
Generation of Novel Tumour-Selective SEA Superantigen-Based Peptides with Improved Safety and Efficacy for Precision Cancer Immunotherapy

Abstract
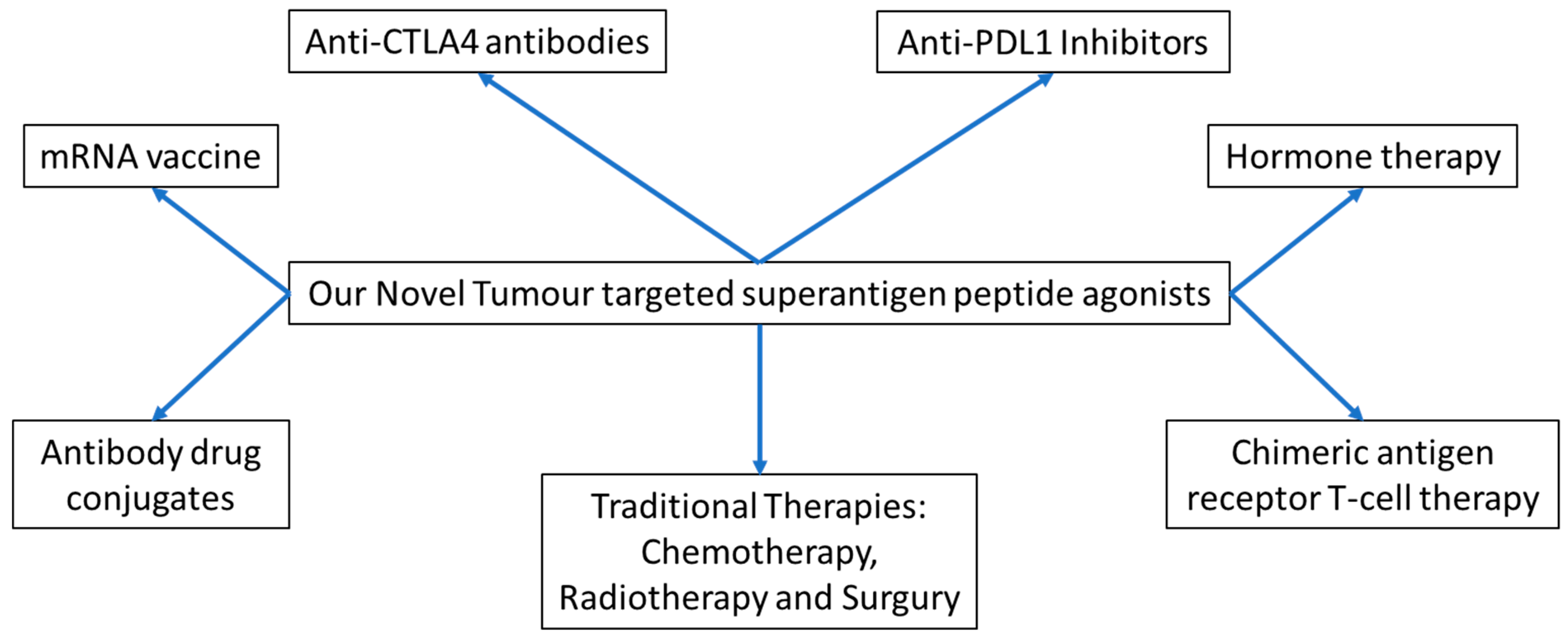
:1. Introduction
2. Results
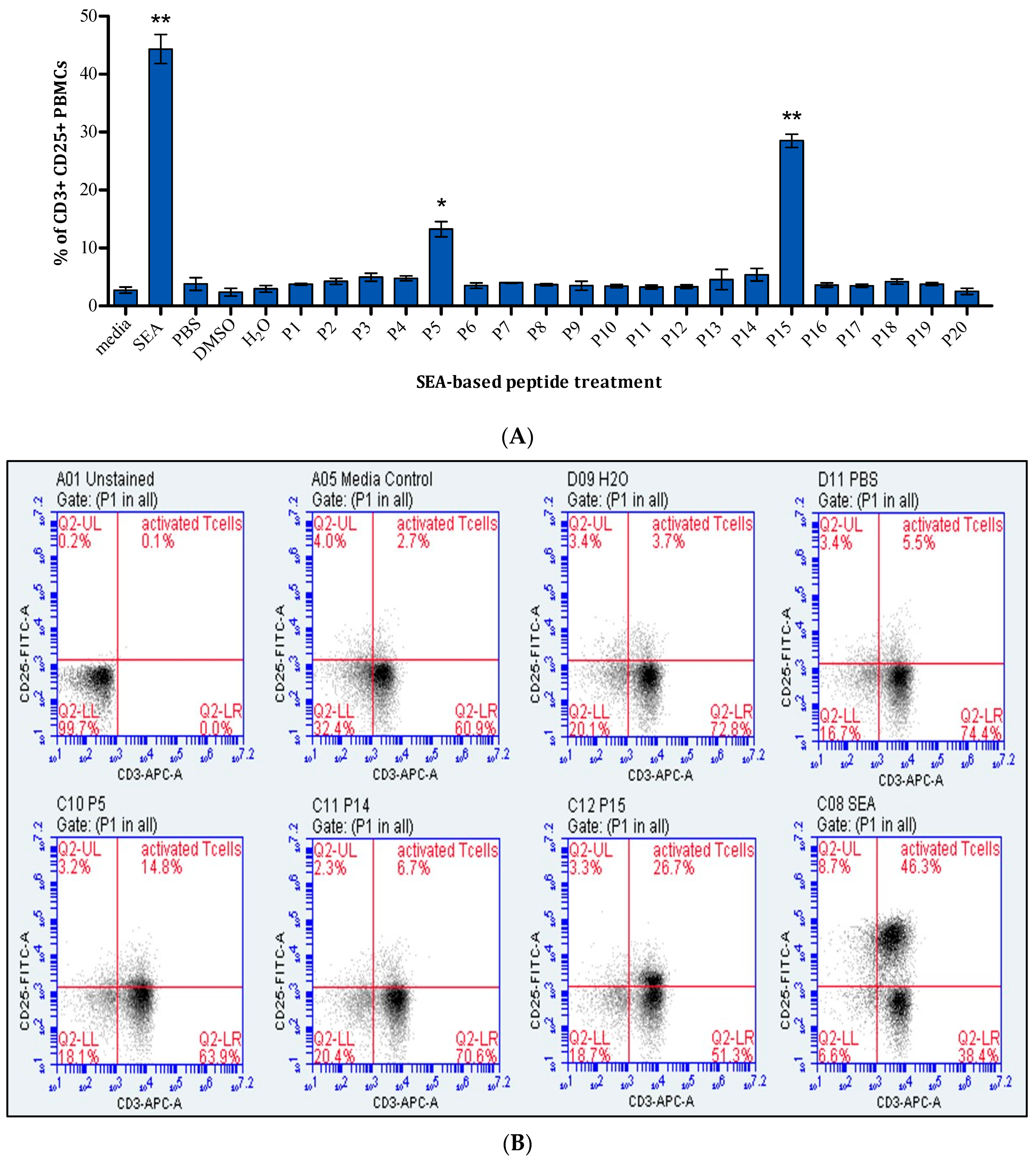
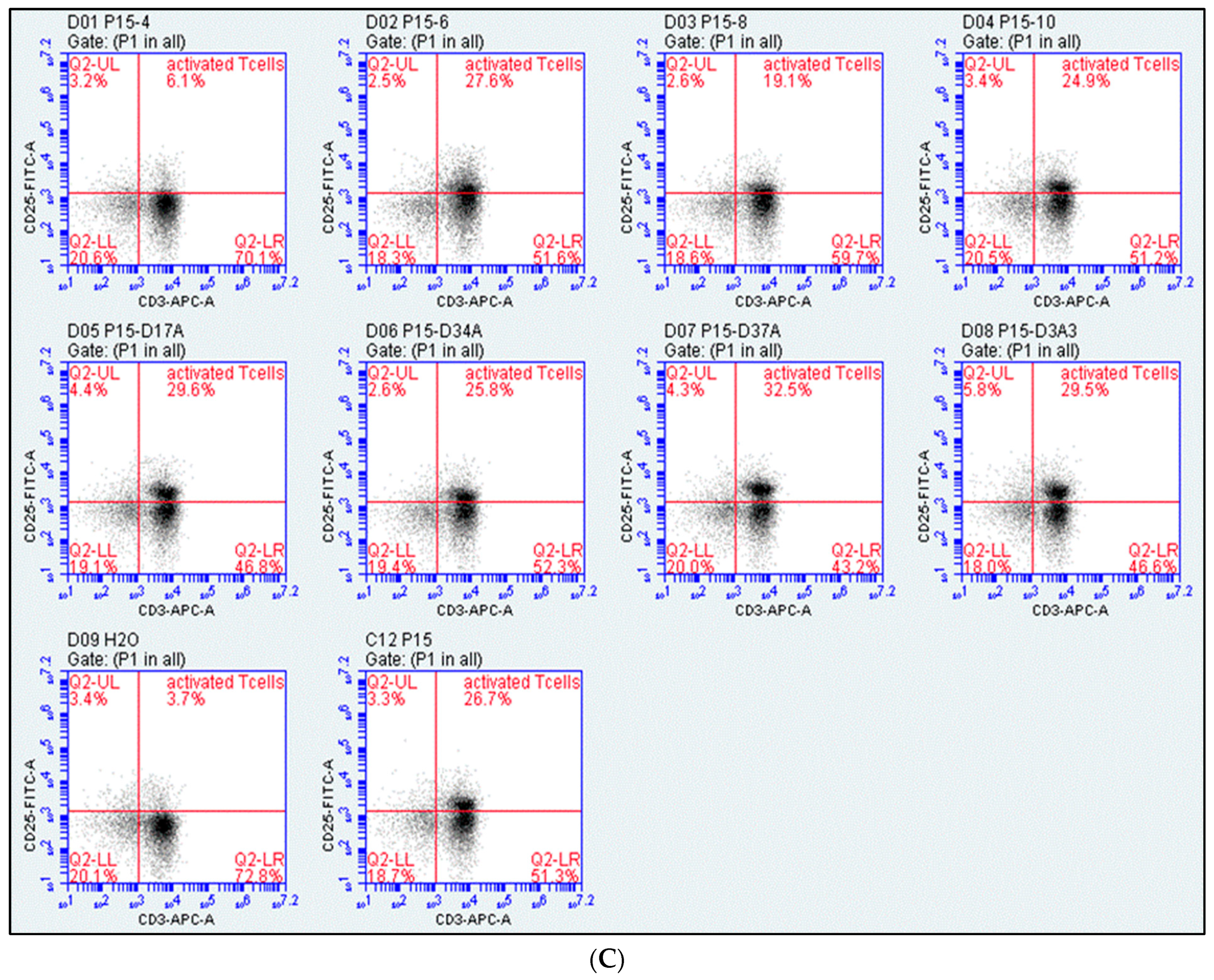
2.1. Assessment of T-Cell Activation
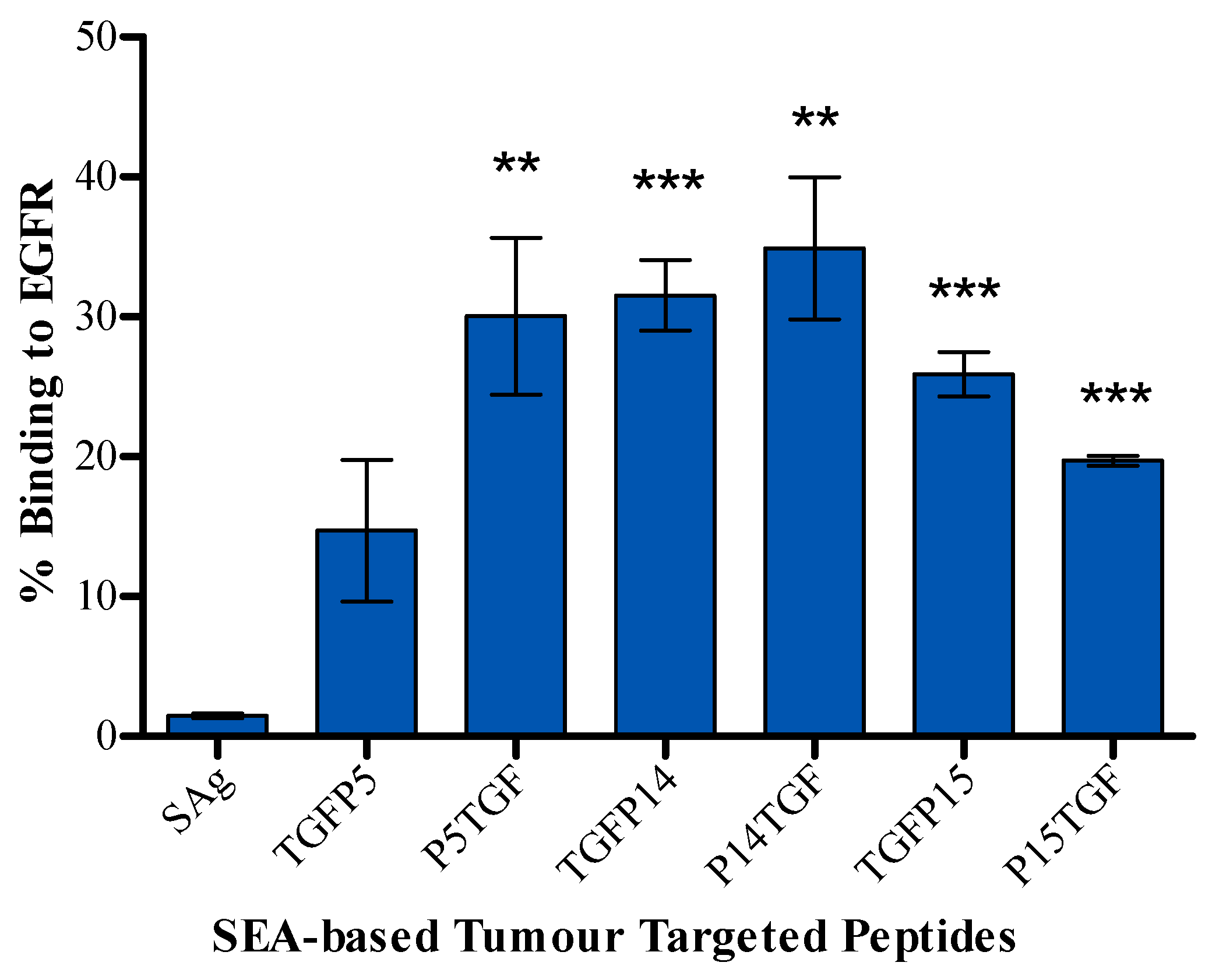
2.2. Tumour Cell Binding Assay
2.3. Antitumour Activity
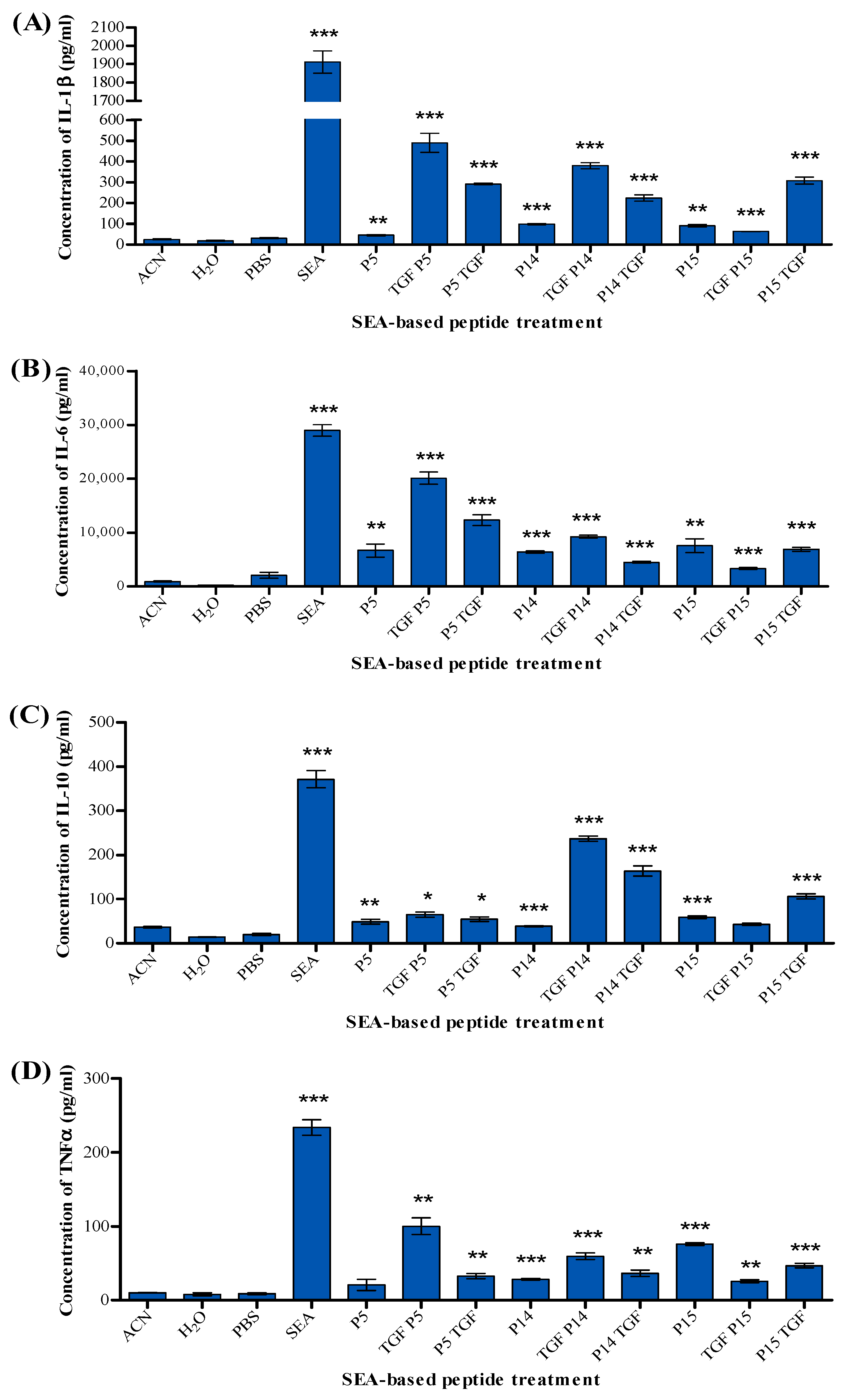
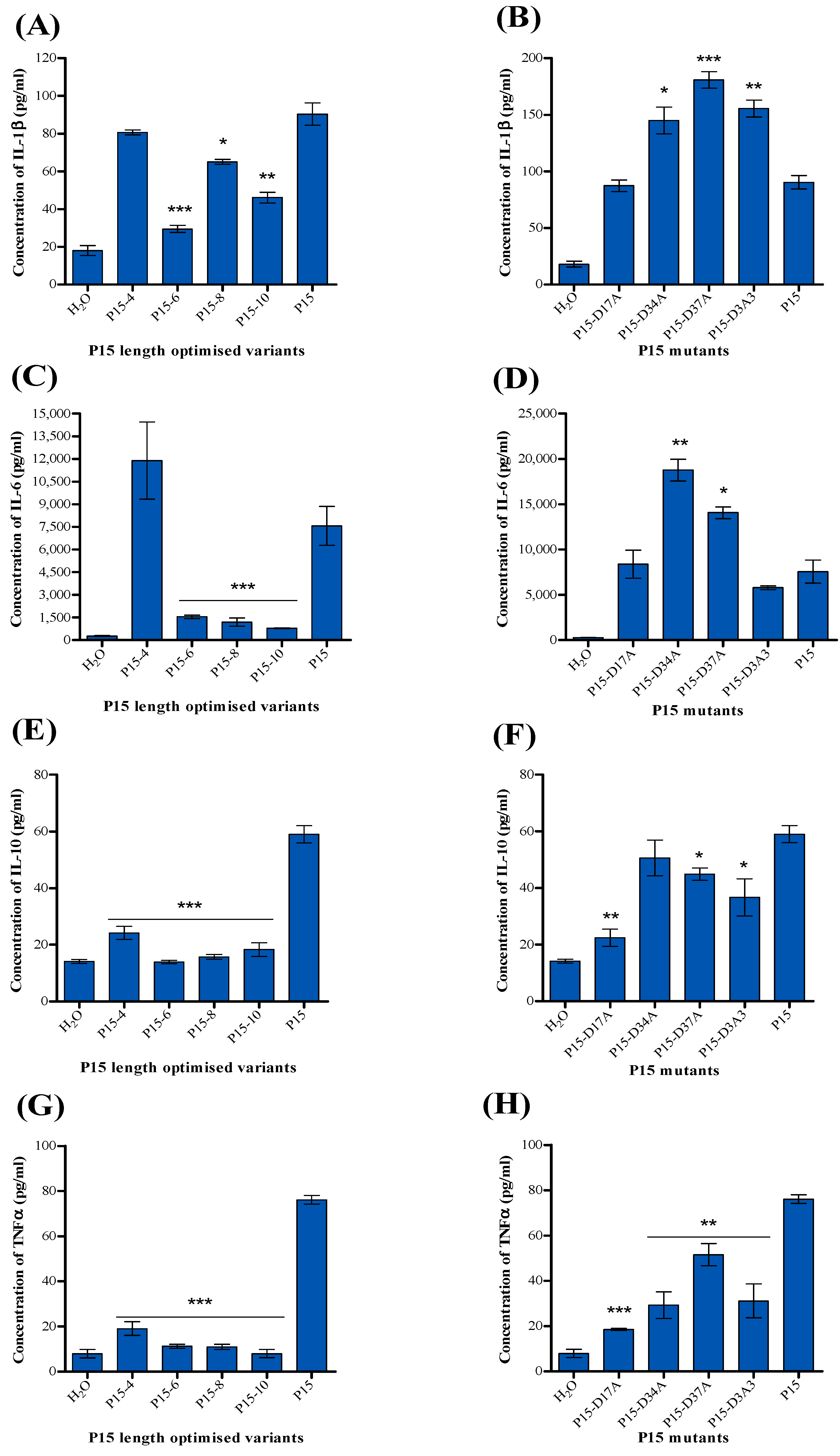
2.4. Detection of Cytokines
2.5. MTT Assay
3. Discussion
4. Method and Materials
4.1. Cells, Reagents, Antibodies, and Kits
4.2. Design and Synthesis of Superantigen Peptides
4.3. Assessment of T-Cell Activation
4.4. Design of Peptide P15 Derivatives
4.5. Synthesis of Tumour-Targeted SEA-Based Peptides
4.6. Tumour Cell Binding Assay
4.7. Antitumour Activity
4.8. Detection of Cytokines
4.9. MTT Assay
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bashraheel, S.S.; Domling, A.; Goda, S.K. Update on targeted cancer therapies, single or in combination, and their fine tuning for precision medicine. Biomed. Pharmacother. 2020, 125, 110009. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xie, T.; Zhou, M.; Sun, Y.; Wang, F.; Tian, Y.; Chen, Z.; Xie, Y.; Wu, R.; Cen, X.; et al. Hsc70 promotes anti-tumor immunity by targeting PD-L1 for lysosomal degradation. Nat. Commun. 2024, 15, 4237. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, C.; Shao, J.; Tang, X.; Wu, J.; Li, P.; Li, W.; Wang, C. The real-world treatment characteristic and efficacy of immune checkpoint inhibitors in non-small cell lung cancer: Data from a retrospective cohort study. Int. Immunopharmacol. 2024, 134, 112152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Hou, Y.; Lin, Y.; Zhao, H.; Shi, Y.; Chen, K.; Nian, C.; Tang, J.; Pan, L.; et al. Osr2 functions as a biomechanical checkpoint to aggravate CD8+ T cell exhaustion in tumor. Cell 2024, 187, 3409–3426. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.; Garcia, J.; Agliardi, G.; Roddie, C. CAR-T cell manufacturing landscape-Lessons from the past decade and considerations for early clinical development. Mol. Ther. Methods Clin. Dev. 2024, 32, 101250. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, G.; Xu, M.; Zhang, X.; Ma, L.; Zhang, H. TRAIL produced by SAM-1-activated CD4+ and CD8+ subgroup T cells induces apoptosis in human tumor cells through upregulation of death receptors. Toxicol. Appl. Pharmacol. 2021, 427, 115656. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, G.; Mechahougui, H.; Ben Aissa, A. Therapeutic cancer vaccines: Challenges and perspectives. Rev. Med. Suisse. 2024, 20, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Kraehenbuehl, L.; Weng, C.H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022, 19, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Larkin, J.; Tabernero, J.; Bonini, C. Enhancing anti-tumour efficacy with immunotherapy combinations. Lancet 2021, 397, 1010–1022. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, M.; Li, Y.; Zhang, Z.; Gu, W.; Halimu, G.; Li, Y.; Zhang, H.; Zhang, C. Induction of CD4+ regulatory T cells by stimulation with Staphylococcal Enterotoxin C2 through different signaling pathways. Biomed. Pharmacother. 2021, 143, 112204. [Google Scholar] [CrossRef] [PubMed]
- Shivaee, A.; Sedighi, M.; Imani Fooladi, A.A. Staphylococcal enterotoxins as good candidates for cancer immunotherapy: A systematic review. Ann. Di Ig. Med. Prev. E Di Comunita 2020, 32, 648–663. [Google Scholar] [CrossRef] [PubMed]
- Knopick, P.; Terman, D.; Riha, N.; Alvine, T.; Larson, R.; Badiou, C.; Lina, G.; Ballantyne, J.; Bradley, D. Endogenous HLA-DQ8αβ programs superantigens (SEG/SEI) to silence toxicity and unleash a tumoricidal network with long-term melanoma survival. J. Immunother. Cancer 2020, 8, e001493. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bashraheel, S.S.; AlQahtani, A.D.; Rashidi, F.B.; Al-Sulaiti, H.; Domling, A.; Orie, N.N.; Goda, S.K. Studies on vascular response to full superantigens and superantigen derived peptides: Possible production of novel superantigen variants with less vasodilation effect for tolerable cancer immunotherapy. Biomed. Pharmacother. 2019, 115, 108905. [Google Scholar] [CrossRef] [PubMed]
- Bashraheel, S.S.; Goda, S.K. Novel SPEA Superantigen Peptide Agonists and Peptide Agonist-TGFαL3 Conjugate. In Vitro Study of Their Growth-Inhibitory Effects for Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2023, 24, 10507. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sakurai, N.; Kudo, T.; Suzuki, M.; Tsumoto, K.; Takemura, S.I.; Kodama, H.; Ebara, S.; Teramae, A.; Katayose, Y.; Shinoda, M.; et al. SEA-scFv as a bifunctional antibody: Construction of a bacterial expression system and its functional analysis. Biochem. Biophys. Res. Commun. 1999, 256, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zeng, L.; Zhao, Z.; He, J.; Xie, Y.; Xiao, L.; Wang, S.; Zhang, J.; Zou, Z.; He, Y.; et al. PBMC activation via the ERK and STAT signaling pathways enhances the anti-tumor activity of Staphylococcal enterotoxin A. Mol. Cell Biochem. 2017, 434, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Yu, H.; Wang, Q.; Jin, H.; Solheim, J.; Labhasetwar, V. A novel approach for cancer immunotherapy: Tumor cells with anchored superantigen SEA generate effective antitumor immunity. J. Clin. Immunol. 2004, 24, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, G.; Dohlsten, M.; Lando, P.A.; Kalland, T. Staphylococcal enterotoxins direct and trigger CTL killing of autologous HLA-DR+ mononuclear leukocytes and freshly prepared leukemia cells. Cell Immunol. 1990, 129, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Dohlsten, M.; Abrahmsen, L.; Björk, P.; Lando, P.A.; Hedlund, G.; Forsberg, G.; Brodin, T.; Gascoigne, N.R.; Förberg, C.; Lind, P. Monoclonal antibody-superantigen fusion proteins: Tumor-specific agents for T-cell-based tumor therapy. Proc. Natl. Acad. Sci. USA 1994, 91, 8945–8949. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dohlsten, M.; Hedlund, G.; Akerblom, E.; Lando, P.A.; Kalland, T. Monoclonal antibody-targeted superantigens: A different class of anti-tumor agents. Proc. Natl. Acad. Sci. USA 1991, 88, 9287–9291. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dohlsten, M.; Lando, P.A.; Björk, P.; Abrahmsén, L.; Ohlsson, L.; Lind, P.; Kalland, T. Immunotherapy of human colon cancer by antibody-targeted superantigens. Cancer Immunol. Immunother. 1995, 41, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Brodin, T.N.; Persson, R.; Soegaard, M.; Ohlsson, L.; d’Argy, R.; Olsson, J.; Molander, A.; Antonsson, P.; Gunnarsson, P.O.; Kalland, T.; et al. Man-made superantigens: Tumor-selective agents for T-cell-based therapy. Adv. Drug Deliv. Rev. 1998, 31, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Bazley, L.A.; Gullick, W.J. The epidermal growth factor receptor family. Endocr. Relat. Cancer 2005, 12 (Suppl. S1), S17–S27. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Canadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Benelli, R.; Costa, D.; Salvini, L.; Tardito, S.; Tosetti, F.; Villa, F.; Zocchi, M.R.; Poggi, A. Targeting of colorectal cancer organoids with zoledronic acid conjugated to the anti-EGFR antibody cetuximab. J. Immunother. Cancer 2022, 10, e005660. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lu, Y.; Huang, J.; Li, F.; Wang, Y.; Ding, M.; Zhang, J.; Yin, H.; Zhang, R.; Ren, X. EGFR-specific single-chain variable fragment antibody-conjugated Fe(3)O(4)/Au nanoparticles as an active MRI contrast agent for NSCLC. MAGMA 2021, 34, 581–591. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, S.C.; Chen, Y.J.; Wang, H.C.; Chou, M.Y.; Chang, T.Y.; Yuan, S.S.; Chen, C.Y.; Hou, M.F.; Hsu, J.T.A.; Wang, Y.M. Bispecific Antibody Conjugated Manganese-Based Magnetic Engineered Iron Oxide for Imaging of HER2/neu- and EGFR-Expressing Tumors. Theranostics 2016, 6, 118–130. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Leung, S.L.; Zha, Z.; Cohn, C.; Dai, Z.; Wu, X. Anti-EGFR antibody conjugated organic-inorganic hybrid lipid nanovesicles selectively target tumor cells. Colloids Surf. B Biointerfaces 2014, 121, 141–149. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maleki, F.; Sadeghifard, N.; Hosseini, H.M.; Bakhtiyari, S.; Goleij, Z.; Behzadi, E.; Sedighian, H.; Fooladi, A.A.I. Growth-inhibitory effects of TGFalphaL3-SEB chimeric protein on colon cancer cell line. Biomed. Pharmacother. 2019, 110, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. 2011, 9, e1001149. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sgagias, M.K.; Kasid, A.; Danforth, D.N., Jr. Interleukin-1 alpha and tumor necrosis factor-alpha (TNF alpha) inhibit growth and induce TNF messenger RNA in MCF-7 human breast cancer cells. Mol. Endocrinol. 1991, 5, 1740–1747. [Google Scholar] [CrossRef] [PubMed]
- Maund, S.L.; Barclay, W.W.; Hover, L.D.; Axanova, L.S.; Sui, G.; Hipp, J.D.; Fleet, J.C.; Thorburn, A.; Cramer, S.D. Interleukin-1alpha mediates the antiproliferative effects of 1,25-dihydroxyvitamin D3 in prostate progenitor/stem cells. Cancer Res. 2011, 71, 5276–5286. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maund, S.L.; Shi, L.; Cramer, S.D. A role for interleukin-1 alpha in the 1,25 dihydroxyvitamin D3 response in mammary epithelial cells. PLoS ONE 2013, 8, e81367. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Azulay, M.; Shahar, M.; Shany, E.; Elbaz, E.; Lifshits, S.; Torngren, M.; Friedmann, A.; Kramer, R.; Hedlund, G. Tumor-targeted superantigens produce curative tumor immunity with induction of memory and demonstrated antigen spreading. J. Transl. Med. 2023, 21, 222. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yap, T.A.; Omlin, A.; de Bono, J.S. Development of therapeutic combinations targeting major cancer signaling pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Analysis of FDA approved anticancer drugs reveals the future of cancer therapy. Cell Cycle 2004, 3, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Zamani, S.; Elrayess, M.A.; Kafienah, W.; Younes, H.M. New Three-Dimensional Poly(decanediol-co-tricarballylate) Elastomeric Fibrous Mesh Fabricated by Photoreactive Electrospinning for Cardiac Tissue Engineering Applications. Polymers 2018, 10, 455. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Sequence | Solvent |
|---|---|---|
| P1 | MSEKSEEINEKDLRKKSELQGTALGNLKQIYYYNEKAKTENKESHDQFLQ | H2O |
| P2 | KDLRKKSELQGTALGNLKQIYYYNEKAKTENKESHDQFLQHTILFKGFFT | H2O |
| P3 | GTALGNLKQIYYYNEKAKTENKESHDQFLQHTILFKGFFTDHSWYNDLLV | H2O |
| P4 | YYYNEKAKTENKESHDQFLQHTILFKGFFTDHSWYNDLLVDFDSKDIVDK | H2O |
| P5 | NKESHDQFLQHTILFKGFFTDHSWYNDLLVDFDSKDIVDKYKGKKVDLYG | H2O |
| P6 | HTILFKGFFTDHSWYNDLLVDFDSKDIVDKYKGKKVDLYGAYYGYQCAGG | DMSO |
| P7 | DHSWYNDLLVDFDSKDIVDKYKGKKVDLYGAYYGYQCAGGTPNKTACMYG | H2O |
| P8 | DFDSKDIVDKYKGKKVDLYGAYYGYQCAGGTPNKTACMYGGVTLHDNNRL | H2O |
| P9 | YKGKKVDLYGAYYGYQCAGGTPNKTACMYGGVTLHDNNRLTEEKKVPINL | H2O |
| P10 | AYYGYQCAGGTPNKTACMYGGVTLHDNNRLTEEKKVPINLWLDGKQNTVP | H2O |
| P11 | TPNKTACMYGGVTLHDNNRLTEEKKVPINLWLDGKQNTVPLETVKTNKKN | H2O |
| P12 | GVTLHDNNRLTEEKKVPINLWLDGKQNTVPLETVKTNKKNVTVQELDLQA | H2O |
| P13 | TEEKKVPINLWLDGKQNTVPLETVKTNKKNVTVQELDLQARRYLQEKYNL | H2O |
| P14 | WLDGKQNTVPLETVKTNKKNVTVQELDLQARRYLQEKYNLYNSDVFDGKV | H2O |
| P15 | LETVKTNKKNVTVQELDLQARRYLQEKYNLYNSDVFDGKVQRGLIVFHTS | H2O |
| P16 | VTVQELDLQARRYLQEKYNLYNSDVFDGKVQRGLIVFHTSTEPSVNYDLF | DMSO |
| P17 | RRYLQEKYNLYNSDVFDGKVQRGLIVFHTSTEPSVNYDLFGAQGQYSNTL | H2O |
| P18 | YNSDVFDGKVQRGLIVFHTSTEPSVNYDLFGAQGQYSNTLLRIYRDNKTI | H2O |
| P19 | QRGLIVFHTSTEPSVNYDLFGAQGQYSNTLLRIYRDNKTINSENMHIDIY | DMSO |
| P20 | TEPSVNYDLFGAQGQYSNTLLRIYRDNKTINSENMHIDIYLYTS | DMSO |
| Label | Sequence |
|---|---|
| P15-4 | KTNKKNVTVQELDLQARRYLQEKYNLYNSDVFDGKVQRGLIV |
| P15-6 | NKKNVTVQELDLQARRYLQEKYNLYNSDVFDGKVQRGL |
| P15-8 | KNVTVQELDLQARRYLQEKYNLYNSDVFDGKVQR |
| P15-10 | VTVQELDLQARRYLQEKYNLYNSDVFDGKV |
| P15-D17A | LETVKTNKKNVTVQELALQARRYLQEKYNLYNSDVFDGKVQRGLIVFHTS |
| P15-D34A | LETVKTNKKNVTVQELDLQARRYLQEKYNLYNSAVFDGKVQRGLIVFHTS |
| P15-D37A | LETVKTNKKNVTVQELDLQARRYLQEKYNLYNSDVFAGKVQRGLIVFHTS |
| P15-D3A3 | LETVKTNKKNVTVQELALQARRYLQEKYNLYNSAVFAGKVQRGLIVFHTS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashraheel, S.S.; Al-Sulaiti, H.; Goda, S.K. Generation of Novel Tumour-Selective SEA Superantigen-Based Peptides with Improved Safety and Efficacy for Precision Cancer Immunotherapy. Int. J. Mol. Sci. 2024, 25, 9423. https://doi.org/10.3390/ijms25179423
Bashraheel SS, Al-Sulaiti H, Goda SK. Generation of Novel Tumour-Selective SEA Superantigen-Based Peptides with Improved Safety and Efficacy for Precision Cancer Immunotherapy. International Journal of Molecular Sciences. 2024; 25(17):9423. https://doi.org/10.3390/ijms25179423
Chicago/Turabian StyleBashraheel, Sara S., Haya Al-Sulaiti, and Sayed K. Goda. 2024. "Generation of Novel Tumour-Selective SEA Superantigen-Based Peptides with Improved Safety and Efficacy for Precision Cancer Immunotherapy" International Journal of Molecular Sciences 25, no. 17: 9423. https://doi.org/10.3390/ijms25179423
APA StyleBashraheel, S. S., Al-Sulaiti, H., & Goda, S. K. (2024). Generation of Novel Tumour-Selective SEA Superantigen-Based Peptides with Improved Safety and Efficacy for Precision Cancer Immunotherapy. International Journal of Molecular Sciences, 25(17), 9423. https://doi.org/10.3390/ijms25179423