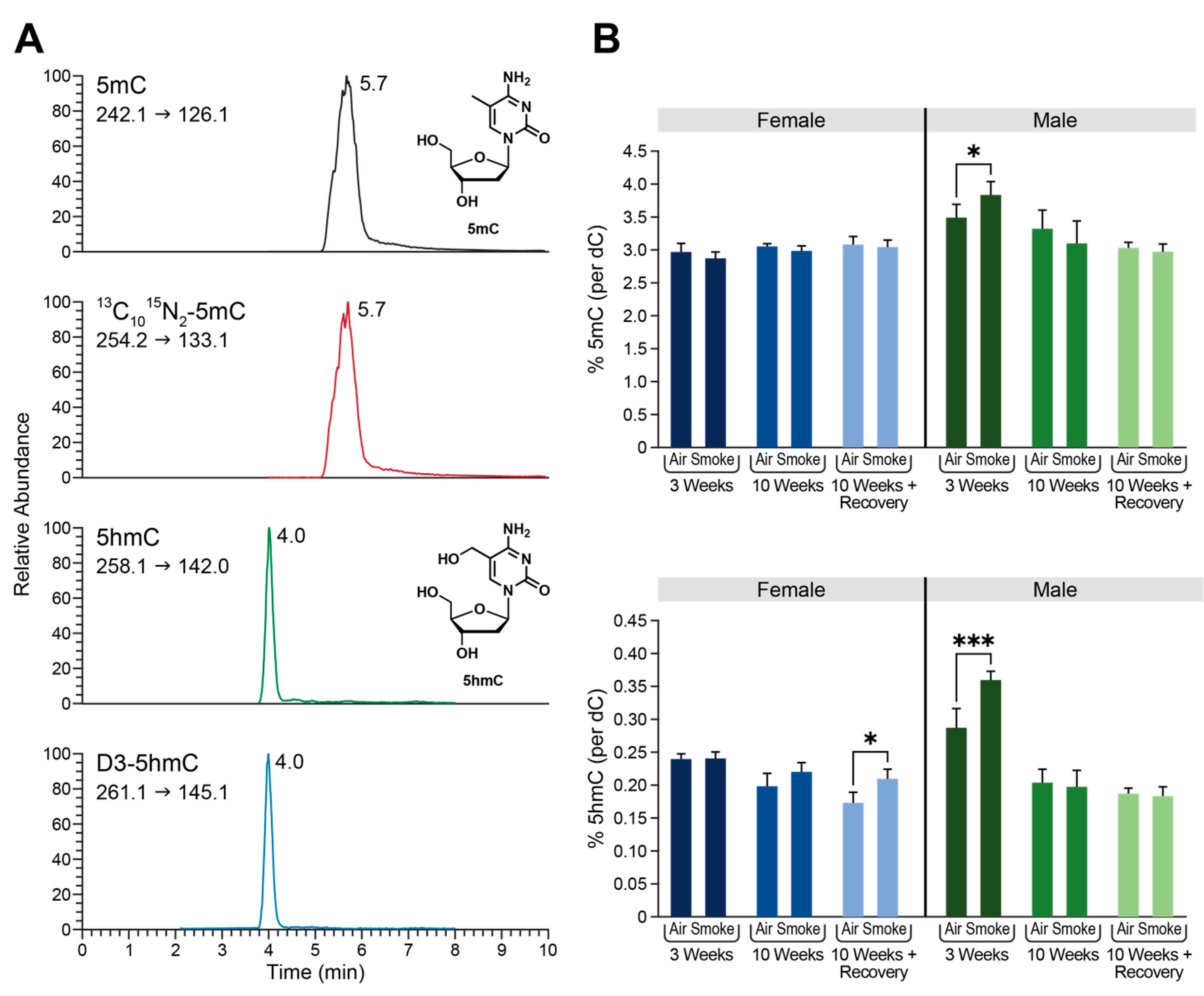
2.3. Global Changes in DNA Methylation and Hydroxymethylation in Type II Alveolar Cells of Mice Exposed to ECS
2.4. Differential Methylation Analysis Reveals Extensive DMRs and DhMRs in Type II Alveolar Epithelial CELLS of Female A/J Mice Exposed to ECS
2.5. Gene Expression Changes in Type II Alveolar Epithelial cells of A/J Mice Exposed to ECS
2.6. Protein Abundance Changes in Type II Alveolar Epithelial Cells of A/J Mice Exposed to ECS
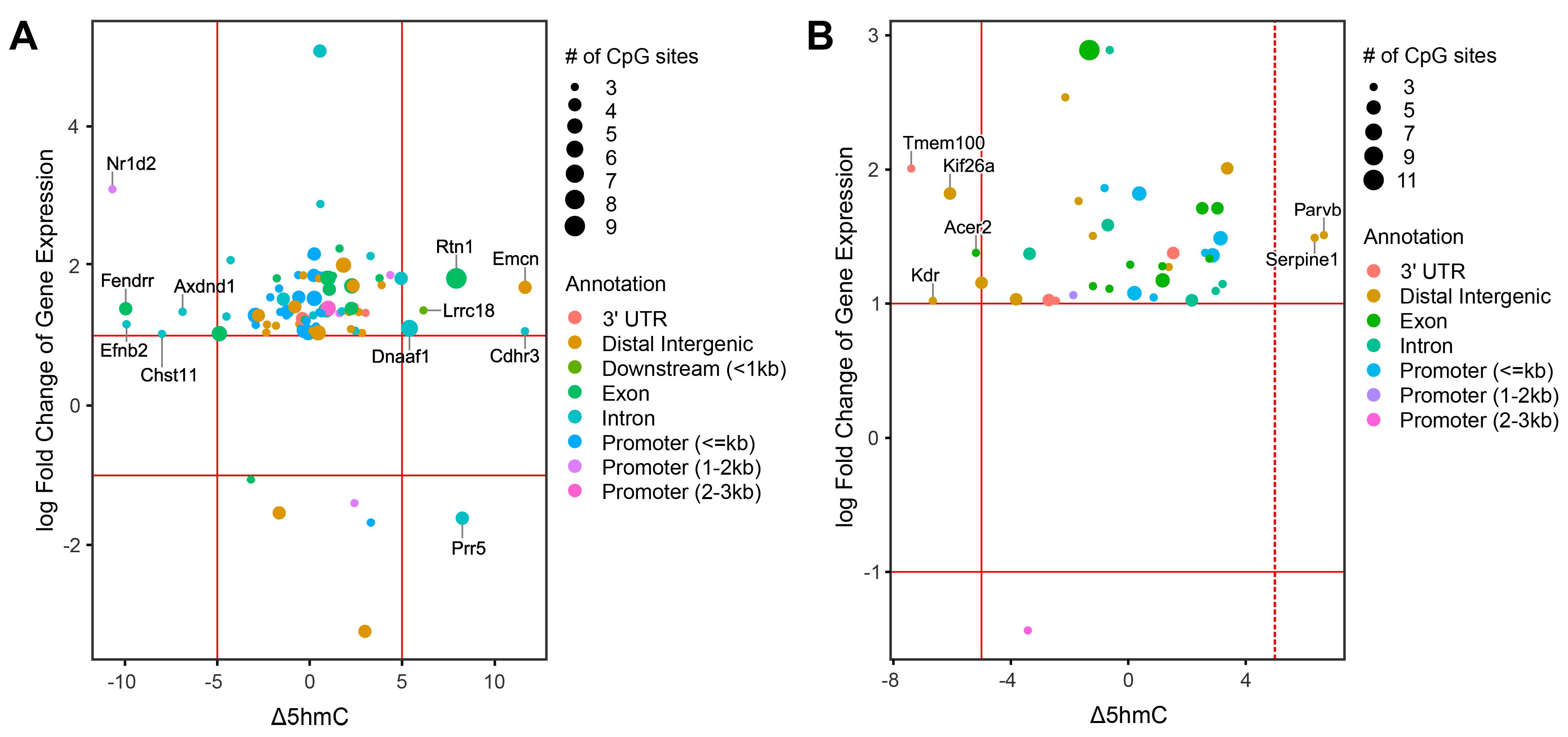
2.7. Integration of Epigenomic and Transcriptomic Data Identifies DEGs Regulated by DNA Methylation and Hydroxymethylation
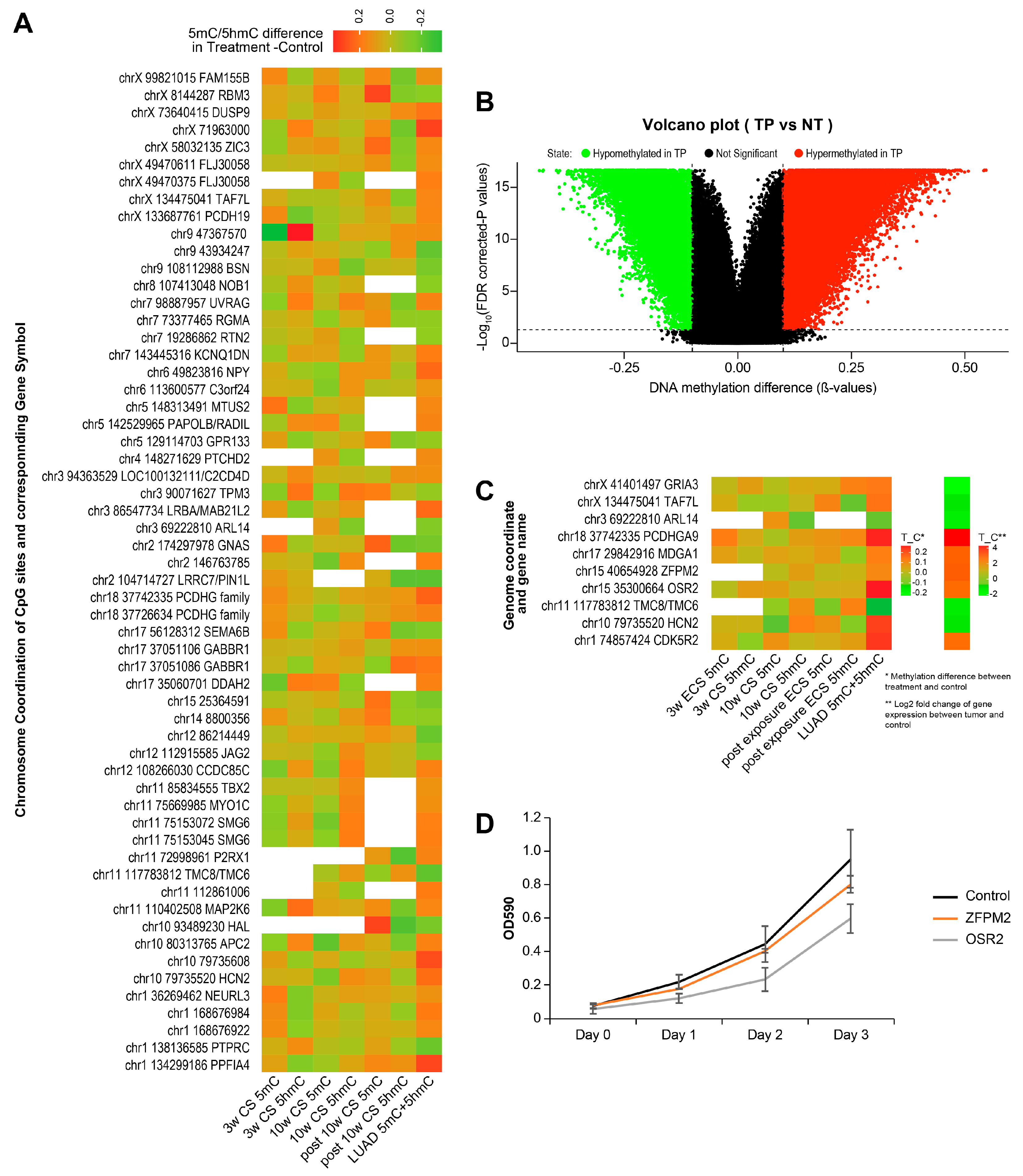
2.8. Identification of Smoking-Induced Early Epigenetic Changes Found in Lung Adenocarcinoma (LUAD)
2.9. Identification of Smoking-Induced Protein Abundance Changes Relevant to Lung Adenocarcinoma (LUAD)
3. Discussion
4. Materials and Methods
4.1. Animal Treatments
4.2. Histopathology Examination
4.3. Isolation of Alveolar Type II Epithelial Cells
4.4. Extraction of DNA, RNA, and Protein from Alveolar Type II Epithelial Cells
4.5. RNA-Seq Analysis of Alveolar Type II Epithelial Cell RNA
4.5.1. RNA-Seq Read Processing
4.5.2. Gene Expression Quantification and Filtering
4.5.3. Differential Gene Expression Testing
4.5.4. Network Analysis
4.6. RNA-Seq Validation via qRT-PCR
4.7. RRBS and Oxo-RRBS
4.7.1. Reduced Representation Bisulfite Sequencing Read Handling
4.7.2. Methylation and Hydroxymethylation Analysis
4.8. DNA Digestion and HPLC Enrichment of mC and hmC
4.9. HPLC-ESI+-MS/MS Quantitation of Global Levels of mC and hmC
4.10. Protein Extraction and Quantitation
4.11. Protein Digestion and Processing
4.12. C18 Stage-Tip TMT Labeling of Peptides
4.13. High pH Fractionation of Peptides
4.14. HPLC-MS/MS Analysis of Proteins
4.15. Proteomics Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Billatos, E.; Vick, J.L.; Lenburg, M.E.; Spira, A.E. The airway transcriptome as a biomarker for early lung cancer detection. Clin. Cancer Res. 2018, 24, 2984–2992. [Google Scholar] [CrossRef]
- Durham, A.L.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat. Rev. Cancer 2004, 4, 707–717. [Google Scholar] [CrossRef]
- Han, Q.; Kono, T.J.Y.; Knutson, C.G.; Parry, N.M.; Seiler, C.L.; Fox, J.G.; Tannenbaum, S.R.; Tretyakova, N.Y. Multi-omics characterization of inflammatory bowel disease-induced hyperplasia/dysplasia in the Rag2−/−/Il10−/− mouse model. Int. J. Mol. Sci. 2020, 22, 364. [Google Scholar] [CrossRef]
- Seiler, C.L.; Song, J.M.; Fernandez, J.; Abrahante, J.E.; Kono, T.J.Y.; Chen, Y.; Ren, Y.; Kassie, F.; Tretyakova, N.Y. Epigenetic changes in alveolar type ii lung cells of A/J mice following intranasal treatment with lipopolysaccharide. Chem. Res. Toxicol. 2019, 32, 831–839. [Google Scholar] [CrossRef]
- Seiler, C.L.; Song, J.U.M.; Kotandeniya, D.; Chen, J.; Kono, T.J.Y.; Han, Q.; Colwell, M.; Auch, B.; Sarver, A.L.; Upadhyaya, P.; et al. Inhalation exposure to cigarette smoke and inflammatory agents induces epigenetic changes in the lung. Sci. Rep. 2020, 10, 11290. [Google Scholar] [CrossRef]
- Melkamu, T.; Qian, X.; Upadhyaya, P.; O’Sullivan, M.G.; Kassie, F. Lipopolysaccharide enhances mouse lung tumorigenesis: A model for inflammation-driven lung cancer. Vet. Pathol. 2013, 50, 895–902. [Google Scholar] [CrossRef]
- Qian, X.; Khammanivong, A.; Song, J.M.; Teferi, F.; Upadhyaya, P.; Dickerson, E.; Kassie, F. RNA-sequencing studies identify genes differentially regulated during inflammation-driven lung tumorigenesis and targeted by chemopreventive agents. Inflamm. Res. 2015, 64, 343–361. [Google Scholar] [CrossRef]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef]
- Kharitonov, S.A.; Sjöbring, U. Lipopolysaccharide challenge of humans as a model for chronic obstructive lung disease exacerbations. Contrib. Microbiol. 2007, 14, 83–100. [Google Scholar] [PubMed]
- Korsgren, M.; Linden, M.; Entwistle, N.; Cook, J.; Wollmer, P.; Andersson, M.; Larsson, B.; Greiff, L. Inhalation of LPS induces inflammatory airway responses mimicking characteristics of chronic obstructive pulmonary disease. Clin. Physiol. Funct. Imaging 2012, 32, 71–79. [Google Scholar] [CrossRef]
- Lin, C.; Song, H.; Huang, C.; Yao, E.; Gacayan, R.; Xu, S.-M.; Chuang, P.-T. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS ONE 2012, 7, e53817. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Rock, J.R.; Lu, Y.; Futtner, C.; Schwab, B.; Guinney, J.; Hogan, B.L.M.; Onaitis, M.W. Evidence for type II cells as cells of origin of K-Ras–induced distal lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 4910–4915. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, A.; Guida, F.; Leffondre, K.; Cenee, S.; Cyr, D.; Schmaus, A.; Radoi, L.; Paget-Bailly, S.; Carton, M.; Menvielle, G.; et al. Heavy smoking and lung cancer: Are women at higher risk? Result of the ICARE study. Br. J. Cancer 2014, 110, 1385–1391. [Google Scholar] [CrossRef]
- Zhang, F.F.; Cardarelli, R.; Carroll, J.; Fulda, K.G.; Kaur, M.; Gonzalez, K.; Vishwanatha, J.K.; Santella, R.M.; Morabia, A. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011, 6, 623–629. [Google Scholar] [CrossRef]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and non-CpG Methylation in epigenetic gene regulation and brain function. Genes 2017, 8, 148. [Google Scholar] [CrossRef]
- Docherty, L.E.; Rezwan, F.I.; Poole, R.L.; Jagoe, H.; Lake, H.; Lockett, G.A.; Arshad, H.; Wilson, D.I.; Holloway, J.W.; Temple, I.K.; et al. Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes. J. Med. Genet. 2014, 51, 229–238. [Google Scholar] [CrossRef]
- Song, Q.; Decato, B.; Hong, E.E.; Zhou, M.; Fang, F.; Qu, J.; Garvin, T.; Kessler, M.; Zhou, J.; Smith, A.D. A reference methylome database and analysis pipeline to facilitate integrative and comparative epigenomics. PLoS ONE 2013, 8, e81148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Gao, C.; Xing, Y.; Qi, Z.; Liu, R.; Wang, Y.; Zhang, X.; Yang, Y.G.; Li, X.; et al. 5-Hydroxymethylome in circulating cell-free DNA as a potential biomarker for non-small-cell lung cancer. Genom. Proteom. Bioinform. 2018, 16, 187–199. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Wang, R.C.; Cheng, K.; Ring, B.Z.; Su, L. Roles of Rap1 signaling in tumor cell migration and invasion. Cancer Biol. Med. 2017, 14, 90–99. [Google Scholar]
- Han, Y. Analysis of the role of the Hippo pathway in cancer. J. Transl. Med. 2019, 17, 116. [Google Scholar] [CrossRef]
- Rapp, J.; Jaromi, L.; Kvell, K.; Miskei, G.; Pongracz, J.E. WNT signaling—Lung cancer is no exception. Respir. Res. 2017, 18, 167. [Google Scholar] [CrossRef]
- Sanaei, M.J.; Razi, S.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/mTOR pathway in lung cancer; oncogenic alterations, therapeutic opportunities, challenges, and a glance at the application of nanoparticles. Transl. Oncol. 2022, 18, 101364. [Google Scholar] [CrossRef]
- Eferl, R.; Wagner, E.F. AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [Google Scholar] [CrossRef]
- Gan, Y.; Li, Y.; Li, T.; Shu, G.; Yin, G. CCNA2 acts as a novel biomarker in regulating the growth and apoptosis of colorectal cancer. Cancer Manag. Res. 2018, 10, 5113–5124. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Wang, X.; Liu, J.; Zhang, M.; Feng, K. Expression and prognostic value of. J. Int. Med. Res. 2021, 49, 300060520980647. [Google Scholar] [PubMed]
- Ni, M.; Liu, X.; Wu, J.; Zhang, D.; Tian, J.; Wang, T.; Liu, S.; Meng, Z.; Wang, K.; Duan, X.; et al. Identification of candidate biomarkers correlated with the pathogenesis and prognosis of non-small cell lung cancer via integrated bioinformatics analysis. Front. Genet. 2018, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Massó-Vallés, D.; Beaulieu, M.E.; Soucek, L. MYC, MYCL, and MYCN as therapeutic targets in lung cancer. Expert Opin. Ther. Targets 2020, 24, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. Entry into mitosis: A solution to the decades-long enigma of MPF. Chromosoma 2015, 124, 417–428. [Google Scholar] [CrossRef]
- Zhang, C.; Min, L.; Zhang, L.; Ma, Y.; Yang, Y.; Shou, C. Combined analysis identifies six genes correlated with augmented malignancy from non-small cell to small cell lung cancer. Tumour Biol. 2016, 37, 2193–2207. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.S.; Luo, M.; Zhao, H.M.; Sun, H. Overexpression of PKMYT1 indicates the poor prognosis and enhances proliferation and tumorigenesis in non-small cell lung cancer via activation of Notch signal pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4210–4219. [Google Scholar]
- Stratmann, J.A.; Sebastian, M. Polo-like kinase 1 inhibition in NSCLC: Mechanism of action and emerging predictive biomarkers. Lung Cancer 2019, 10, 67–80. [Google Scholar] [CrossRef]
- Tsai, Y.M.; Wu, K.L.; Chang, Y.Y.; Hung, J.Y.; Chang, W.A.; Chang, C.Y.; Jian, S.F.; Tsai, P.H.; Huang, Y.C.; Chong, I.W.; et al. Upregulation of Thr/Tyr kinase increases the cancer progression by neurotensin and dihydropyrimidinase-like 3 in lung cancer. Int. J. Mol. Sci. 2020, 21, 1640. [Google Scholar] [CrossRef]
- Hwang, J.W.; Sundar, I.K.; Yao, H.; Sellix, M.T.; Rahman, I. Circadian clock function is disrupted by environmental tobacco/cigarette smoke, leading to lung inflammation and injury via a SIRT1-BMAL1 pathway. Vet. Pathol. 2014, 28, 176–194. [Google Scholar] [CrossRef]
- Yu, J.; Peng, W.; Xue, Y.; Li, Y.; Yang, L.; Geng, Y. FUBP1 promotes the proliferation of lung squamous carcinoma cells and regulates tumor immunity through PD-L1. Allergol. Immunopath. 2022, 50, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Xinlu, C.; Lu, L.; Yanqiao, Z.; Wei, Z.; Chonggao, Y.; Hongli, L. miR-30b-3p Inhibits the proliferation and invasion of lung adenocarcinoma by targeting COX6B1. Zhongguo Fei Ai Za Zhi 2022, 25, 567–574. [Google Scholar]
- Schweikert, E.; Devarajan, A.; Witte, I.; Wilgenbus, P.; Amort, J.; Förstermann, U.; Shabazian, A.; Grijalva, V.; Shih, D.; Farias-Eisner, R. PON3 is upregulated in cancer tissues and protects against mitochondrial superoxide-mediated cell death. Cell Death Differ. 2012, 19, 1549–1560. [Google Scholar] [CrossRef]
- Di, X.; Jin, X.; Ma, H.; Wang, R.; Cong, S.; Tian, C.; Liu, J.; Zhao, M.; Li, R.; Wang, K. The oncogene IARS2 promotes non-small cell lung cancer tumorigenesis by activating the AKT/MTOR pathway. Front. Oncol. 2019, 9, 393. [Google Scholar] [CrossRef]
- Zamay, T.N.; Zamay, G.S.; Kolovskaya, O.S.; Zukov, R.A.; Petrova, M.M.; Gargaun, A.; Berezovski, M.V.; Kichkailo, A.S. Current and prospective protein biomarkers of lung cancer. Cancers 2017, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Laffin, B.; Petrash, J.M. Expression of the aldo-ketoreductases AKR1B1 and AKR1B10 in human cancers. Front. Pharmacol. 2012, 3, 104. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, L.; Ma, R.; Wang, Y.; Chen, X.; Liu, H.; Ji, Y.; Liu, T.; Gao, P. E2F1 induces KIF26A transcription and promotes cell cycle progression. Front. Oncol. 2020, 10, 530933. [Google Scholar]
- Mao, C.; Xu, R.; Obeid, L.M. Alkaline ceramidase 2 (ACER2) promotes tumor angiogenesis and growth via sphingosine-1-phosphate. FASEB J. 2011, 25, 739.10. [Google Scholar]
- Arroyo-Solera, I.; Pavón, M.; León, X.; López, M.; Gallardo, A.; Céspedes, M.V.; Casanova, I.; Pallarès, V.; López-Pousa, A.; Mangues, M.A.; et al. Effect of serpinE1 overexpression on the primary tumor and lymph node, and lung metastases in head and neck squamous cell carcinoma. Head Neck 2019, 41, 429–439. [Google Scholar] [CrossRef]
- Yu, M.; Li, W.; Wang, Q.; Wang, Y.; Lu, F. Circadian regulator NR1D2 regulates glioblastoma cell proliferation and motility. Oncogene 2018, 37, 4838–4853. [Google Scholar] [CrossRef]
- Abéngozar, M.A.; de Frutos, S.; Ferreiro, S.; Soriano, J.; Perez-Martinez, M.; Olmeda, D.; Marenchino, M.; Cañamero, M.; Ortega, S.; Megias, D.; et al. Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood 2012, 119, 4565–4576. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-M.; Chiu, I.-A.; Jan, Y.-H.; Hsiao, M. CHST11 Promotes lung cancer metastasis through changing intracellular iron metabolism. FASEB J. 2019, 33, 127.3. [Google Scholar] [CrossRef]
- Herman, D.; Leakey, T.I.; Behrens, A.; Yao-Borengasser, A.; Cooney, C.A.; Jousheghany, F.; Phanavanh, B.; Siegel, E.R.; Safar, A.M.; Korourian, S.; et al. CHST11 gene expression and DNA methylation in breast cancer. Int. J. Oncol. 2015, 46, 1243–1251. [Google Scholar] [CrossRef]
- Lawrence, M.; Gentleman, R.; Carey, V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics 2009, 25, 1841–1842. [Google Scholar] [CrossRef]
- Han, S.; Cao, D.; Sha, J.; Zhu, X.; Chen, D. LncRNA ZFPM2-AS1 promotes lung adenocarcinoma progression by interacting with UPF1 to destabilize ZFPM2. Mol. Oncol. 2020, 14, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, P.A.; Markey, S.P.; Roth, J.; Mirokhin, Y.; Yan, X.; Tchekhovskoi, D.V.; Edwards, N.J.; Thangudu, R.R.; Ketchum, K.A.; Kinsinger, C.R.; et al. A Description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) common data analysis pipeline. J. Proteome Res. 2016, 15, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Walser, T.; Cui, X.; Yanagawa, J.; Lee, J.M.; Heinrich, E.; Lee, G.; Sharma, S.; Dubinett, S.M. Smoking and lung cancer: The role of inflammation. Proc. Am. Thorac. Soc. 2008, 5, 811–815. [Google Scholar] [CrossRef]
- Alberg, A.J.; Brock, M.V.; Ford, J.G.; Samet, J.M.; Spivack, S.D. Epidemiology of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2013, 143 (Suppl. S5), e1S–e29S. [Google Scholar] [CrossRef]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef] [PubMed]
- Hasday, J.D.; Bascom, R.; Costa, J.J.; Fitzgerald, T.; Dubin, W. Bacterial endotoxin is an active component of cigarette smoke. Chest 1999, 115, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.; Kavakli, I.H.; Goel, N.; Cardinale, C.J.; Dinges, D.F.; Kuna, S.T.; Maislin, G.; Van Dongen, H.P.; Tufik, S.; Hogenesch, J.B.; et al. A novel BHLHE41 variant is associated with short sleep and resistance to sleep deprivation in humans. Sleep 2014, 37, 1327–1336. [Google Scholar] [CrossRef]
- Lopez-Molina, L.; Conquet, F.; Dubois-Dauphin, M.; Schibler, U. The DBP gene is expressed according to a circadian rhythm in the suprachiasmatic nucleus and influences circadian behavior. EMBO J. 1997, 16, 6762–6771. [Google Scholar] [CrossRef]
- Katamune, C.; Koyanagi, S.; Hashikawa, K.I.; Kusunose, N.; Akamine, T.; Matsunaga, N.; Ohdo, S. Mutation of the gene encoding the circadian clock component PERIOD2 in oncogenic cells confers chemoresistance by up-regulating the Aldh3a1 gene. J. Biol. Chem. 2019, 294, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Hayashi, S.; Isagawa, T.; Oshima, M.; Iwama, A.; Shimba, S.; Okamura, H.; Manabe, I. Bmal1 regulates inflammatory responses in macrophages by modulating enhancer RNA transcription. Sci. Rep. 2017, 7, 7086. [Google Scholar] [CrossRef] [PubMed]
- Lamb, T.; Muthumalage, T.; Meehan-Atrash, J.; Rahman, I. Nose-only exposure to cherry- and tobacco-flavored e-cigarettes induced lung inflammation in mice in a sex-dependent manner. Toxics 2022, 10, 471. [Google Scholar] [CrossRef]
- Sainz de Aja, J.; Dost, A.F.M.; Kim, C.F. Alveolar progenitor cells and the origin of lung cancer. J. Intern. Med. 2021, 289, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Gereke, M.; Autengruber, A.; Grobe, L.; Jeron, A.; Bruder, D.; Stegemann-Koniszewski, S. Flow cytometric isolation of primary murine type II alveolar epithelial cells for functional and molecular studies. J. Vis. Exp. 2012, 70, 4322. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Baller, J.; Kono, T.; Herman, A.; Zhang, Y. CHURP: A lightweight CLI framework to enable novice users to analyze sequencing datasets in parallel. In Proceedings of the Practice and Experience in Advanced Research Computing on Rise of the Machines (Learning), Chicago, IL, USA, 28 July–1 August 2019; Association for Computing Machinery: Chicago, IL, USA, 2019; pp. 1–5. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Hoffman, G.E.; Schadt, E.E. variancePartition: Interpreting drivers of variation in complex gene expression studies. BMC Bioinform. 2016, 17, 483. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Qu, J.; Zhou, M.; Song, Q.; Hong, E.E.; Smith, A.D. MLML: Consistent simultaneous estimates of DNA methylation and hydroxymethylation. Bioinformatics 2013, 29, 2645–2646. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Johnston, H.E.; Yadav, K.; Kirkpatrick, J.M.; Biggs, G.S.; Oxley, D.; Kramer, H.B.; Samant, R.S. Solvent Precipitation SP3 (SP4) enhances recovery for proteomics sample preparation without magnetic beads. Anal. Chem. 2022, 94, 10320–10328. [Google Scholar] [CrossRef]
- Myers, S.A.; Rhoads, A.; Cocco, A.R.; Peckner, R.; Haber, A.L.; Schweitzer, L.D.; Krug, K.; Mani, D.R.; Clauser, K.R.; Rozenblatt-Rosen, O.; et al. Streamlined protocol for deep proteomic profiling of fac-sorted cells and its application to freshly isolated murine immune cells. Mol. Cell. Proteom. 2019, 18, 995–1009. [Google Scholar] [CrossRef]
- Dimayacyac-Esleta, B.R.; Tsai, C.F.; Kitata, R.B.; Lin, P.Y.; Choong, W.K.; Lin, T.D.; Wang, Y.T.; Weng, S.H.; Yang, P.C.; Arco, S.D.; et al. Rapid high-pH reverse phase StageTip for sensitive small-scale membrane proteomic profiling. Anal. Chem. 2015, 87, 12016–12023. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Dan, K.; Shin, H.; Lee, J.; Wang, J.I.; Han, D. An efficient method for high-pH peptide fractionation based on C18 StageTips for in-depth proteome profiling. Anal. Methods 2019, 11, 4693–4698. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Yu, S.H.; Ferretti, D.; Schessner, J.P.; Rudolph, J.D.; Borner, G.H.H.; Cox, J. Expanding the Perseus software for omics data analysis with custom plugins. Curr. Protoc. Bioinform. 2020, 71, e105. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Kumar, P.; Panigrahi, P.; Johnson, J.; Weber, W.J.; Mehta, S.; Sajulga, R.; Easterly, C.; Crooker, B.A.; Heydarian, M.; Anamika, K.; et al. QuanTP: A Software resource for quantitative proteo-transcriptomic comparative data analysis and informatics. J. Proteome Res. 2019, 18, 782–790. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Q.; Fernandez, J.; Rajczewski, A.T.; Kono, T.J.Y.; Weirath, N.A.; Rahim, A.; Lee, A.S.; Seabloom, D.; Tretyakova, N.Y. A Multi-Omics Study of Epigenetic Changes in Type II Alveolar Cells of A/J Mice Exposed to Environmental Tobacco Smoke. Int. J. Mol. Sci. 2024, 25, 9365. https://doi.org/10.3390/ijms25179365
Han Q, Fernandez J, Rajczewski AT, Kono TJY, Weirath NA, Rahim A, Lee AS, Seabloom D, Tretyakova NY. A Multi-Omics Study of Epigenetic Changes in Type II Alveolar Cells of A/J Mice Exposed to Environmental Tobacco Smoke. International Journal of Molecular Sciences. 2024; 25(17):9365. https://doi.org/10.3390/ijms25179365
Chicago/Turabian StyleHan, Qiyuan, Jenna Fernandez, Andrew T. Rajczewski, Thomas J. Y. Kono, Nicholas A. Weirath, Abdur Rahim, Alexander S. Lee, Donna Seabloom, and Natalia Y. Tretyakova. 2024. "A Multi-Omics Study of Epigenetic Changes in Type II Alveolar Cells of A/J Mice Exposed to Environmental Tobacco Smoke" International Journal of Molecular Sciences 25, no. 17: 9365. https://doi.org/10.3390/ijms25179365