
Functional Characterization of Accessible Chromatin in Common Wheat

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
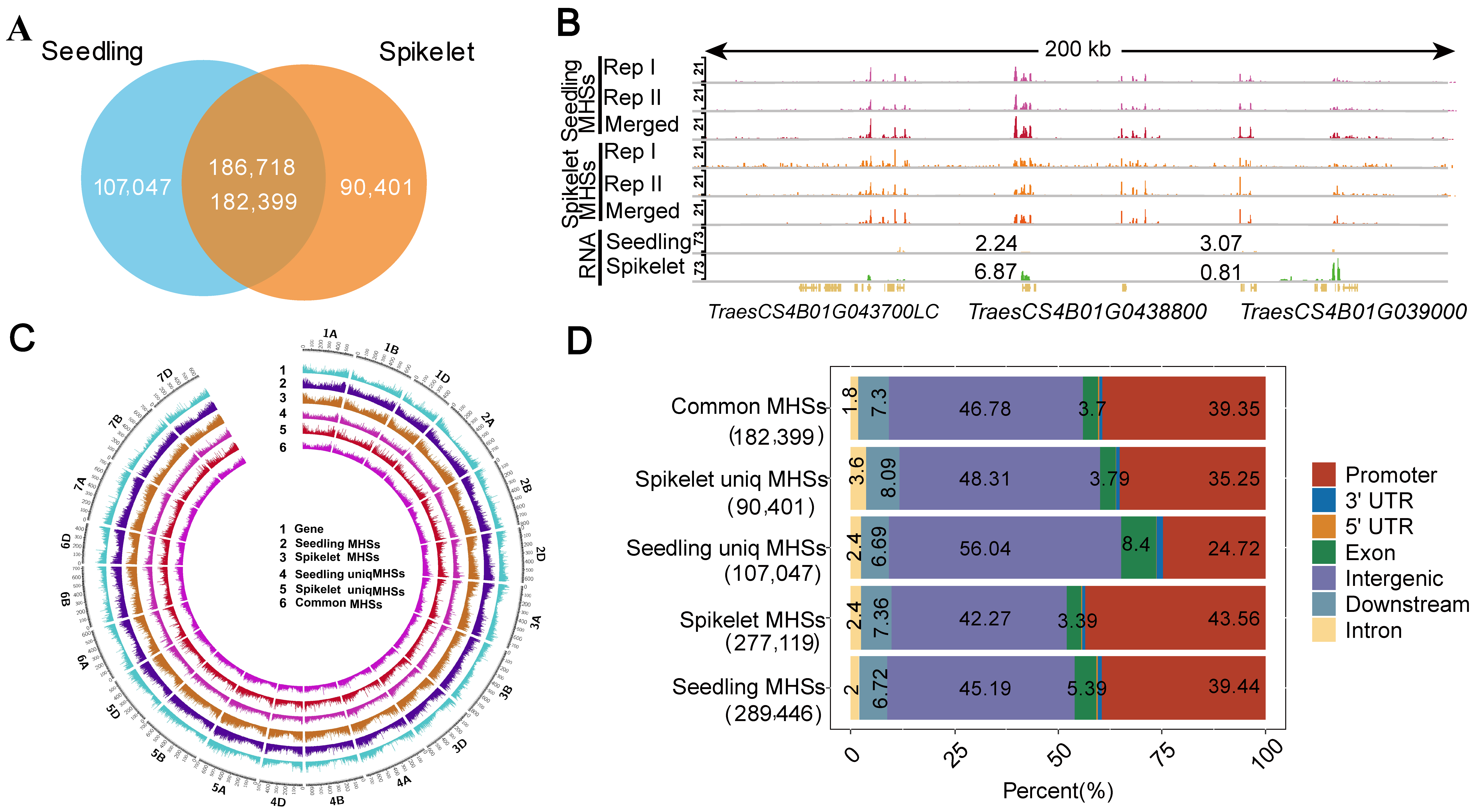
2.1. Profiling of Open Chromatin in the Seedling and Spikelet of Common Wheat Using MH-Seq
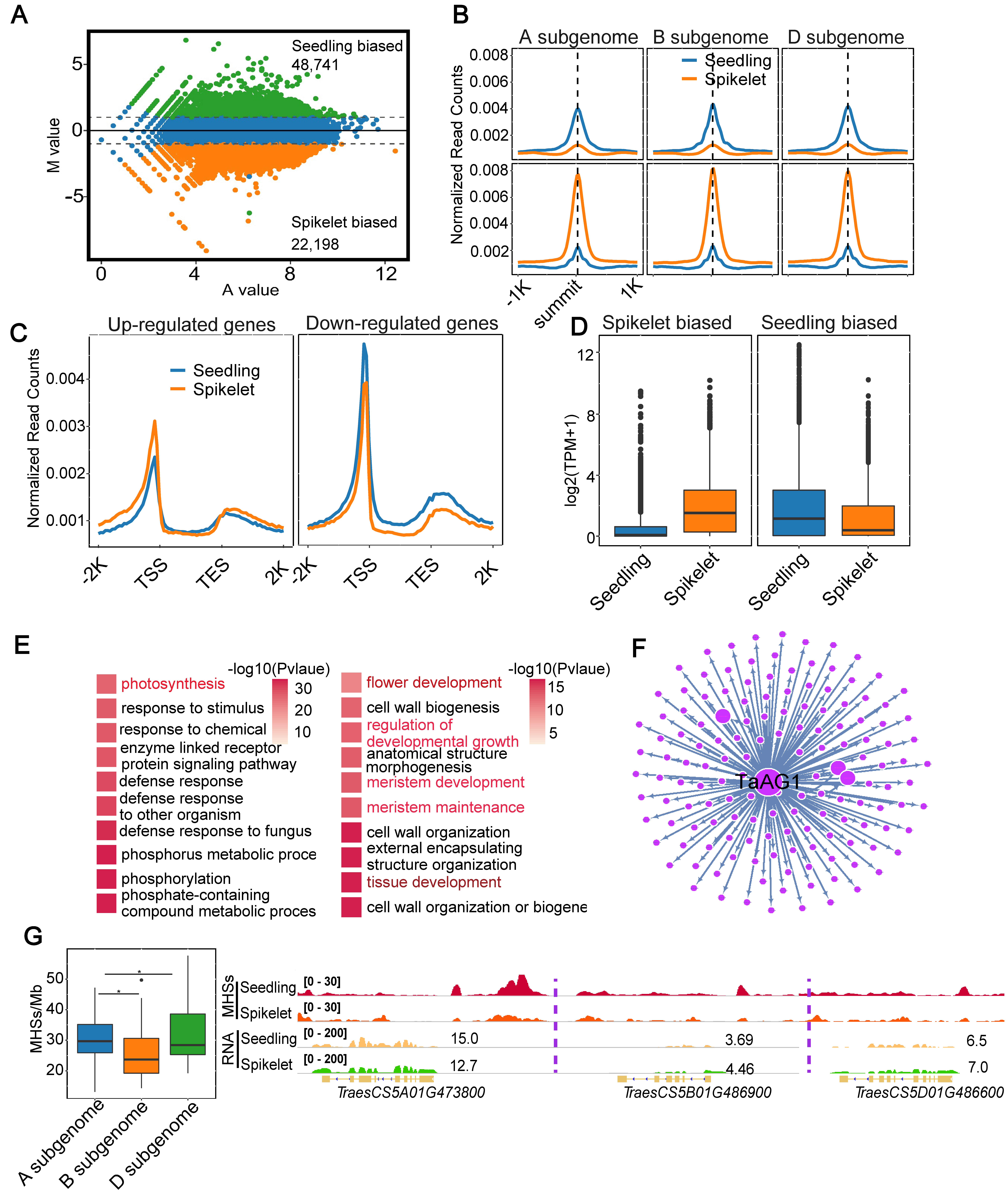
2.2. Biological Functions of Differential Promoter MHSs
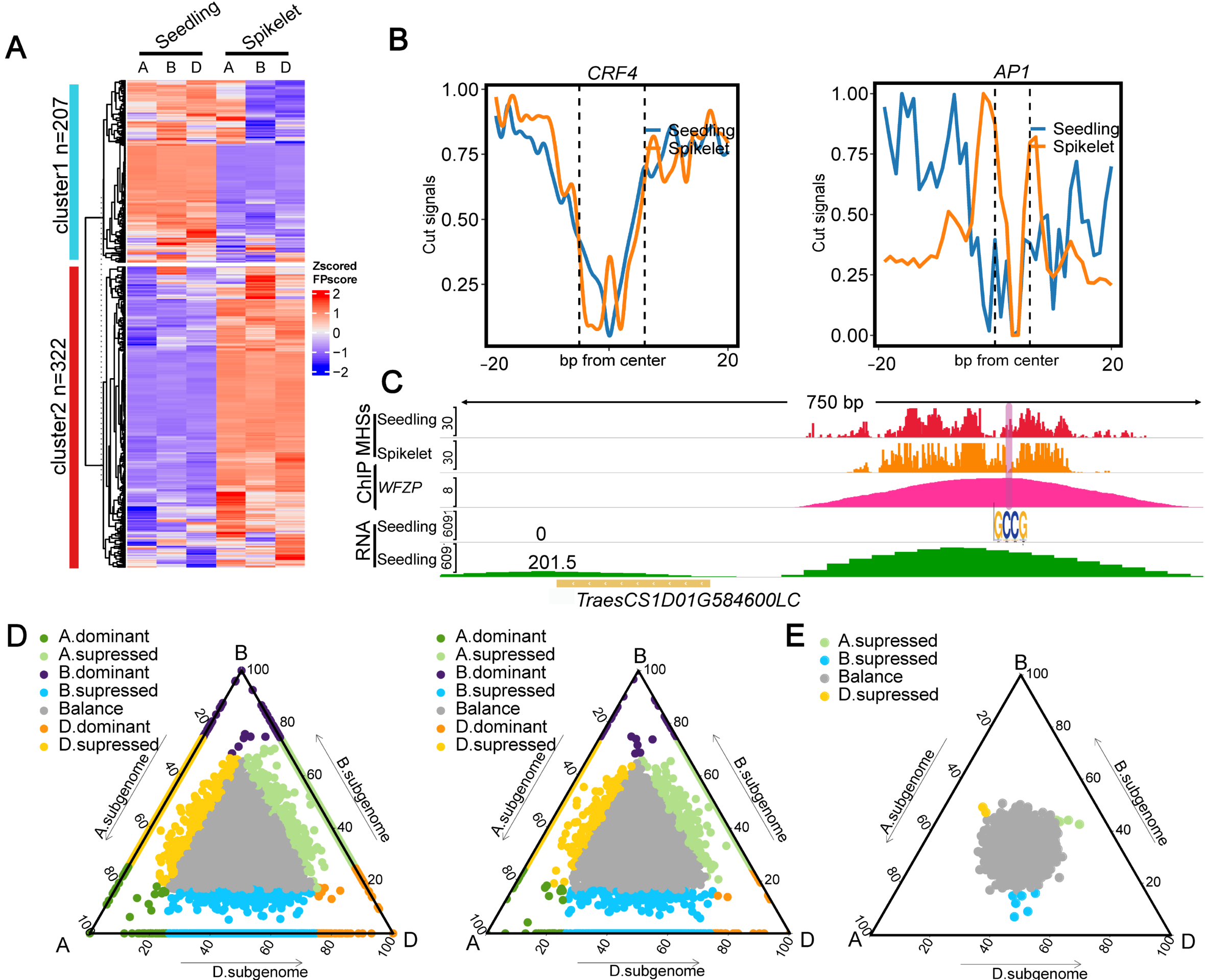
2.3. Differential TF Binding between Seedling and Spikelet
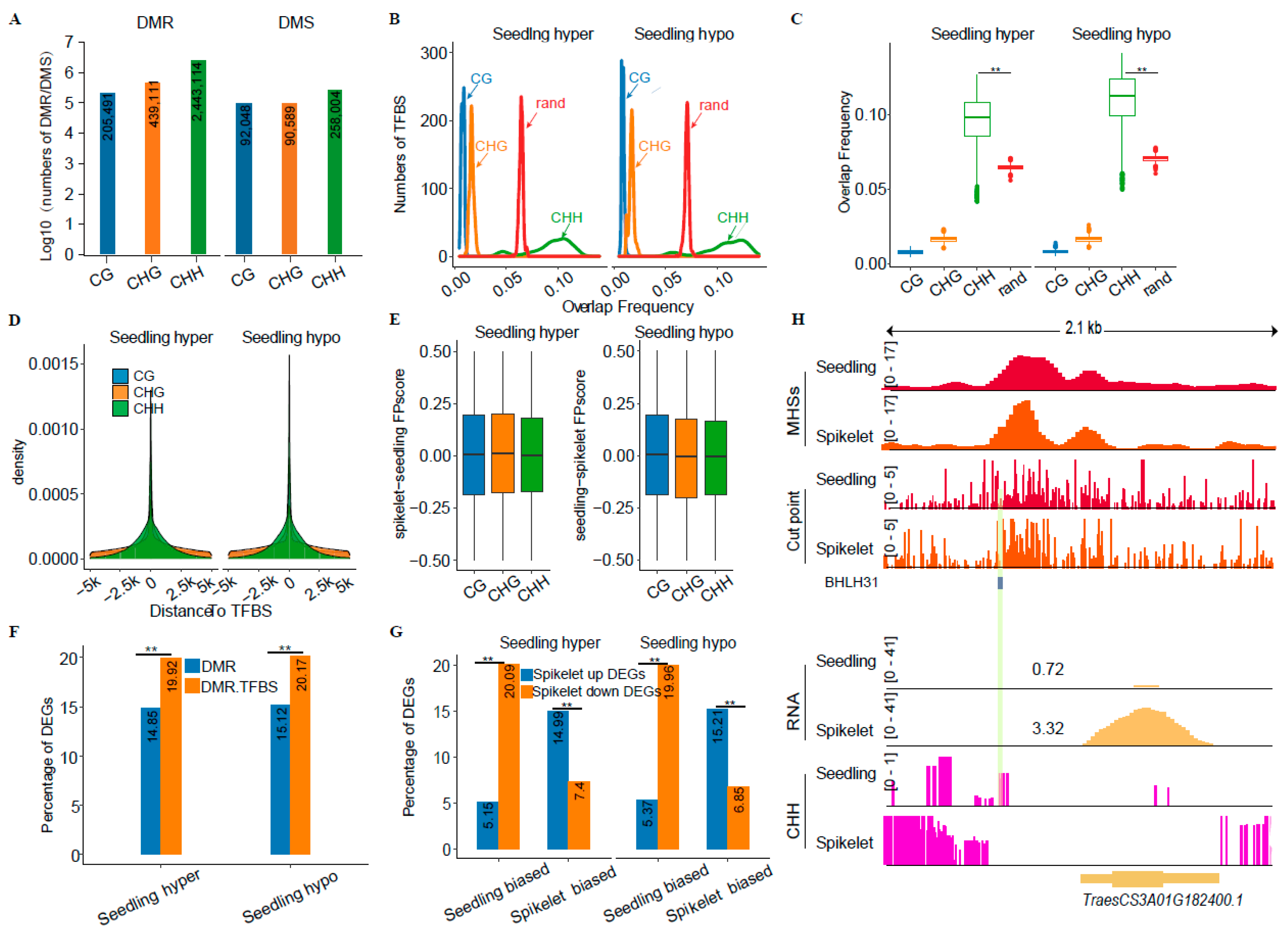
2.4. Interrelationship between CHH Methylation and TFBSs
3. Discussion
3.1. Involvement of CREs in Regulating Tissue-Related Differential Gene Expression and Biased Expression of Homoeologs in Common Wheat
3.2. Impacts of CHH Methylation on Tissue-Related Differential Gene Expression in Common Wheat
4. Materials and Methods
4.1. Plant Materials
4.2. RNA-Seq Data Analyses
4.3. MH-Seq Library Preparation and Data Analyses
4.4. Identification of Differential TF Binding Sites
4.5. Bisulfite Sequencing Data Analyses
4.6. Resequencing Data Analyses and Linkage Disequilibrium Calculation
4.7. Normalization of Read Counts
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.T.; Corces, V.G. Enhancer function: New insights into the regulation of tissue-specific gene expression. Nat. Rev. Genet. 2011, 12, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Wong, Y.C.; Elgin, S.C. The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell 1979, 16, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Gross, D.S.; Garrard, W.T. Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem. 1988, 57, 159–197. [Google Scholar] [CrossRef]
- Henikoff, S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat. Rev. Genet. 2008, 9, 15–26. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Hesselberth, J.R.; Chen, X.; Zhang, Z.; Sabo, P.J.; Sandstrom, R.; Reynolds, A.P.; Thurman, R.E.; Neph, S.; Kuehn, M.S.; Noble, W.S.; et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods 2009, 6, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, Y.; Schnable, J.C.; Zeng, Z.; Freeling, M.; Crawford, G.E.; Jiang, J. High-resolution mapping of open chromatin in the rice genome. Genome Res. 2012, 22, 151–162. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Lu, Z.; Hofmeister, B.T.; Vollmers, C.; DuBois, R.M.; Schmitz, R.J. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017, 45, e41. [Google Scholar] [CrossRef] [PubMed]
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885. [Google Scholar] [CrossRef]
- Tao, S.; Lin, K.; Zhu, Q.; Zhang, W. MH-seq for Functional Characterization of Open Chromatin in Plants. Trends Plant Sci. 2020, 25, 618–619. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, W.; Zhang, T.; Lin, Y.; Hu, Y.; Fang, C.; Jiang, J. Genome-wide MNase hypersensitivity assay unveils distinct classes of open chromatin associated with H3K27me3 and DNA methylation in Arabidopsis thaliana. Genome Biol. 2020, 21, 24. [Google Scholar] [CrossRef] [PubMed]
- Nicieza, R.G.; Huergo, J.; Connolly, B.A.; Sanchez, J. Purification, characterization, and role of nucleases and serine proteases in Streptomyces differentiation. Analogies with the biochemical processes described in late steps of eukaryotic apoptosis. J. Biol. Chem. 1999, 274, 20366–20375. [Google Scholar] [CrossRef] [PubMed]
- Rodgers-Melnick, E.; Vera, D.L.; Bass, H.W.; Buckler, E.S. Open chromatin reveals the functional maize genome. Proc. Natl. Acad. Sci. USA 2016, 113, E3177–E3184. [Google Scholar] [CrossRef]
- Li, M.; Feng, Y.; Han, Q.; Yang, Y.; Shi, Y.; Zheng, D.; Zhang, W. Genomic variations combined with epigenetic modifications rewire open chromatin in rice. Plant Physiol. 2023, 193, 1880–1896. [Google Scholar] [CrossRef]
- Han, J.; Wang, P.; Wang, Q.; Lin, Q.; Chen, Z.; Yu, G.; Miao, C.; Dao, Y.; Wu, R.; Schnable, J.C.; et al. Genome-Wide Characterization of DNase I-Hypersensitive Sites and Cold Response Regulatory Landscapes in Grasses. Plant Cell 2020, 32, 2457–2473. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.W.; He, F.; de Soto, M.F.; Akhunova, A.; Akhunov, E. Differential chromatin accessibility landscape reveals structural and functional features of the allopolyploid wheat chromosomes. Genome Biol. 2020, 21, 176. [Google Scholar] [CrossRef] [PubMed]
- Maher, K.A.; Bajic, M.; Kajala, K.; Reynoso, M.; Pauluzzi, G.; West, D.A.; Zumstein, K.; Woodhouse, M.; Bubb, K.; Dorrity, M.W.; et al. Profiling of Accessible Chromatin Regions across Multiple Plant Species and Cell Types Reveals Common Gene Regulatory Principles and New Control Modules. Plant Cell 2018, 30, 15–36. [Google Scholar] [CrossRef]
- Marand, A.P.; Zhang, X.; Nelson, J.; Braga dos Reis, P.A.; Schmitz, R.J. Profiling single-cell chromatin accessibility in plants. STAR Protoc. 2021, 2, 100737. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.M.; Arsovski, A.A.; Lempe, J.; Bubb, K.L.; Weirauch, M.T.; Sabo, P.J.; Sandstrom, R.; Thurman, R.E.; Neph, S.; Reynolds, A.P.; et al. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014, 8, 2015–2030. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, W.; Marand, A.P.; Zhu, B.; Buell, C.R.; Jiang, J. Cold stress induces enhanced chromatin accessibility and bivalent histone modifications H3K4me3 and H3K27me3 of active genes in potato. Genome Biol. 2019, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, T.; Wu, Y.; Jiang, J. Genome-wide identification of regulatory DNA elements and protein-binding footprints using signatures of open chromatin in Arabidopsis. Plant Cell 2012, 24, 2719–2731. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing, C. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef]
- El Baidouri, M.; Murat, F.; Veyssiere, M.; Molinier, M.; Flores, R.; Burlot, L.; Alaux, M.; Quesneville, H.; Pont, C.; Salse, J. Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol. 2017, 213, 1477–1486. [Google Scholar] [CrossRef]
- Marcussen, T.; Sandve, S.R.; Heier, L.; Spannagl, M.; Pfeifer, M.; Jakobsen, K.S.; Wulff, B.B.; Steuernagel, B.; Mayer, K.F.; Olsen, O.A. Ancient hybridizations among the ancestral genomes of bread wheat. Science 2014, 345, 1250092. [Google Scholar] [CrossRef]
- Akhunova, A.R.; Matniyazov, R.T.; Liang, H.; Akhunov, E.D. Homoeolog-specific transcriptional bias in allopolyploid wheat. BMC Genom. 2010, 11, 505. [Google Scholar] [CrossRef]
- Liu, Z.; Qin, J.; Tian, X.; Xu, S.; Wang, Y.; Li, H.; Wang, X.; Peng, H.; Yao, Y.; Hu, Z.; et al. Global profiling of alternative splicing landscape responsive to drought, heat and their combination in wheat (Triticum aestivum L.). Plant Biotechnol. J. 2018, 16, 714–726. [Google Scholar] [CrossRef]
- Ramírez-González, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Quilichini, T.D.; Liu, Z.; Gao, P.; Pan, Y.; Li, Q.; Nilsen, K.T.; Venglat, P.; Esteban, E.; Pasha, A.; et al. The Transcriptional Landscape of Polyploid Wheats and Their Diploid Ancestors during Embryogenesis and Grain Development. Plant Cell 2019, 31, 2888–2911. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, M.; Lin, K.; Xie, Y.; Guo, J.; Ye, L.; Zhuang, Y.; Teng, W.; Ran, X.; Tong, Y.; et al. The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements. Genome Biol. 2019, 20, 139. [Google Scholar] [CrossRef]
- Chaudhary, B.; Flagel, L.; Stupar, R.M.; Udall, J.A.; Verma, N.; Springer, N.M.; Wendel, J.F. Reciprocal silencing, transcriptional bias and functional divergence of homeologs in polyploid cotton (gossypium). Genetics 2009, 182, 503–517. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Marand, A.P.; Ricci, W.A.; Ethridge, C.L.; Zhang, X.; Schmitz, R.J. The prevalence, evolution and chromatin signatures of plant regulatory elements. Nat. Plants 2019, 5, 1250–1259. [Google Scholar] [CrossRef]
- Ricci, W.A.; Lu, Z.; Ji, L.; Marand, A.P.; Ethridge, C.L.; Murphy, N.G.; Noshay, J.M.; Galli, M.; Mejía-Guerra, M.K.; Colomé-Tatché, M.; et al. Widespread long-range cis-regulatory elements in the maize genome. Nat. Plants 2019, 5, 1237–1249. [Google Scholar] [CrossRef]
- Song, L.; Zhang, Z.; Grasfeder, L.L.; Boyle, A.P.; Giresi, P.G.; Lee, B.K.; Sheffield, N.C.; Gräf, S.; Huss, M.; Keefe, D.; et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011, 21, 1757–1767. [Google Scholar] [CrossRef]
- Vera, D.L.; Madzima, T.F.; Labonne, J.D.; Alam, M.P.; Hoffman, G.G.; Girimurugan, S.B.; Zhang, J.; McGinnis, K.M.; Dennis, J.H.; Bass, H.W. Differential nuclease sensitivity profiling of chromatin reveals biochemical footprints coupled to gene expression and functional DNA elements in maize. Plant Cell 2014, 26, 3883–3893. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Mena, M.; Ambrose, B.A.; Meeley, R.B.; Briggs, S.P.; Yanofsky, M.F.; Schmidt, R.J. Diversification of C-function activity in maize flower development. Science 1996, 274, 1537–1540. [Google Scholar] [CrossRef]
- Feldman, M.; Levy, A.A.; Fahima, T.; Korol, A. Genomic asymmetry in allopolyploid plants: Wheat as a model. J. Exp. Bot. 2012, 63, 5045–5059. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, A.; Song, G.; Geng, S.; Gill, B.S.; Faris, J.D.; Mao, L. Comprehensive analysis of Q gene near-isogenic lines reveals key molecular pathways for wheat domestication and improvement. Plant J. 2020, 102, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Bentsen, M.; Goymann, P.; Schultheis, H.; Klee, K.; Petrova, A.; Wiegandt, R.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 2020, 11, 4267. [Google Scholar] [CrossRef] [PubMed]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.L.; Walavalkar, N.M.; Anderson, W.D.; Zang, C.; Guertin, M.J. Universal correction of enzymatic sequence bias reveals molecular signatures of protein/DNA interactions. Nucleic Acids Res. 2018, 46, e9. [Google Scholar] [CrossRef] [PubMed]
- Varala, K.; Marshall-Colón, A.; Cirrone, J.; Brooks, M.D.; Pasquino, A.V.; Léran, S.; Mittal, S.; Rock, T.M.; Edwards, M.B.; Kim, G.J.; et al. Temporal transcriptional logic of dynamic regulatory networks underlying nitrogen signaling and use in plants. Proc. Natl. Acad. Sci. USA 2018, 115, 6494–6499. [Google Scholar] [CrossRef]
- Zwack, P.J.; Compton, M.A.; Adams, C.I.; Rashotte, A.M. Cytokinin response factor 4 (CRF4) is induced by cold and involved in freezing tolerance. Plant Cell Rep. 2016, 35, 573–584. [Google Scholar] [CrossRef]
- Irish, V.F.; Sussex, I.M. Function of the apetala-1 gene during Arabidopsis floral development. Plant Cell 1990, 2, 741–753. [Google Scholar]
- Du, D.; Zhang, D.; Yuan, J.; Feng, M.; Li, Z.; Wang, Z.; Zhang, Z.; Li, X.; Ke, W.; Li, R.; et al. FRIZZY PANICLE defines a regulatory hub for simultaneously controlling spikelet formation and awn elongation in bread wheat. New Phytol. 2021, 231, 814–833. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, L.; Zhao, M.; Guo, L.; Guo, X.; Zhao, D.; Batool, A.; Dong, B.; Xu, H.; Cui, S.; et al. Wheat FRIZZY PANICLE activates VERNALIZATION1-A and HOMEOBOX4-A to regulate spike development in wheat. Plant Biotechnol. J. 2021, 19, 1141–1154. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Héberlé, É.; Bardet, A.F. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [PubMed]
- Lazarovici, A.; Zhou, T.; Shafer, A.; Dantas Machado, A.C.; Riley, T.R.; Sandstrom, R.; Sabo, P.J.; Lu, Y.; Rohs, R.; Stamatoyannopoulos, J.A.; et al. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc. Natl. Acad. Sci. USA 2013, 110, 6376–6381. [Google Scholar] [CrossRef]
- O’Malley, R.C.; Huang, S.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 2016, 165, 1280–1292. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef]
- Carroll, S.B. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell 2008, 134, 25–36. [Google Scholar] [CrossRef]
- Lemmon, Z.H.; Bukowski, R.; Sun, Q.; Doebley, J.F. The role of cis regulatory evolution in maize domestication. PLoS Genet. 2014, 10, e1004745. [Google Scholar] [CrossRef]
- Lango Allen, H.; Estrada, K.; Lettre, G.; Berndt, S.I.; Weedon, M.N.; Rivadeneira, F.; Willer, C.J.; Jackson, A.U.; Vedantam, S.; Raychaudhuri, S.; et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 2010, 467, 832–838. [Google Scholar] [CrossRef]
- Swinnen, G.; Goossens, A.; Pauwels, L. Lessons from Domestication: Targeting Cis-Regulatory Elements for Crop Improvement. Trends Plant Sci. 2019, 24, 1065. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.; Wang, S.; Wang, Y.; Chen, X.; Zhang, Y.; Gao, C.; et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat. Genet. 2015, 47, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhao, H.; Zhu, B.; Zhang, T.; Yang, M.; Li, Y.; Han, Y.; Jiang, J. Genomic editing of intronic enhancers unveils their role in fine-tuning tissue-specific gene expression in Arabidopsis thaliana. Plant Cell 2021, 33, 1997–2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gallagher, J.; Arevalo, E.D.; Chen, R.; Skopelitis, T.; Wu, Q.; Bartlett, M.; Jackson, D. Enhancing grain-yield-related traits by CRISPR-Cas9 promoter editing of maize CLE genes. Nat. Plants 2021, 7, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Hendelman, A.; Zebell, S.; Rodriguez-Leal, D.; Dukler, N.; Robitaille, G.; Wu, X.; Kostyun, J.; Tal, L.; Wang, P.; Bartlett, M.E.; et al. Conserved pleiotropy of an ancient plant homeobox gene uncovered by cis-regulatory dissection. Cell 2021, 184, 1724–1739.e16. [Google Scholar] [CrossRef]
- He, X.; Futterer, J.; Hohn, T. Sequence-specific and methylation-dependent and -independent binding of rice nuclear proteins to a rice tungro bacilliform virus vascular bundle expression element. J. Biol. Chem. 2001, 276, 2644–2651. [Google Scholar] [CrossRef]
- Han, T.; Wang, F.; Song, Q.; Ye, W.; Liu, T.; Wang, L.; Chen, Z.J. An epigenetic basis of inbreeding depression in maize. Sci. Adv. 2021, 7, eabg5442. [Google Scholar] [CrossRef]
- Urbano, A.; Smith, J.; Weeks, R.J.; Chatterjee, A. Gene-Specific Targeting of DNA Methylation in the Mammalian Genome. Cancers 2019, 11, 1515. [Google Scholar] [CrossRef]
- Wang, M.; Li, Z.; Zhang, Y.; Zhang, Y.; Xie, Y.; Ye, L.; Zhuang, Y.; Lin, K.; Zhao, F.; Guo, J.; et al. An atlas of wheat epigenetic regulatory elements reveals subgenome divergence in the regulation of development and stress responses. Plant Cell 2021, 33, 865–881. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; He, Q.Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef]
- Shao, Z.; Zhang, Y.; Yuan, G.C.; Orkin, S.H.; Waxman, D.J. MAnorm: A robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012, 13, R16. [Google Scholar] [CrossRef]
- Guo, W.; Zhu, P.; Pellegrini, M.; Zhang, M.Q.; Wang, X.; Ni, Z. CGmapTools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 2018, 34, 381–387. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Ma, S.; Wang, M.; Wu, J.; Guo, W.; Chen, Y.; Li, G.; Wang, Y.; Shi, W.; Xia, G.; Fu, D.; et al. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant 2021, 14, 1965–1968. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, X.; Li, Y.; Xu, J.; Bi, A.; Kang, L.; Xu, D.; Chen, H.; Wang, Y.; Wang, Y.-G.; et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 2020, 52, 1412–1422. [Google Scholar] [CrossRef]
- Dong, S.S.; He, W.M.; Ji, J.J.; Zhang, C.; Guo, Y.; Yang, T.L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2021, 22, bbaa227. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, D.; Lin, K.; Yang, X.; Zhang, W.; Cheng, X. Functional Characterization of Accessible Chromatin in Common Wheat. Int. J. Mol. Sci. 2024, 25, 9384. https://doi.org/10.3390/ijms25179384
Zheng D, Lin K, Yang X, Zhang W, Cheng X. Functional Characterization of Accessible Chromatin in Common Wheat. International Journal of Molecular Sciences. 2024; 25(17):9384. https://doi.org/10.3390/ijms25179384
Chicago/Turabian StyleZheng, Dongyang, Kande Lin, Xueming Yang, Wenli Zhang, and Xuejiao Cheng. 2024. "Functional Characterization of Accessible Chromatin in Common Wheat" International Journal of Molecular Sciences 25, no. 17: 9384. https://doi.org/10.3390/ijms25179384