
Applying Molecular Modeling to the Design of Innovative, Non-Symmetrical CXCR4 Inhibitors with Potent Anticancer Activity

 , ,
, ,  , , ,
, , ,  ,
,  , and
, and
Abstract
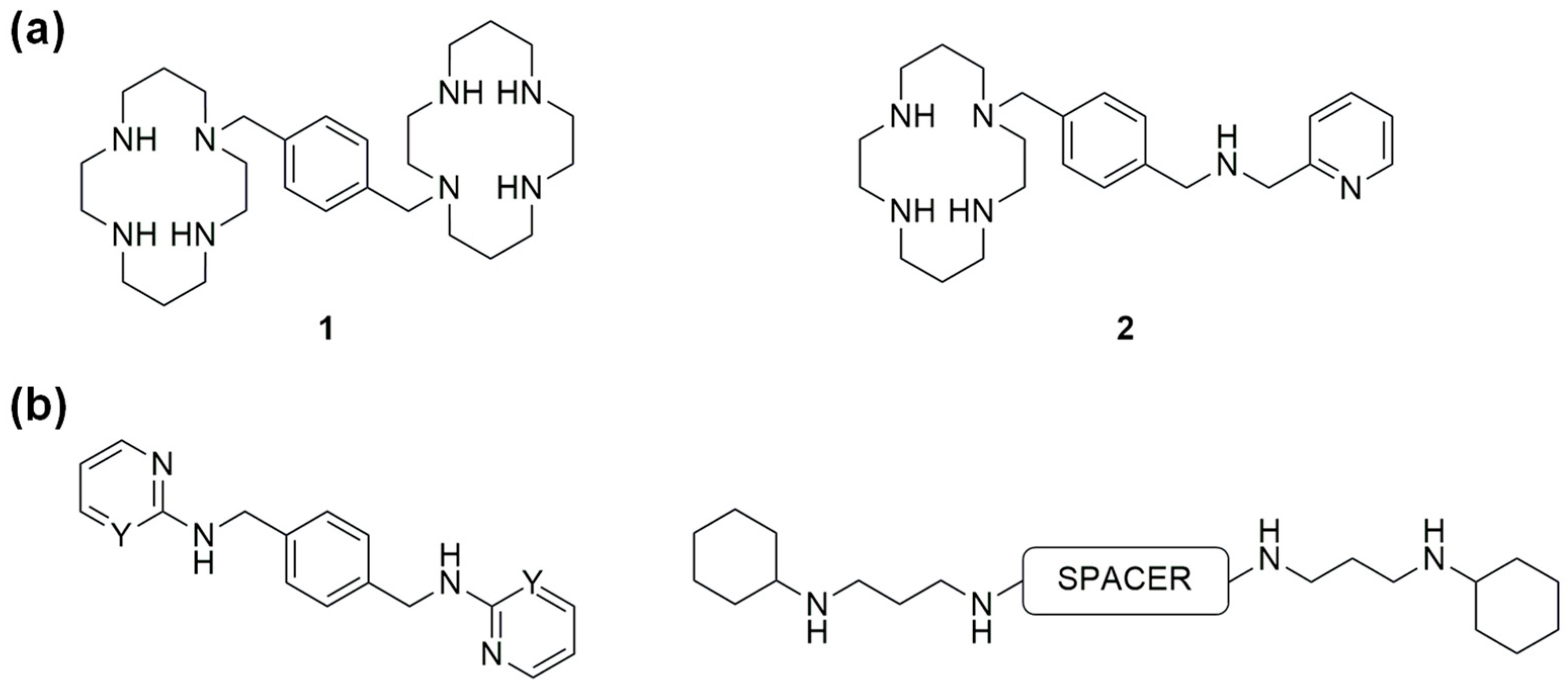
1. Introduction
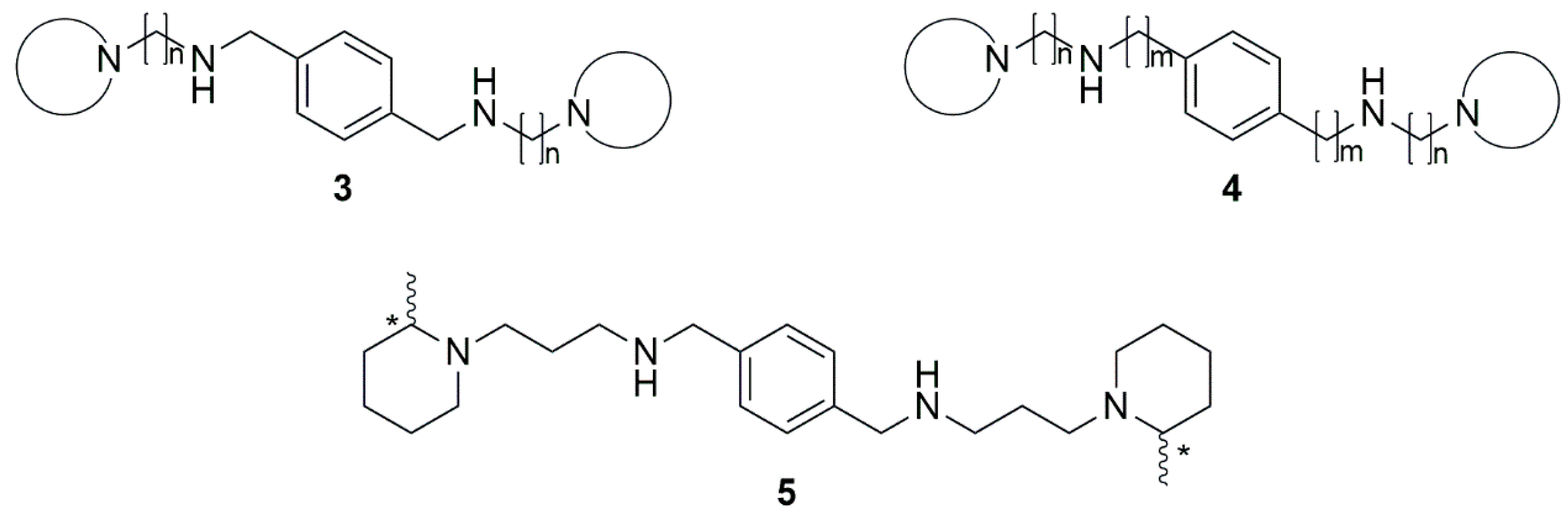
2. Results and Discussion
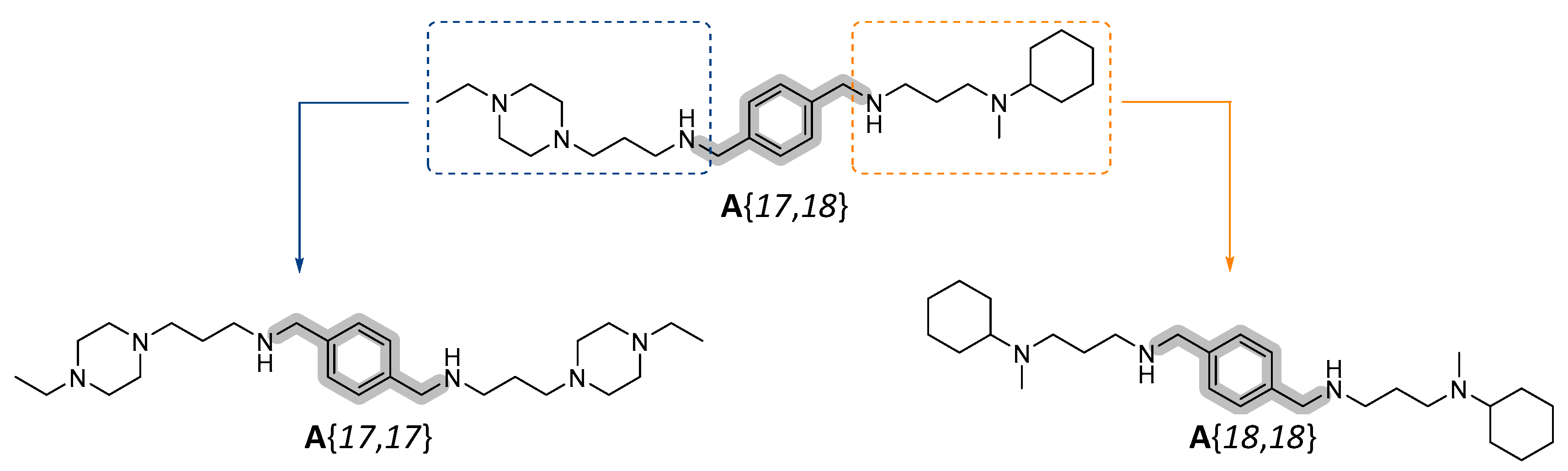
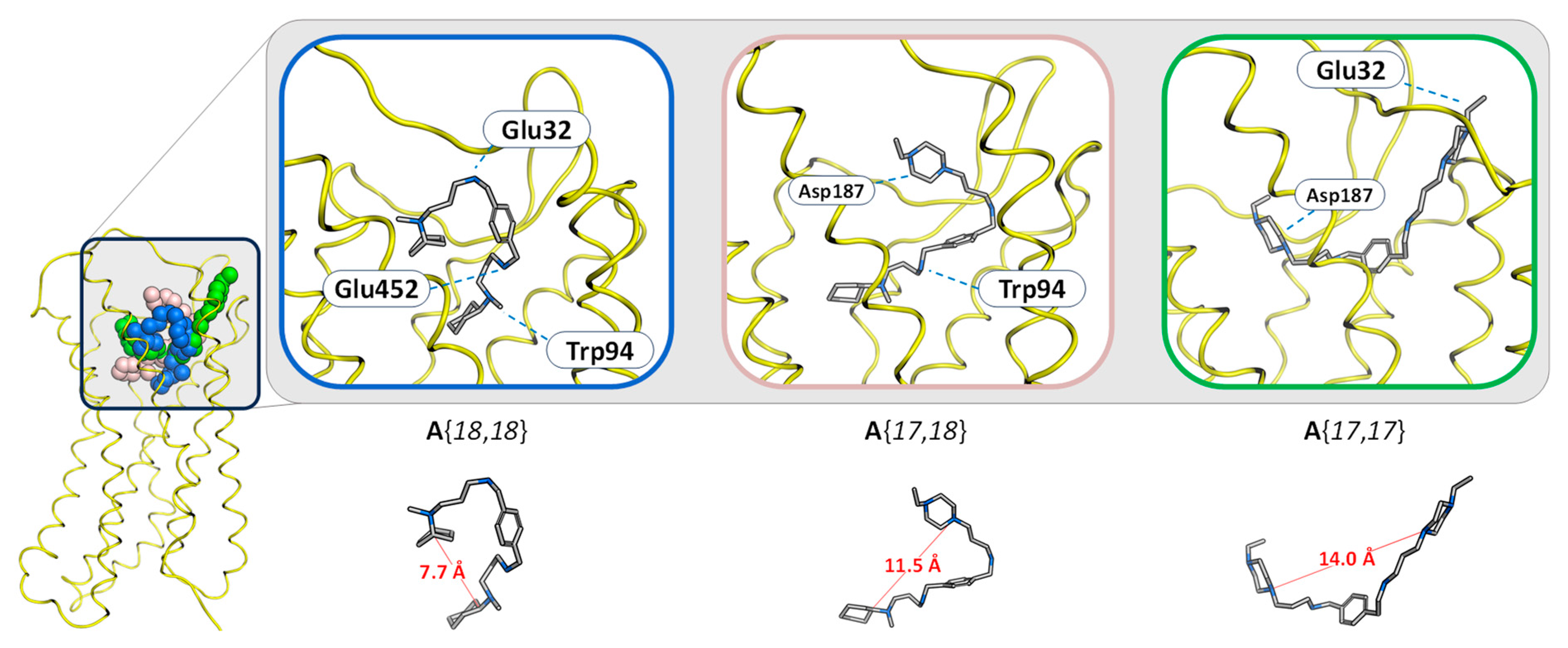
2.1. Prediction of the Binding Mechanism
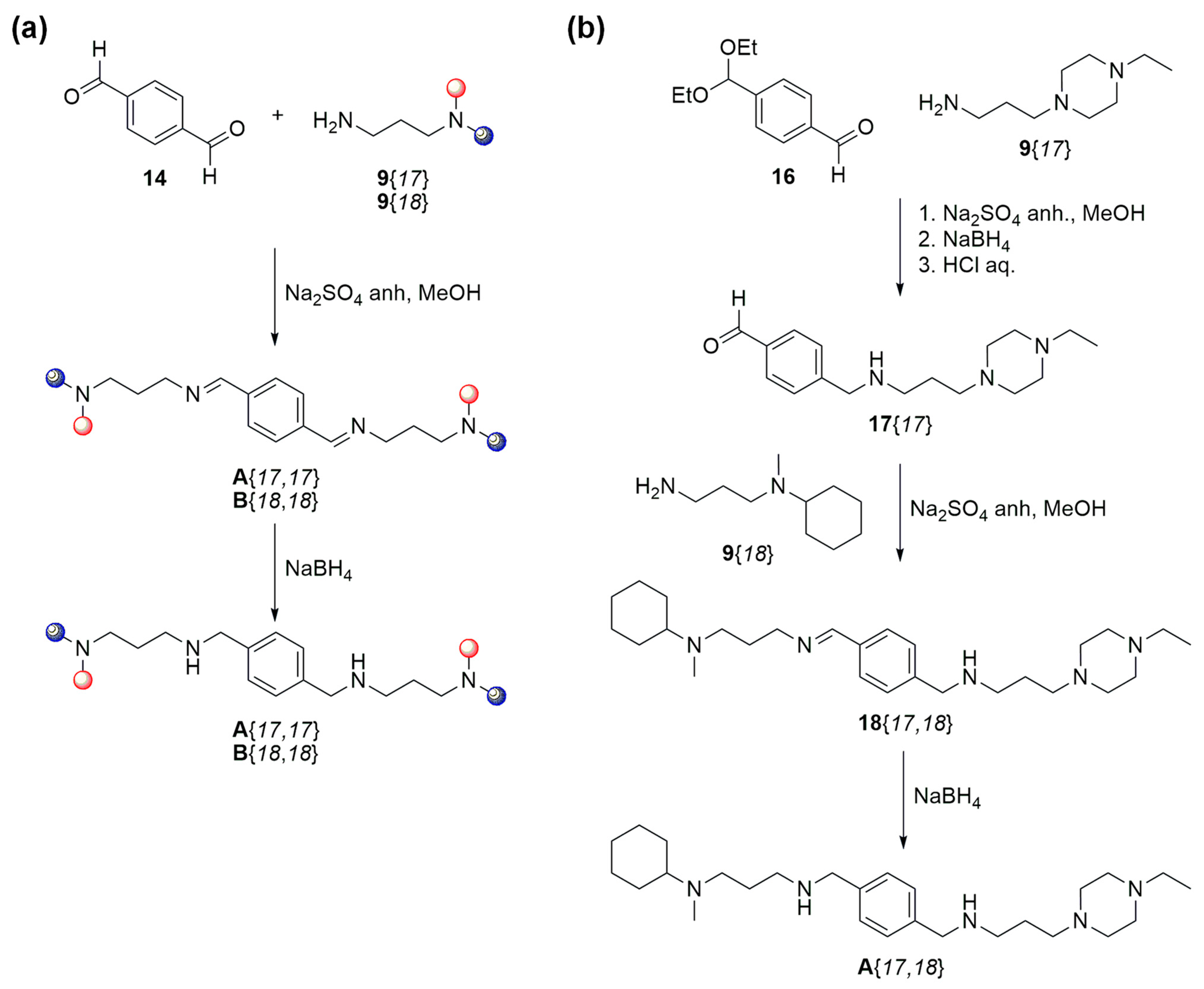
2.2. Synthesis of the Selected Compounds
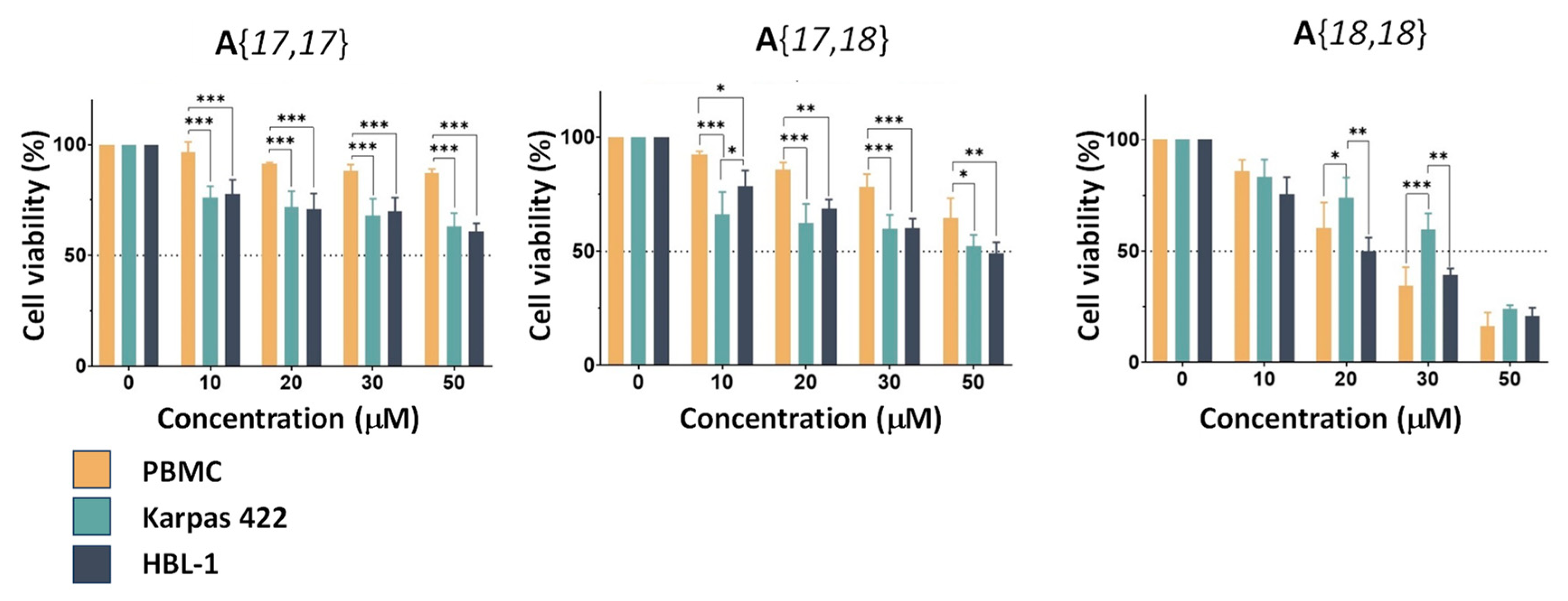
2.3. Assessment of the Biological Activity of Selected Candidates
3. Materials and Methods
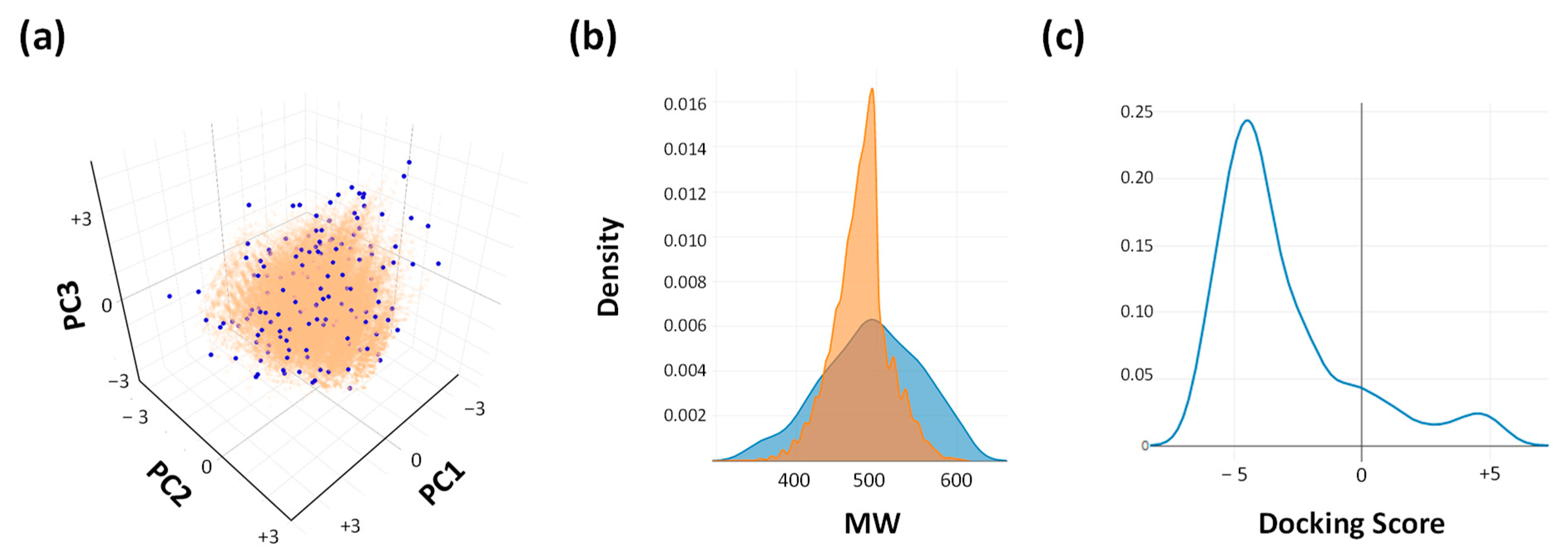
3.1. Combinatorial Library Enumeration
3.2. Molecular Docking
3.3. Molecular Dynamics Simulations
3.4. General
3.5. Synthesis
3.6. Cell Lines and Primary Cultures
3.7. MTT Proliferation Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Esté, J.A.; Telenti, A. HIV entry inhibitors. Lancet 2007, 370, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Furusato, B.; Mohamed, A.; Uhlén, M.; Rhim, J.S. CXCR4 and cancer. Pathol. Int. 2010, 60, 497–505. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Ping, Y.F.; Yao, X.H.; Jiang, J.Y.; Zhao, L.T.; Yu, S.C.; Jiang, T.; Lin, M.C.M.; Chen, J.-H.; Wang, B.; Zhang, R.; et al. The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J. Pathol. 2011, 224, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.J.; Bosch, R.; Dieguez-Gonzalez, R.; Novelli, S.; Mozos, A.; Gallardo, A.; Pavón, M.Á.; Céspedes, M.V.; Grañena, A.; Alcoceba, M.; et al. CXCR4 expression enhances diffuse large B cell lymphoma dissemination and decreases patient survival. J. Pathol. 2015, 235, 445–455. [Google Scholar] [CrossRef]
- Donzella, G.A.; Schols, D.; Lin, S.W.; Esté, J.A.; Nagashima, K.A.; Maddon, P.J.; Allaway, G.P.; Sakmar, T.P.; Henson, G.; De Clercq, E.; et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 1998, 4, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Plerixafor: A review of its use in stem-cell mobilization in patients with lymphoma or multiple myeloma. Drugs 2011, 71, 1623–1647. [Google Scholar] [CrossRef]
- Calandra, G.; Bridger, G.; Fricker, S. CXCR4 in clinical hematology. Curr. Top. Microbiol. Immunol. 2010, 341, 173–191. [Google Scholar] [CrossRef]
- Hatse, S.; Princen, K.; De Clercq, E.; Rosenkilde, M.M.; Schwartz, T.W.; Hernandez-Abad, P.E.; Skerlj, R.T.; Bridger, G.J.; Schols, D. AMD3465, a monomacrocyclic CXCR4 antagonist and potent HIV entry inhibitor. Biochem. Pharmacol. 2005, 70, 752–761. [Google Scholar] [CrossRef]
- Zhan, W.; Liang, Z.; Zhu, A.; Kurtkaya, S.; Shim, H.; Snyder, J.P.; Liotta, D.C. Discovery of Small Molecule CXCR4 Antagonists. J. Med. Chem. 2007, 50, 5655–5664. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Meng, Q.; Zhang, H.; Liang, B.; Zhu, S.; Wang, J.; Zhang, C.; Huang, L.S.; Zhang, X.; Schooley, R.T.; et al. Design, synthesis, and biological characterization of a new class of symmetrical polyamine-based small molecule CXCR4 antagonists. Eur. J. Med. Chem. 2020, 200, 112410. [Google Scholar] [CrossRef]
- Pettersson, S.; Pérez-Nueno, V.I.; Ros-Blanco, L.; Puig de La Bellacasa, R.; Rabal, M.O.; Batllori, X.; Clotet, B.; Clotet-Codina, I.; Armand-Ugón, M.; Esté, J.; et al. Discovery of novel non-cyclam polynitrogenated CXCR4 coreceptor inhibitors. ChemMedChem 2008, 3, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, S.; Pérez-Nueno, V.I.; Mena, M.P.; Clotet, B.; Esté, J.A.; Borrell, J.I.; Teixidó, J. Novel monocyclam derivatives as HIV entry inhibitors: Design, synthesis, anti-HIV evaluation, and their interaction with the CXCR4 co-receptor. ChemMedChem 2010, 5, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Nueno, V.I.; Ritchie, D.W.; Rabal, O.; Pascual, R.; Borrell, J.I.; Teixidó, J. Comparison of ligand-based and receptor-based virtual screening of HIV entry inhibitors for the CXCR4 and CCR5 receptors using 3D ligand shape matching and ligand-receptor docking. J. Chem. Inf. Model. 2008, 48, 509–533. [Google Scholar] [CrossRef]
- Puig de la Bellacasa, R.; Gibert, A.; Planesas, J.M.; Ros-Blanco, L.; Batllori, X.; Badía, R.; Clotet, B.; Esté, J.; Teixidó, J.; Borrell, J.I. Nitrogen positional scanning in tetramines active against HIV-1 as potential CXCR4 inhibitors. Org. Biomol. Chem. 2016, 14, 1455–1472. [Google Scholar] [CrossRef]
- Ros-Blanco, L.; Anido, J.; Bosser, R.; Esté, J.; Clotet, B.; Kosoy, A.; Ruíz-Ávila, L.; Teixidó, J.; Seoane, J.; Borrell, J.I. Noncyclam tetraamines inhibit CXC chemokine receptor type 4 and target glioma-initiating cells. J. Med. Chem. 2012, 55, 7560–7570. [Google Scholar] [CrossRef]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef]
- Planesas, J.M.; Pérez-Nueno, V.I.; Borrell, J.I.; Teixidó, J. Impact of the CXCR4 structure on docking-based virtual screening of HIV entry inhibitors. J. Mol. Graph. Model. 2012, 38, 123–136. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Kleandrova, V.; Scotti, M.; Cordeiro, M. 3D-QSAR Methodologies and Molecular Modeling in Bioinformatics for the Search of Novel Anti-HIV Therapies: Rational Design of Entry Inhibitors. Curr. Bioinform. 2013, 8, 452–464. [Google Scholar] [CrossRef]
- Karaboga, A.S.; Planesas, J.M.; Petronin, F.; Teixidó, J.; Souchet, M.; Pérez-Nueno, V.I. Highly specific and sensitive pharmacophore model for identifying CXCR4 antagonists. Comparison with docking and shape-matching virtual screening performance. J. Chem. Inf. Model. 2013, 53, 1043–1056. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Staropoli, I.; Brelot, A.; Suzanne, P.; Lesnard, A.; Fontaine, F.; Perato, S.; Rault, S.; Helynck, O.; Arenzana-Seisdedos, F.; et al. Discovery of Bis-Imidazoline Derivatives as New CXCR4 Ligands. Molecules 2023, 28, 1156. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.H.; Chen, X.M.; Zhang, F.B.; Zhao, C.; Tu, S.S. Inhibition of CXCR4 regulates epithelial mesenchymal transition of NSCLC via the Hippo-YAP signaling pathway. Cell Biol. Int. 2018, 42, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput.-Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Mo-tion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Sagui, C.; Pedersen, L.G.; Darden, T.A. Towards an Accurate Representation of Electrostatics in Clas-sical Force Fields: Efficient Implementation of Multipolar Interactions in Biomolecular Simulations. J. Chem. Phys. 2004, 120, 73–87. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | LD50 (µM) | ||
|---|---|---|---|
| Karpas422 | HBL-1 | PBMCs | |
| A{17,17} | >50 | >50 | >50 |
| A{17,18} | 39 | 45 | >50 |
| A{18,18} | 36 | 20 | 23 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Asensio, M.; Sàrrias, L.; Gorjón-de-Pablo, G.; Fernández-Serrano, M.; Camaló-Vila, J.; Gibert, A.; Puig de la Bellacasa, R.; Teixidó, J.; Roué, G.; Borrell, J.I.; et al. Applying Molecular Modeling to the Design of Innovative, Non-Symmetrical CXCR4 Inhibitors with Potent Anticancer Activity. Int. J. Mol. Sci. 2024, 25, 9446. https://doi.org/10.3390/ijms25179446
Martínez-Asensio M, Sàrrias L, Gorjón-de-Pablo G, Fernández-Serrano M, Camaló-Vila J, Gibert A, Puig de la Bellacasa R, Teixidó J, Roué G, Borrell JI, et al. Applying Molecular Modeling to the Design of Innovative, Non-Symmetrical CXCR4 Inhibitors with Potent Anticancer Activity. International Journal of Molecular Sciences. 2024; 25(17):9446. https://doi.org/10.3390/ijms25179446
Chicago/Turabian StyleMartínez-Asensio, Miquel, Lluís Sàrrias, Gema Gorjón-de-Pablo, Miranda Fernández-Serrano, Judith Camaló-Vila, Albert Gibert, Raimon Puig de la Bellacasa, Jordi Teixidó, Gaël Roué, José I. Borrell, and et al. 2024. "Applying Molecular Modeling to the Design of Innovative, Non-Symmetrical CXCR4 Inhibitors with Potent Anticancer Activity" International Journal of Molecular Sciences 25, no. 17: 9446. https://doi.org/10.3390/ijms25179446
APA StyleMartínez-Asensio, M., Sàrrias, L., Gorjón-de-Pablo, G., Fernández-Serrano, M., Camaló-Vila, J., Gibert, A., Puig de la Bellacasa, R., Teixidó, J., Roué, G., Borrell, J. I., & Estrada-Tejedor, R. (2024). Applying Molecular Modeling to the Design of Innovative, Non-Symmetrical CXCR4 Inhibitors with Potent Anticancer Activity. International Journal of Molecular Sciences, 25(17), 9446. https://doi.org/10.3390/ijms25179446