Abstract
Purinergic signaling has emerged as an important paracrine–autocrine intercellular system that regulates physiological and pathological processes in practically all organs of the body. Although this system has been thoroughly defined since the nineties, recent research has made substantial advances regarding its role in aspects of liver physiology. However, most studies have mainly targeted the entire organ, 70% of which is made up of parenchymal cells or hepatocytes. Because of its physiological role, the liver is exposed to toxic metabolites, such as xenobiotics, drugs, and fatty acids, as well as to pathogens such as viruses and bacteria. Under injury conditions, all cell types within the liver undergo adaptive changes. In this context, the concentration of extracellular ATP has the potential to increase dramatically. Indeed, this purinergic response has not been studied in sufficient detail in non-parenchymal liver cells. In the present review, we systematize the physiopathological adaptations related to the purinergic system in chronic liver diseases of non-parenchymal liver cells, such as hepatic stellate cells, Kupffer cells, sinusoidal endothelial cells, and cholangiocytes. The role played by non-parenchymal liver cells in these circumstances will undoubtedly be strategic in understanding the regenerative activities that support the viability of this organ under stressful conditions.
1. Introduction
Although the more recognized tasks in the liver are supported by hepatocytes, non-parenchymal liver cells (NPLCs) play essential roles in the tissular homeostasis and in the structure and maintenance of the organ. For example, hepatic stellate cells (HSCs) support the extracellular matrix production, while Kupffer cells (KCs) play a role in managing infections. Moreover, NPLCs play an essential role in pathological conditions, where they present notable phenotypic adaptations. The objective of the present review was to systematically summarize the experimental evidence on the expression and function of purinergic signaling elements on NPLCs, and its relevance in physiological and pathological conditions. Although the available data are mainly from basic research, we expect that the landscape of NLPC and purinergic signaling opens new ways for new research programs to propose innovative therapeutic targets with clinical impact.
2. The Liver and Chronic Disease
The liver is the organ that controls several metabolic pathways that are indispensable in maintaining homeostasis. Some functions of these pathways include urea synthesis, fatty acid metabolism, glycogen storage, albumin, and complement-system component synthesis, among many others [1]. It also coordinates the activity of parenchymal and non-parenchymal cells.
Furthermore, the liver is a complex anatomical structure consisting of hexagonal units (called “lobules”) arranged in a honeycomb-like pattern [2]. Histologically, these units are delimited by vascular points of reference: six portal triads around a central vein. The triad includes branches of the hepatic artery, the portal vein, and the bile duct, and it is connected by a system of liver sinusoids to the central vein. Hepatocytes create cords along the sinusoids, but they differ from those surrounding the central vein in that their metabolic patterns are noticeably different. This cellular arrangement underlies three discrete functional areas termed elsewhere as “liver zonation” [3]. Periportal zone 1 is rich in oxygen and nutrient supply. Its main functions are glycogen metabolism and ammonia handling by urea formation. Intermediate perivenous zone 2 is specialized in xenobiotic metabolism. Perivenous zone 3 is smaller than the others and surrounds the central vein. Glutamine synthetase, increased glycolytic activity, and biotransformative reactions occur in this area. It has been suggested that liver zonation is established as a response of the biochemical gradients of oxygen, hormones, and nutrients implicated in hepatic blood circulation [4], as well as in selective Wnt ligand secretion, acting as morphogens from liver endothelial cells [5].
The liver performs important functions, including absorption, synthesis, storage, and nutrient metabolism. However, it is exposed to toxic metabolites such as xenobiotics, alcohol, and excess fatty acids, as well as to pathogenic factors such as viruses, bacteria, and other microbial components derived from feeding activity. Despite these circumstances, the liver remains in homeostasis thanks to the high plasticity of its cells and the regenerative capacity that allows this organ to continue functioning. For example, mice with a ~70% hepatectomy regain 100% of their size 7 to 10 days after surgery. Under these conditions, total liver regeneration occurs in humans in 3 months [6,7]. However, when there is chronic exposure to toxic agents, this regenerative capacity is insufficient, leading to multiple complications, which trigger liver diseases. The first complication is liver inflammation. If not controlled, liver inflammation generates fibrosis, which can lead to cirrhosis and, eventually, hepatocellular carcinoma [8,9].
2.1. Inflammation
When the liver is challenged by metabolic stress due to exposure to toxic agents (xenobiotics, paracetamol, alcohol, fatty acids) or to the presence of pathogens, it promotes an inflammatory response [8]. As a result of the damage, cells release molecules that serve as alarming cellular signals to favor survival and attract immune cells. These are known as danger-associated molecular patterns (DAMPs) and comprise various molecules, such as nucleic acids, nuclear and cytosolic proteins, and nucleotides (mainly ATP) [10]. DAMPs are recognized by pattern recognition receptors (PRR) of resident immune cells of the liver, such as Kupffer cells (KCs), dendritic cells, hepatocytes, and stellate cells (HSCs). Resident immune cells secrete proinflammatory cytokines and chemokines (e.g., IL-1β, IL-6, IL-32, IL-33, and TNF-α [11,12,13]). These cells promote the hepatic extravasation of neutrophils and monocytes, which differentiate into macrophages and magnify the inflammatory response [14]. In addition, ATP, through the P2X7 receptor, can also activate the NLRP3 inflammasome in endothelial sinusoidal cells, hepatocytes, stellate cells, KCs, and neutrophils [15,16]. Stimulation of NLRP3 activates caspase-1, which induces proteolytic processing and the release of interleukins IL-1β and IL-18, further enhancing the proinflammatory response [17]. It has recently been shown that murine neutrophil-specific NLRP3 activation can independently initiate liver inflammation, alter liver development, and promote liver fibrosis [18].
Hepatitis, or liver inflammation, can be beneficial or pathological to the liver, depending on the intensity and duration of the inflammation. When it is controlled, it supports repair, but when inflammation is disproportionate, it becomes pathological.
2.2. Fibrosis
In vertebrates, the liver is a soft and well-defined organ. Hepatocytes are the most abundant cells in the liver and are arranged in plates. Among the plates, non-parenchymal cell populations are located in the sinusoids and the Disse space, which lies between the endothelial cells and hepatocytes (Figure 1) [12]. The components of the extracellular matrix (ECM) provide the liver with the appropriate stiffness to fulfill its functions. The ECM is a critical component in the liver, since it provides a surface for cell adhesion, growth, and migration, and serves as a reservoir for signaling molecules [19,20]. When the ECM components build up, and their degradation slows down, fibrosis occurs. Type I collagen, elastin, hyaluronan, fibronectin, and some proteoglycans are primarily involved in this process [21]. Fibrosis causes changes in the structure of the liver and results in the formation of fibrous scars and collagen cords [22].
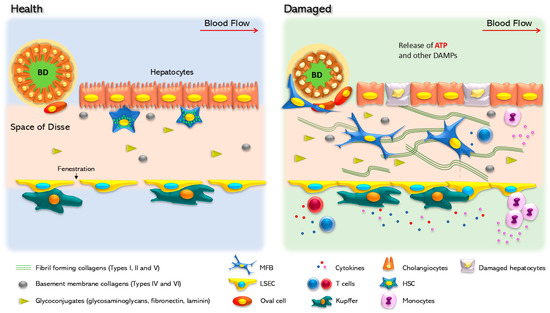
Figure 1.
Cellular changes in healthy and damaged liver. In the healthy liver, parenchymal cells are well organized, following a portal/central zonation in basal contact with the extracellular matrix. In this condition, hepatocytes show cilium and nuclear integrity. Hepatic stellate cells (HSCs), located in the Disse space, between the parenchymal layer and the liver sinusoidal endothelial cells (LSECs), show a quiescent phenotype characterized by vitamin A (retinol) storage. LSEC surrounds the Disse space and in specific zones form specialized structures named fenestra that regulate the flux of metabolites to the acinus. Kupffer cells are in contact with LSECs in an inactivated state, and oval cells are in a quiescent state surrounding the biliary duct (BD), which is made up of cholangiocytes. In response to a chronic injury, the liver suffers continuous scarring, until fibrosis is established as a result of the modification and accumulation of the extracellular matrix. Hepatocytes are the first cells to respond and adapt to stressful stimuli. Non-parenchymal cells undergo a set of phenotypic changes. Experimental HSC activation is a transdifferentiation process that promotes HSC conversion into myofibroblasts, a phenotype characterized by the production of extracellular matrix components, mainly collagen Iα. Kupffer cells are also activated and initiate the secretion of proinflammatory cytokines. In addition, LSEC loses the fenestra and releases inhibitors of platelet aggregation, such as nitric oxide and prostaglandin G12 (NO and PGI2). Cholangiocytes within the biliary duct lose the cilia and change their secretory profile.
Although various cell types can alter the ECM, myofibroblasts (MFBs) play a central role in this process, since they are absent from healthy livers [22,23]. Activated HSCs transdifferentiate and generate approximately 90% of MFBs in experimental hepatotoxic models, thus producing profibrogenic factors such as transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF), as well as proinflammatory cytokines [24]. Hence, the stellate cells lose their characteristic basal shape and their vitamin A storages, becoming active producers of type I collagen, α-smooth muscle actin (αSMA), and tissue inhibitor of metalloproteinase 1 (TIMP1) (Figure 1). The accumulation of these components causes phenotypic changes in the liver and reprograms cellular metabolic pathways that affect nutrient exchange [25].
Liver fibrosis may present reversible and irreversible states. Fibrosis is reversible if the promoting etiological agent is removed, as it has been observed in samples from patients with autoimmune inflammatory liver disease, non-alcoholic steatohepatitis (NASH), and alcoholic steatohepatitis (ASH) [22]. Although the reversal of fibrosis is a complex process, macrophages support fibrogenesis by secreting TGF-β and proinflammatory cytokines such as IL-6, IL-1β, and TNF-α, but when there is no inflammatory stimulus, they stop producing proinflammatory molecules and secrete a large amount of collagenase and metalloproteinase enzymes MMP12 and MMP13, which dissolve fibrotic complex aggregates [26]. Furthermore, activated HSCs revert their phenotype in the absence of TGF-β stimulation. Although they do this differently from quiescent cells, HSCs manage to reestablish some of their basic characteristics, such as the accumulation of vitamin A droplets [27]. It has been reported that 50% of these HSCs undergo apoptosis after the cessation of CCl4 stimulation [28]. HSCs that revert their phenotype are more susceptible to reactivation by proinflammatory stimuli, as if they had an epigenetic memory [27]. If fibrosis is not reversed, it can damage liver function and increase the risk of developing hepatocellular carcinoma.
2.3. Cirrhosis
Currently, cirrhosis is a leading cause of death, with 2 million annually worldwide, accounting for 4% of all deaths [29]. The most common causes of cirrhosis are related to viral hepatitis, alcohol intake, and the spectrum of non-alcoholic fatty liver diseases [29]. Cirrhosis is a consequence of fibrosis. Chronic inflammation causes excessive production of fibrotic scars, which generates plastic and dynamic changes in the stiffness, flexibility, and density of the ECM, thus reducing the parenchymal mass of the liver [30]. Regenerative hepatic nodules replace the typical hepatic architecture; the fenestrations of the liver that allow the exchange of essential molecules are lost, eventually leading to liver failure [31]. In addition, hepatic structural abnormalities prevent proper blood circulation, causing portal hypertension [32], altered bile flow (cholestasis), acidic intestinal pH, as well as changes in the intestinal microbiota and hepatic immune response [33,34]. The chronic inflammation of the liver does not progress to cirrhosis in all patients. However, when it does, the speed at which it occurs can vary from weeks to years. Moreover, cirrhosis often presents asymptomatically [35]. In most cases, symptoms are detected in the advanced stages of the disease, which seriously compromises the survival of the patients.
2.4. Hepatocellular Carcinoma
Hepatocellular carcinoma is one of the most lethal and prevalent cancers in the world, according to Global Cancer Observatory (GLOBOCAN). It is usually related to advanced liver disease, with cirrhosis [36] and hepatitis B and C as pathognomonic risk factors for the development of this type of cancer [37]. The mechanisms by which hepatocellular carcinoma is generated are diverse and complex. For example, it is suggested that the continuous renewal of hepatocytes promotes the shortening of telomeres, which generates chromosomal instability [38]. On the other hand, reactive oxygen species (ROS) production and a persistent inflammatory response can be correlated with the activation of genes that favor cell proliferation, thereby sustaining damaged cells that will ultimately lead to cancer [39].
3. Purines and Cell Signaling
Chemical communication is a widespread evolutionary strategy to coordinate metabolic and physiological adaptations in a variety of biological systems, which encompass everything, from individual cells to complex organisms. This type of cellular interaction involves the synthesis and release of molecular messengers, as well as the presence of specific receptors in target cells. These receptors are tangled proteins that can undergo concerted conformational changes to allow signal transduction responses. Purines, especially the nucleotide ATP and the nucleoside adenosine (ADO), are ubiquitous adenine-related molecules that play important roles as ligands of an extensive family of membrane receptors. Some relevant physiological processes that involve purinergic signaling are sleep, immunological response, renal electrolyte reabsorption, and platelet aggregation. In addition, it is well accepted that ATP and ADO are valuable mediators in pathological situations such as cancer and inflammation [40,41]. Indeed, the extracellular actions of ATP and ADO as chemical messengers are coordinated with their relevant intracellular roles as enzymatic allosteric factors and energy metabolites [42].
Purinergic signaling is distinctive in one particular aspect: ATP can transform into ADO by consecutive phosphate removal, with ADP and AMP as intermediates in this conversion. Since ATP and ADO recognize their own receptors, the final purinergic communication in a given cellular or tissular system will depend on the expression of selective ATP and ADO receptors, but more importantly, it will depend on the enzymatic machinery of ectonucleotidases and phosphatases, which dictate the proportion of ATP and ADO in the extracellular milieu [43].
Purines are an extensive family of aromatic nitrogen-containing heterocyclic compounds formed by the fusion of pyrimidine and imidazole rings. They are important biochemical, metabolic, and informative entities that function as energy metabolites in the structure of coenzymes and within genetic material. They are also psychoactive factors and chemical messengers. In a cellular context, the two principal purines are adenine (6-aminopurine) and guanine (2-amino-6-hydroxypurine). This review focuses on adenine-related molecules, mainly ATP and ADO, both of which present a ribose as a furanose ring attached to position 9.
ATP is the most abundant adenine nucleotide within cellular systems and is found in the millimolar range. The proportion of the other two nucleotides, ADP and AMP, dictates the equilibrium between anabolic and catabolic reactions [44]. Usually, the extracellular presence of ATP serves to accomplish its messenger/ligand role. For example, ATP can access the extracellular space via exocytosis (secretory vesicles), microvesicles derived from the plasma membrane, transporters (ABC cassettes), pannexin-1 or connexins acting as channels, or purinergic receptor channels such as the P2X7 receptor [45]. However, under cellular stress, ATP flows out of the cells and reaches much higher external concentrations. In this scenario, ATP acts similarly to alarmins or DAMPs, inducing a characteristic inflammatory response [46]. It has also been reported that high levels of ATP are present in the extracellular milieu of cancerous tumors [47].
The appearance of ADO as a ligand in the ECM mainly depends on ATP conversion by a set of dephosphorylation events that remove phosphates to form ADO. ADP and AMP are intermediates in this process. Originally, two ectoenzymes were postulated to dephosphorylate ATP: clusters of differentiation 39 and 73 (CD39 and CD73), which are intrinsic membrane proteins [48]. CD39 turns ATP and ADP into AMP, whereas CD73 converts AMP into ADO. More recently, the family of these enzymes has been expanded to include eight members of the ectonucleoside triphosphate diphosphohydrolase (NTPDase) family, seven mammalian paralogs of the ectonucleotide pyrophosphatases/phosphodiesterases (ENPPase) family, ecto-alkaline phosphatases, and ecto-5′-nucleotidases [41]. The presence of extracellular ADO can end by means of two principal mechanisms: ADO conversion to inosine by the activity of an ecto-adenosine deaminase or ADO internalization to the cytoplasmic milieu by ENT and CNT (equilibrative and concentrative nucleoside transporters, respectively), two types of membrane transporters [49].
ATP and ADO interact with a set of specific receptors. These proteins, which are present essentially in every tissue, appeared early in the phylogenetic evolution, since eukaryotic protozoa and simple algae express purinergic receptors [50]. There are two types of receptors for ATP: channel receptors (P2X) and G-protein-coupled receptors (P2Y). P2X receptors are ATP-gated ion channels that are permeable to cations (Na+, K+, and Ca2+). Molecular cloning studies have reported seven genes coding for the P2X1-7 subunits. Each protein is characterized by two transmembrane domains, and the amino and carboxy ends are located on the intracellular side. A functional receptor is a homo- or heterotrimer of P2X subunits [51]. P2Y receptors belong to the family of seven transmembrane regions and influence the levels of second messengers, such as cAMP and Ca2+. So far, eight different P2Y receptors have been reported (P2Y1,2,4,6 and 11–14). Apart from ATP, some of these receptors interact with other ligands; for example, ADP with P2YR1, 6, 12–14; UTP with P2YR2 and 4; UDP-glucose with P2YR14 [52]. ADO has four different receptors (A1, A2a, A2b, and A3), all of which belong to the G-protein-coupled receptor family. The signal transduction of ADO receptors usually downregulates the cAMP levels [53].
In summary, purinergic transmission is a recurrent and complex form of cellular communication. Studies have described the ATP and ADO signaling pathways as both complementary and sometimes antagonistic; for example, in some cellular systems, ATP functions as an immunostimulant, while ADO acts as an immunosuppressive agent [54]. Hence, the specific types of ATP and ADO receptors, along with the set of ectonucleotidases in the extracellular space, determine the final purinergic response in a given physiological or pathological event. These parameters, acting in coordination, dictate the proportion of ATP and ADO as ligands and underpin the signaling transduction processes that these purines promote [55].
4. Purines in Liver Pathology
Extracellular nucleotides play critical roles in maintaining normal homeostasis and modulate many physiological processes in the liver. However, certain biological stressors alter the release of these nucleotides and the activities of ectonucleotidase enzymes [56,57,58]. In hepatic regeneration, it has been demonstrated that P2Y2 [59], P2X4 [60], and P2X receptors have a global positive impact on the regulation of physiological processes [61]. Additionally, some studies have found that the A3 receptor activation has a protective effect against damage and leads to an increased rate of liver regeneration [62,63].
In experimental liver fibrosis, when the damage was induced by chemical toxins such as diethylnitrosamine, carbon tetrachloride (CCl4), or thioacetamide, it was demonstrated that P2 [64], P2X7 [65,66], P2Y2 [67], A2, and A1 receptors [68,69,70] are involved in the onset of the fibrotic damage, mediating the activation of various profibrotic factors and the accumulation of collagen, probably through HSC activation. Furthermore, the P2X7 receptor has been shown to mediate the oxidative stress-induced exacerbation of inflammatory liver injury via an NADPH oxidase-dependent mechanism [71]. In addition, a more aggressive effect of hepatotoxic CCl4 has also been demonstrated in ectonucleoside triphosphate diphosphohydrolase-2 null mice [72].
Regarding N-acetyl-para-aminophenol (APAP)-induced hepatotoxicity, it was observed that P2X receptors [73], especially the P2X7 receptor, may be involved in necrosis, and that stimulating both of these receptors may enhance cell death independently of immune system activation [74,75,76]. Additionally, the P2Y2 receptor was demonstrated to be involved in increasing the liver damage mediated by APAP and also when hepatitis was induced by the administration of concanavalin A [77].
On the other hand, the P2X4 receptor signaling pathway was responsible for raising the profibrotic profile during bile duct ligation and under a high-fat diet [78]. Furthermore, A1 receptors have been shown to regulate the signaling pathways that lead to fibrosis, as the induction of liver fibrosis associated with CCl4 administration was attenuated in mice lacking this receptor [70].
Concerning liver damage induced by lipotoxicity in experimental NASH, it was demonstrated that A2A receptor activation is necessary to induce fibrosis [79]. Moreover, studies have shown that the P2X7 receptor is a key regulator of autophagy induced by metabolic oxidative stress in NASH and therefore, it also modulates hepatic inflammation [80] and the activation of the NLRP3 inflammasome [81,82,83]. In addition, recent studies have proposed that P2X7 receptor activation promotes autophagic flux, thereby improving the outcome of non-alcoholic fatty liver disease (NAFLD) through lipophagy [84].
Moreover, the P2Y2 receptor was shown to play a critical role in the pathogenesis of NAFLD by regulating AMPK [85]. Furthermore, the A2a receptor was demonstrated to halt NASH progression in mice with established steatohepatitis by means of its immunoinhibitory and cytoprotective effects [86,87]. In addition, pharmacological stimulation of the A2a receptor prevents hepatocyte lipotoxicity and inhibits the progression from NAFLD to NASH by interfering with JNK-1/2 activation [79]. On the other hand, the A3 receptor’s pharmacological activation reduced NAFLD by anti-steatotic, anti-inflammatory, and anti-fibrotic actions [88].
According to reports, the P2X4 receptor may boost the progression of alcoholic liver fibrosis by modulating the PI3K/AKT pathway in parenchymal cells or by influencing macrophages to regulate HSC activation [89]. Additionally, the P2X4 receptor promotes the inflammatory response in alcoholic steatohepatitis, while CD39 plays a protective role in regulating P2X4 receptor expression by hydrolyzing ATP [90]. Reports have also indicated that mice lacking the A1a receptor were more susceptible to acute ethanol-induced liver damage than the wild-type mice. This sensitivity was related to increased lipogenesis and lipid peroxidation, as well as to the depletion of superoxide dismutase, which resulted in hepatic steatosis [91].
Regarding the role of purinergic signaling in the activation and recruitment of immune cells in the liver, an increase in CD8+ T-cells was reported in a mouse model of primary sclerosing cholangitis (multidrug resistance protein 2 knockout) with CD39 deletion. This increase exacerbated liver injury and fibrosis, suggesting that extracellular ATP signaling favors CD8+ T-cell infiltration into the damaged region [92]. In agreement, CD39 expression limited the proinflammatory signaling of the P2X7 receptor and cytokine production in a sepsis-induced liver injury model [93]. Additionally, the P2Y2 receptor has also been related to immune response activation in acute liver damage. The P2Y2 receptor −/− mice expressed fewer chemoattractants compared to the wild-type mice in response to concanavalin A, which had a direct impact on hepatic immune cell infiltration [77]. On the other hand, in cholestatic injury, P2X7 receptor activity was reported during NLRP3 inflammasome activation and in the subsequent secretion of potent proinflammatory cytokines [94].
The P2Y2 receptor has been shown to play a fundamental role in hepatocarcinogenesis by stimulating DNA damage responses and hepatocyte proliferation [95]. Moreover, A3 receptor agonists have been utilized to treat hepatocellular carcinoma because of their anti-cancer and hepatoprotective effects [96].
This body of evidence emphasizes how crucial the purinergic system is in regulating different liver diseases. Most of the research has focused on the liver as a whole, without distinguishing between the functional effects observed and the different cells that make up the liver. Generally, such effects and functions are attributed to parenchymal cells; however, some studies have shown that non-parenchymal cells are also essential in the regulation of various pathological conditions of the liver. In this sense, in the following sections, we set out to describe and highlight in detail the importance of the purinergic system in the main non-parenchymal cells of the liver (i.e., HSCs, KCs, LSECs, and cholangiocytes). Note that, although oval cells were considered, no information about purinergic signaling in this cell type is currently available.
5. Purinergic Signaling in Non-Parenchymal Liver Cells
5.1. Hepatic Stellate Cells
HSCs or Ito cells, are a small population of mesenchymal liver cells whose main structural characteristic is the storage of retinoids, such as vitamin A. In the 19th century, Wilhelm von Kupffer identified these cells using a gold chloride-staining method. HSCs are residents of the perisinusoidal region known as Disse space, located between the hepatocyte cords and endothelial cells [97] (Figure 1). Classical works [98] and recent approaches [99,100,101] support the notion that HSCs represent between 1% and 15% of the total number of liver cells. HSCs are of great biomedical interest because they play a fundamental role in liver fibrogenesis [102,103]. The quiescent HSCs in the normal liver are characterized by cytoplasmic lipid droplets containing retinoids (vitamin A and its metabolites) [97]. These HSCs have a similar genetic program to that of mature adipocytes, such as high expression levels of peroxisome proliferator-activated receptor gamma (PPARγ) and the sterol regulatory element-binding protein 1c (SREBP-1c) [104,105]. Importantly, PPARγ knockdown promotes HSC activation [106].
In response to injury, HSCs undergo a transdifferentiation process known as “activation”, whereby they lose the lipocyte phenotype and acquire MFB characteristics [107]. Cells with the MFB phenotype lose their lipid droplets and express αSMA and collagen 1a1 (Col1a1), a pivotal step for fibrosis induction. Recent studies support the idea that HSCs are the main source of MFB in the fibrotic liver [108,109]. When HSCs are cultured in plastic Petri dishes, they go through spontaneous and progressive activation within 5 to 7 days [97].
Interestingly, evidence suggests that HSC activation is reversible. In hepatocellular damage induced by CCl4, the removal of the hepatotoxic stimulus is enough to trigger the reversion of a fraction of the fibrotic MFB phenotype, by downregulating fibrotic genes and inhibiting apoptotic death [27]. Furthermore, the administration of PPARγ or exogenous expression of SREBP-1c have also been reported to revert the MFB phenotype to quiescent HSCs [104,105]. Novel therapeutic approaches to treat hepatic fibrosis are made possible by the reversal mechanisms of the HSC-activated phenotype. In the next section, we will review the direct actions of purines on HSCs mediated by purinergic receptors or ectonucleotidase activities.
5.1.1. Nucleotide Signaling in Hepatic Stellate Cells
A fundamental study conducted on rat HSCs before the cloning of purinergic receptors described the production of IP3—the intermediate metabolite that mobilizes intracellular Ca2+—and the induction of cell contraction in response to purinergic substances. Stimulation with ATP, UTP, ADP, or 5′-O-(3-thiotriphosphate) incremented intracellular levels of inositol phosphates and cytosolic Ca2+ ([Ca2+]c) and induced cellular contraction [110]. These observations suggested that purines regulate HSC physiology through their action on specific receptors (Figure 2, Table 1).
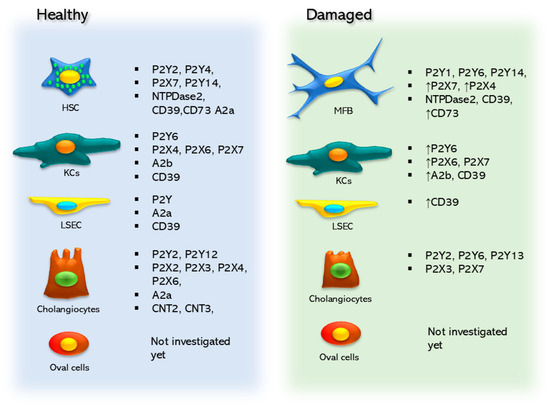
Figure 2.
Purinergic elements are expressed in the non-parenchymal cell types of the liver, and these elements change and are modified between healthy and damaged states.

Table 1.
Purinergic signaling in hepatic stellate cells (HSCs).
In 2004, Rebeca Wells and collaborators characterized the expression and function of P2YRs in quiescent and activated rat HSCs. Quiescent HSCs express transcripts coding for P2Y2 and P2Y4 receptors, whereas activated HSCs express P2Y1 and P2Y6 receptors. Pharmacological characterization of [Ca2+]c mobilization assays mediated by purinergic agonists confirmed these findings. These findings show that, in addition to the P2Y6 receptor, activated HSCs express an ATP-sensitive receptor that is not a P2Y1, P2Y2, or P2Y4 receptor.
Importantly, the stimulation of activated HSCs with UDP increased procollagen-1 expression levels threefold, suggesting a pro-fibrotic role for the P2Y6 receptor [111]. On the other hand, in a model of alcoholic liver toxicity, administering acetaldehyde to rat quiescent HSCs for 48 h increased the expression of the P2X7 receptor, along with the fibrotic markers αSMA and Col1a1. Furthermore, P2X7 receptor activation with 2′(3′)-O-(4-Benzoylbenzoyl) adenosine-5′-triphosphate (BzATP) in quiescent HSC potentiated acetaldehyde-dependent proliferation, cyclin D1 expression, and the upregulation of inflammation and activation markers. Conversely, knocking down the P2X7 receptor expression or incubation with the antagonist A438079 inhibited acetaldehyde-dependent HSC activation [112], suggesting a role for the P2X7 receptor and extracellular ATP in rat HSC activation. The P2X4 receptor, for its part, is overexpressed in the liver of mice with fibrosis induced by hepatotoxic damage (ethanol plus CCl4), as well as in the immortalized cell line HSC-T6, in response to acetaldehyde administration. Receptor blocking with 5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one (5-BDBD) prevented the activation of HSC-T6 cells [89].
A study on the contribution of DAMPs and their receptors to hepatic fibrogenesis found that the P2Y14 receptor is enriched in HSCs. Accordingly, their ligands (UDP-glucose and UDP-galactose) are released by dying hepatocytes. An robust ligand–receptor interaction analysis, combining proteomics and single-cell transcriptomics, confirmed the relationship between both components. Importantly, the stimulation of the P2Y14 receptor by specific agonists or co-culture with dying hepatocytes activated HSCs through a pathway involving extracellular signal-regulated kinase (ERK) and Yes-associated protein (YAP). Moreover, the systemic or HSC-specific P2Y14 receptor abrogation diminished fibrosis induction in different models of liver damage [113].
5.1.2. Ectonucleotidases in Hepatic Stellate Cells
Regarding ectonucleotidase expression and function in HSCs (Table 1), an early study determined that HSC activation is associated with NTPDase2 expression [111]. The NTPDase family has also been analyzed in other studies. HSCs cultured for 1, 4, or 7 days expressed the transcripts coding for NTPDase1 (CD39) and NTPDase2. The comparative analysis of ectonucleotidase activity between quiescent (1 day) and activated (7 days) HSCs showed that both types can hydrolyze ATP in ADP and AMP, suggesting that NTPDase1 is expressed similarly in both conditions. In hepatic fibrosis induced by the administration of CCl4 for 6 weeks, NTPDase2 co-localized with activated HSC [111]. Recently, it has been shown that the pharmacological blocking or genetic inhibition of CD39 expression in the T6 rat HSC line abolished acetaldehyde-dependent proliferation and expression of fibrosis markers [114]. The ENPP family (ENPPs: NPP2-3) was also detected in the mouse GRX HSC line. ENPP activity was higher in quiescent HSCs compared to activated HSCs, and during activation, the NPP2 expression increased, whereas the NPP3 expression decreased [115].
CD73 (5′-NT) is the enzyme that catalyzes the conversion of AMP into ADO. Interestingly, CD73 knockout mice are resistant to experimental fibrosis, indicating that ADO plays a part in fibrosis induction [116]. Pivotal studies in rat HSC primary cultures have shown that the CD73 expression is very low in the quiescent phenotype. However, the activation process triggers a strong increase in the expression levels of this enzyme, which is consistent with activated HSCs. Similar findings were made in immortalized and primary cultured human HSCs. Furthermore, it was determined that CD73 overexpression is transcriptionally regulated by the canonical TGF-β pathway via the SP1 and SMAD response elements in the NT5E promoter [117].
Other reports have supported a role for CD73 in HSC activation [118] mainly using alcohol-related liver fibrosis models [119]. Thus, silencing CD73 in the HSC-T6 line blocked the acetaldehyde-dependent accumulation of αSMA and Col1a1 and induced apoptotic cell death and cell cycle arrest [119]. Similarly, the expression of fibrotic markers and cell proliferation was inhibited by knocking down the ectonucleotidase with a siRNA or by the inhibitor APCP in LX2 and HSC-T6 lines [120]. In agreement, the overexpression of CD73 potentiates acetaldehyde-dependent fibrosis [119]. Between the possible mechanisms explaining the findings described above, it has been proposed that alcoholic liver damage potentiates autophagy induction in activated HSC in a CD73-dependent way by an AMPK/AKT/mTOR pathway [121]. CD73 regulates HSC senescence throughout the p53 pathway by regulating aurora kinase A (AURKA) expression [119]. In addition, a role for the Wnt/β-catenin pathway has been suggested in the execution of the HSC activation program [118].
5.1.3. Adenosine Signaling in Hepatic Stellate Cells
Adenosinergic signaling has been investigated in HSCs (Table 1). Diverse evidence highlights the importance of A2a-mediated ADO signaling in these cells [122]. Thus, in the human HSC line LX2, the activation of the A2a receptor inhibited the PDGF-induced chemotactic response by modulating a pathway that involves adenylate cyclase and cAMP. This led to the upregulation of TGF-β and collagen I RNA production [122]. In this context, it was reported that the A2a-dependent collagen type I and type III overexpression is mediated by a pathway that involves protein kinase A (PKA), src kinase, and mitogen-activated protein kinases ERK and p38 [123], suggesting a role for the ADO/A2a pathway in fibrosis onset. The latter effect can be modulated by interferon γ (IFN-γ), which downregulates adenylate cyclase expression and inhibits A2a-dependent signaling [124].
In addition, in LX2 cells, it was described that the A2a receptor mediates the loss of actin stress fibers via a pathway that comprises cAMP, PKA, and Rho inhibition and suppresses endothelin-1 and lysophosphatidic acid-induced cell contraction [125], suggesting a role in the control of mesenchymal phenotype acquisition. In the same model and rat primary HSCs, A2a activation with NECA or CGS21680 increased cell proliferation and inhibited senescence entry; these changes were accompanied by the downregulation of p53, Rb, and Rac1 and by the inhibition of ERK phosphorylation. These effects were blocked by the A2a antagonist ZM241385 and a PKA inhibitor and reproduced by a cAMP analog and by forskolin, an adenylate cyclase activator [126]. ADO was unable to induce increments in [Ca2+]c in LX2 cells. However, the nucleoside prevented Ca2+ mobilization induced by ATP or PDGF [122].
Interestingly, the A2a receptor antagonist caffeine and ZM241385 inhibited the acetaldehyde-dependent expression of fibrotic markers in HSC-T6 cells. The pathway regulated by A2a involved cAMP, PKA, Src, and ERK in pro-collagen I regulation and P38 in collagen III regulation [127]. On the other hand, caffeine inhibited the activation of primary mice HSCs acting on ADO receptors; however, in this model, the mechanisms were independent of intracellular Ca2+, phosphodiesterase activity, cAMP levels, and ERK phosphorylation, but they relied on Akt1 signaling [128]. In agreement, in systemic models of alcoholic liver disease in rats, caffeine administration decreased fibrosis markers induced by alcohol in isolated HSCs [130].
It was also demonstrated that ADO and the aspartate salt of ADO (IFC305 compound) reversed CCl4-induced hepatic fibrosis and cirrhosis in rats. IFC305 prevented the activation of primary rat HSCs and inhibited the proliferative effect of PDGF on HSCs. However, the participation of ADO receptors in this protocol has yet to be elucidated [129].
5.2. Kupffer Cells
KCs are the resident macrophages of the liver; they are located in the Disse space, surrounding the sinusoid. KCs were first described in 1873 by von Kupffer and constitute approximately 15% of the liver cell population, 30–35% of the non-parenchymal cells [131], and 95% of the macrophages in the mammalian body [132]. KCs are an essential component of innate immunity, acting as the first line of response against potentially harmful elements [133]. They are also the first macrophage population to come in contact with microbes, endotoxins, and microbial debris from the gastrointestinal tract [133]. KCs originate from bone marrow-derived monocytes that migrate to various organs to differentiate into macrophages [134]. In adult organs, they can originate from stem cells.
KCs are differentially distributed throughout the liver; 43% are located in the periportal zone 1, 28% in the intermedial perivenous zone 2, and 29% in zone 3 [135]. Moreover, KCs are the first to be exposed to substances absorbed by the intestinal tract and, consequently, have been considered scavengers of the liver for their ability to eliminate microbes, endotoxins, dead cells, and xenobiotics [136]. Evidence demonstrates that KCs can also function as antigen-presenting and essential regulators of the liver’s inflammatory response. This has various pathophysiological implications, including the fact that they are able to modulate tumor growth and fibrosis onset.
Nucleotide Signaling in Kupffer Cells
The role of purinergic receptors has been widely described in circulating and resident macrophages. Monocytes or macrophages express all the P2Y receptor subtypes [137]. An early study identified the P2Y2 receptor as the main purinergic receptor mediating nucleotide-dependent Ca2+ signaling in peritoneal macrophages. After activation with LPS, the P2Y6 receptor elicited Ca2+ mobilization from the endoplasmic reticulum [138]. In macrophages, P2Y receptors mediate proinflammatory or anti-inflammatory responses [137]. For example, the macrophage-expressed P2Y2 receptor detects nucleotides released by dying cells, enabling their phagocytosis and thereby maintaining tissue homeostasis [139]. In human macrophages, the P2Y2 receptor and the P2Y12 receptor cooperate to kill and engulf bacteria [140]. In the peritoneal macrophages, the P2Y2 receptor mediates the release of proinflammatory mediators, such as IL-1β, in response to LPS stimulation [141].
The P2X7 receptor, for its part, is the best characterized receptor in macrophages [142]. Its expression is upregulated during the differentiation from monocytes to macrophages. The protein level of P2X7 and its ability to induce megapore formation is greater (tenfold) in macrophages compared to monocytes, but it is clearly expressed in both phenotypes [143,144]. In macrophages, P2X7 receptor activation induces ion currents and membrane depolarization [143,144], large pore activation [145], the secretion of inflammatory mediators such as IL-1β [146,147], NLRP3 inflammasome activation [146,147,148,149], and pyroptosis [146].
Purinergic receptor expression and function have also been explicitly characterized in KCs (Table 2). An early study using immunofluorescence to analyze the expression of P2X receptor subunits in KCs (ED1+) identified P2X4 receptor and P2X6 receptor subunits. In the same study, intraperitoneal injection of LPS upregulated the P2X6 receptor expression [150]. The P2X7 receptor has also been found to induce inflammation in response to diverse stimuli. In isolated KCs, extracellular ATP acts on the P2X7 receptor, leading to an NLRP3 inflammasome assembly and, consequently, IL-1β release [76]. In mouse primary KCs, treatment with the environmental contaminant imidacloprid, a compound that causes liver toxicity, induced P2X7 receptor-dependent pyroptosis [151].
Table 2.
Purinergic signaling in Kupffer cells (KCs).
Similar observations have been made in immortalized KC lines. In the mouse KUP5 cell line primed with LPS, stimulation with 3 mM of ATP induced channel aperture, the formation of large pores, and MAPK activation, all of which are hallmarks of the P2X7 receptor activation [152]. Furthermore, the P2X7 receptor activation also induced the release of inflammatory messengers, such as prostaglandin E2, IL-1β, and high mobility group box-1 protein (HMGB1) [152]. In KUP5 cells, whether primed with LPS or not, exposure to silica nanoparticles (30 nm diameter) induced ATP release, activating the P2X7 receptor and eliciting IL-1ẞ production [153].
On the other hand, a study reported that the P2Y6 receptor is overexpressed in the liver, specifically in the KCs of mice with alcohol-induced steatohepatitis. In the liver of these animals, the administration of UDP, a P2Y6 receptor agonist, elevated the levels of damage markers and the release of proinflammatory cytokines TNF-α, IL-1β, and IL-6 induced by alcohol treatment. However, administering MRS2678, an antagonist of this receptor, counteracted these effects. The study also showed that the P2Y6 receptor is functional in the mouse macrophage line RAW264.7, and that blocking its activity attenuated the ethanol-dependent inflammatory response [154].
The ADO receptor expression in KCs was reported in the context of liver grafts. Reperfusion of the organ induces the release of TNF-α by KCs, affecting graft function and causing rejection. However, ADO administration in ischemic preconditioning, which involves a transitory occlusion of the portal triad followed by reperfusion prior to ischemia, protects graft viability [155,156]. In cultured rat KCs, ADO inhibited LPS-dependent TNF release. This effect was also reproduced by the A2 receptor agonists 5-N-ethyl carboxamido adenosine, 2-chloro-adenosine, and R-phenylisopropyl adenosine. CGS15943A, a selective antagonist of the A2 receptor, reverses the inhibition exerted by the aforementioned agonists. Furthermore, the suppression of TNF alpha was also reproduced by dibutyryl-cAMP [157]. According to a recent study, A2bR expression increased in liver homogenates and primary cultures of KCs in alcohol-fed mice, indicating that A2b plays a role in the inflammatory response that underlies alcoholic hepatitis. Similar results were obtained with the human macrophage line RAW264.7 in response to ethanol. Knocking down the A2b receptor in RAW264.7 cells promoted an increment in the release of inflammatory cytokines (IL-6, IL-1β, and TNF-α). In agreement, the A2b receptor overexpression results in an attenuation of the inflammatory response [158]. These results suggest that ADO, acting through the A2b receptor, modulates the inflammatory response in KCs in alcoholic steatosis.
The concentration and proportion of nucleotides and ADO in the extracellular space are critical for regulating inflammation in tissue damage conditions, such as steatosis and fibrosis. Ectonucleotidases are critical elements of these mechanisms. CD39 is one of the main enzymes regulating ATP concentration in the extracellular space. Systemic or myeloid cell-specific deletion of this enzyme increased the sensibility of mice to pharmacologically induced biliary fibrosis, as well as the expression levels of the transcripts for inflammation markers (i.e., TGF-β1 and TNF-α) [159]. CD39−/− mice also show greater inflammation and liver damage in response to sepsis, probably due to macrophage activity by deficient extracellular ATP scavenging [93].
5.3. Purinergic Signaling in Liver Sinusoidal Endothelial Cells
Liver sinusoidal endothelial cells (LSECs) are highly specialized vascular cells surrounding the hepatic lobule [160,161]. They have pores, called fenestrae, with diameters that range from 50 to 300 nm. Fenestrae travel along the cytoplasm and form clusters called “sieve plates”. These pores regulate the solute flux towards the hepatic lobule. The fenestration in sieve plates has distinctive patterns according to the zone of the lobule. The periportal fenestrae are large and scarce, the centrilobular ones are small and abundant, and the pericentral LSECs are few and scattered. LSECs are different from other endothelial cells in that they lack a basal membrane and thus allow solutes to move along the space of Disse and into the sinusoidal circulation. LSECs also have high endocytic capacity with specific receptors, functioning as scavengers for several types of collagen, hyaluronan, chondroitin sulfate, oxidized low-density lipoproteins, and immune complexes formed with IgG [162,163,164,165].
Purinergic signaling was first explored in vascular endothelial cells before the entire family of P2 receptors was fully described. Subsequent studies showed that ATP and ADP stimulate the release of prostacyclin (PGI2) and nitric oxide (NO), both of which inhibit platelet aggregation via a mechanism involving Ca2+ mobilization. These studies helped to demonstrate that extracellular nucleotides modulate intravascular platelet aggregation when acting through their receptors [166,167]. In LSECS (Table 3), it was shown that prostaglandin E2 (PGE2) is the main prostanoid secreted by primary cultures in response to extracellular nucleotides. Pharmacological approaches suggested that this response was mediated by P2YR and ADO receptors [168].
Table 3.
Purinergic signaling in liver sinusoidal endothelial cells (LSECs).
CD39 is the most expressed ectonucleotidase in the vascular endothelium, and it has been demonstrated that this enzyme is a regulator of platelet activation [172]. CD39 null mice show alterations in blood coagulation and angiogenesis [173]. In the liver, partial hepatectomy induces a specific increment in the expression level of CD39 in LSECs, which was detected from 6 h to 5 days’ post-surgery. In CD39−/− mice, hepatic regeneration was impaired, and it was observed that throughout the regenerative process, angiogenesis decreased, while hepatic damage increased. LSECs exhibited a decrease in HGF release and an attenuation in the activation of VEGF receptor 2. The latter was attributed to a lack of extracellular nucleotide transactivation [169]. Interestingly, a co-culture of mouse LSECs expressing CD39, but not LSECs from CD39−/− mice, boosted the proliferation of tumor cells (B16/F10 from melanoma) and limited the induction of extracellular ATP-dependent cell death [170].
Activation of the A2a receptor with the agonist CGS21680 protected LSECs from the damage induced by ischemia reperfusion, favoring the expression of genes associated with cytoprotection, regeneration, energy metabolism, and response to oxidative stress [171].
5.4. Purinergic Signaling in Cholangiocytes
Cholangiocytes, or biliary epithelial cells, are specialized cells forming the biliary epithelium and lining the bile ducts. Although they represent less than 5% of the nuclear mass of the liver, they form an extensive branching network and are thought to account for up to 40% of the bile volume in humans [174].
Cholangiocytes are polarized cells with apical and basal membranes that maintain bile flow via the cilium system; they also maintain intraductal homeostasis by active biomolecule transport; modify bile by secreting bicarbonate (HCO3−) through the plasma membrane domain; maintain cross-ductal interaction in the liver, depending on their tight junctions and immunoglobulin A (IgA) secretion; and reabsorb different molecules, including bile salts and acids, glucose, amino acids, and ions [175,176,177]. Cholangiocytes are classified as small or large, according to their morphology [178]. Large cholangiocytes line the larger branches of the biliary tree and form more complex structures than those of small cholangiocytes. In addition, large cholangiocytes engage in hormone-modulated bile secretion, while small cholangiocytes can proliferate and exhibit functional plasticity during diseases [179,180,181]. Small cholangiocytes are capable of self-replication during liver injury, confirming their potential in liver regeneration and ductular reaction [182]. Ductular reaction is a complex of dynamic interactions among liver parenchymal cells, stromal cells, and immune cells, which serves a crucial machinery during liver injury–regeneration, fibrogenesis, and malignant transformation processes [183].
Cholangiocytes have crucial functions in liver pathobiology. They interact with both intra- and extrahepatic ductal environments and are exposed to both hepatic- and gut-derived stimuli, such as pathogen-associated molecular patterns (PAMPS), DAMPs, and microorganisms) [184,185]. As a result, cholangiocytes are collateral targets of various liver disorders, such as NAFLD, NASH, and alcohol-related liver disease. Cholangiocytes are also directly injured in chronic cholestatic liver diseases, including primary biliary cholangitis, primary sclerosing cholangitis, biliary atresia, and cholangiocarcinoma (CCA) [186,187]. Furthermore, stimulated cholangiocytes can adopt varying secretory phenotypes. Some findings suggest that the bile duct-derived ductular cells play active roles in liver regeneration. Interestingly, activated cholangiocytes exhibit a peculiar secretory phenotype that shapes their surrounding microenvironment by modulating immune cell recruitment and mesenchymal cell migration and activation [188]. The release of cholangiokines (cholangiocyte-secreted cytokines, including chemokines, growth factors, and others) is associated with cellular responses, which are affected by tissue inflammation, infection, and metabolic dysregulations. Both acute and chronic liver disorders have been shown to alter the secretory profiles of cholangiocytes [189,190,191].
5.4.1. Nucleotide Signaling in Cholangiocytes
Cholangiocytes exhibit constitutive ATP efflux. In the monolayers of rat cholangiocytes, it was demonstrated that ATP release is polarized with apical concentrations (lumenal) of approximately 250 nM, which are fivefold greater than basolateral concentrations (approximately 50 nM) [192]. These values are within the range required for the half-maximal stimulation of Cl− secretion in biliary epithelium. Accordingly, the ATP in bile is most likely derived from release across the canalicular membrane of hepatocytes and the apical membrane of cholangiocytes. The most potent stimulator of hepatic ATP release is an increase in cell volume. Moreover, exposure to exogenous ATP causes a rapid decrease in cell volume due to K+ and Cl− channel activation. These and other findings suggest that extracellular ATP plays a key role in recovery from cell swelling in different liver and biliary cell models [193,194,195].
Additionally, cholangiocytes express MDR-1 and CFTR ATP-transporting proteins [196]. By modulating the function of these proteins, it is possible to regulate ATP release, and recent studies indicate that these transporters are more likely to serve as ATP channel regulators, increasing the amount of ATP released through an as-yet-unidentified ATP channel [197].
Furthermore, increases in cell volume cause activation of phosphoinositide 3-kinase (PI 3-kinase) [198], which plays a central role in the regulation of hepatocyte and cholangiocyte ATP release [199,200]. PI3-kinase signaling could lead to the insertion of new ATP channels or, alternatively, could directly modulate the opening of pre-existing ATP channels through the generation of secondary messengers.
On the other hand, the degradation of extracellular nucleotides is an essential step in the overall process of purinergic signaling. In vitro studies of polarized biliary epithelia demonstrated substantial differences in the kinetics of ATP degradation between apical and basolateral surfaces. In the apical compartment, the time course of nucleotide degradation can be described by a single exponential, resulting in the clearance of 13% of ATP within the first minute. In the basolateral compartment, the time course was more complex, suggesting the presence of more than one degradation pathway. These differences in degradation kinetics are likely because of location-specific biliary ecto-ATPases [192].
Current research is aiming to fully elucidate the roles of ADO, P2X, and P2Y receptor signaling in hepatic and biliary function. Some functions have been demonstrated in cell volume regulation, paracrine signaling, and bile formation.
The maintenance of cell volume is crucial for liver cells that receive dual perfusion from both the systemic and portal circulations and are exposed to unusually large changes in the concentrations of solutes, such as amino acids, bile acids, and glucose, between the fasted and fed states. Extracellular ATP plays a key role in restoring the cell volume to basal values in a process referred to as regulatory volume decrease [193,195].
In the liver, ATP release appears to represent a paracrine signal that is responsible for the coordination of calcium signaling among cells, suggesting a role in cell-to-cell coupling along the sinusoid. Specifically, the ATP released by one cell binds to P2 receptors on neighboring cells, stimulating a rise in cytosolic calcium [201]. Simultaneously, ATP released from the canalicular hepatocyte membrane or the apical cholangiocyte membrane may significantly contribute to bile formation by stimulating the apical purinergic receptors.
ATP is released into bile by both hepatocytes and cholangiocytes, and functions as a potent autocrine/paracrine stimulus for cholangiocyte secretion [202,203]. When the cell volume increases, the ATP membrane permeability is enhanced by PKC activation, resulting in the elevation of extracellular ATP [204]. Once in the bile, ATP has direct access to the apical cholangiocyte membrane, where it induces increases in intracellular Ca2+ concentration by purinergic receptor activation and activates the membrane Cl− and K+ channels [205]. The resulting transepithelial transport of Cl− significantly contributes to the alkalinization and dilution of the bile [206]. Thus, the description of proteins implicated in cholangiocyte secretion by purines represents an excellent tool for understanding the entire mechanism implicated in the purinergic regulation of bile physiology and pathophysiology.
P2YRs are recognized as the prototype effector pathway responsible for Ca2+-dependent secretory responses [205,206]. ATP binding to these G-protein-coupled receptors stimulates phospholipase C, generates IP3, and releases calcium from intracellular stores. The P2Y2 receptor represents the best characterized member of this family. cDNAs, mRNAs, and protein corresponding to the P2Y2 receptor are readily detectable in cholangiocytes, and the P2Y2 receptor-preferring agonist UTP effectively stimulates cholangiocyte secretion, which is consistent with an important physiological role for this receptor [205,206].
The P2Y12 receptor is also localized in the apical membrane of cholangiocytes [192,205,207]. This receptor appears to be restricted to the cilium, a mechanosensory, osmosensory, and chemosensory organelle that detects and transmits different stimuli from the bile into the cell, modifying cellular functions [208,209]. P2 receptors have also been observed, although less frequently, at the basolateral domain of cholangiocytes [206,210]. By acting on these P2 receptors, ATP triggers an increase in intracellular Ca2+, which results in the activation of K+ and Cl− channels [211].
On the other hand, transcripts for P2XR 2, 3, 4, and 6 have been detected in cultured rat cholangiocytes and cholangiocyte lysates. The P2X4 receptor protein was readily detected, and immunohistochemical staining of intact rat liver revealed that the P2X4 receptor concentrated in intrahepatic bile ducts. The functional significance of the P2X4 receptor was assessed in isolated Mz-ChA-1 cells using the P2X4 receptor-preferring agonist BzATP. Moreover, BzATP elicited robust Cl− secretory responses, demonstrating that the P2X4 receptor is an important component of the purinergic signaling complex that modulates biliary secretion [210] (Table 4).
Table 4.
Purinergic signaling in cholangiocytes.
5.4.2. Adenosine Signaling in Cholangiocytes
The functional expression of ADO receptors and transporters has been characterized in polarized rat cholangiocytes. Studies demonstrated that biliary cells exclusively express the efficient ADO transporters CNT3 (located only in the apical membrane) and CNT2 (located in the apical and basolateral domains). In both domains, cholangiocytes also express the high-affinity ADO receptor A2a, whose activation may modulate the activity of apical CNT3 in a domain-specific manner. The A2a receptor-mediated regulation of CNT3 is dependent upon the cAMP/PKA/ERK/CREB axis, intracellular trafficking mechanisms, and AMPK phosphorylation. Moreover, extracellular ATP (an ADO precursor) is able to exert an inhibitory effect on the apical activity of both CNT3 and CNT2, which seemingly involves different P2 receptors [212].
Purinergic signaling has also been investigated in CCA, a rare malignancy of the epithelial cells from various areas of the biliary tree, including intrahepatic, perihilar, and extrahepatic bile ducts. Studies demonstrated that CCA cell lines and MMNK-1, an immortalized CCA cell line, share P2Y2 and P2X3 receptors. On the other hand, P2Y6, P2Y13, and P2X7 receptors were expressed in CCA cells but not in immortalized CCA cells. MMNK-1 cells showed some resistance to cell proliferation and cell motility inhibition by ATP and ADO. Therefore, it is suggested that the receptors expressed in CCA cells but not in MMNK-1 cells might be responsible for the aforementioned inhibition. In addition, ADO was shown to suppress cell proliferation and motility [213,214]. However, further studies are required in order to examine the mechanism of inhibition caused by ADO on CCA cells.
6. Therapeutic Targeting of Purinergic Signaling in Non-Parenchymal Liver Cells
Taking advantage of the abundant knowledge accumulated regarding the importance of ATP and ADO signaling in diverse tissues, as well as the reported 3D structures of a variety of purinergic receptors, research groups in recent years have postulated the translational use of drugs that interact through elements of purinergic signaling. For example, a number of purinergic drugs are commercially available, including P2Y12 receptor antagonists for stroke and thrombosis, P2Y2 receptor agonists for dry eye, A1 receptor agonists for supraventricular tachycardia, and P2X3 receptor antagonists for the treatment of chronic cough, visceral pain, and hypertension [215,216].
As for liver pathologies, some promising therapeutic advances have been reported [58]. For example, MRS1754, an A2B ADO receptor antagonist, has shown positive actions as an antifibrotic agent [58,217]; A438079, a P2X7R antagonist, promoted a reduction in inflammation and collagen accumulation [65]; 5-BDBD, a P2XR antagonist, alleviated liver fibrosis in an experimental model of methionine-choline deficient diet [78]; NF340, a P2Y11R antagonist, counteracted HCC cells [78,218]; CF102, an A3AR agonist, reduced HCC cell availability [219].
Among these studies, one well-characterized effect is the targeting of A3 receptors to address non-alcoholic fatty liver disease/non-alcoholic steatohepatitis (NAFLD/NASH). The expression of the A3 receptor in NAFLD patients is significantly lower than in healthy persons; in addition, a high fat diet in mice lacking A3-receptor expression results in an enhanced expression of genes involved in hepatic inflammation and steatosis [220]. Moreover, the administration of the A3-receptor agonist prodrug MRS7476 protects against NASH development in STAM mice [221]. Another A3-receptor agonist, C1-IBMECA (namodenoson), was tested in the treatment of NASH in mice. Namodenoson is currently in a Phase 2 clinical trial for NASH therapeutics (ClinicalTrials.gov Identifiers: NCT02927314 and NCT04697810, accessed on 31 May 2021) [88]. Moreover, another A3 agonist, named CF102, was administered to patients with advanced HCC in a phase I/II trial. It was observed that this drug is safe, has a linear pharmacokinetics, and reverses the metastatic process, demonstrating clinical applicability [222].
Another case is IFC305, an analog of adenosine, which is able to reverse fibrosis in pre-established rat cirrhosis [223]. Mechanistic studies demonstrated that IFC305 prevents HSC activation [129] and modulates the inflammatory response dependent on Kupffer cells [224]. Clinical trials and pharmaceutical developments are currently in development [225].
Finally, the scarcity of reports regarding the purinergic system in well-recognized experimental models of hepatic disorders is notable. For example, data in Pubmed (August, 2024) did not show any record of purinergic signaling and liver injury in relation to hemochromatosis, Wilson disease, and herbal-induced hepatitis, whereas from among 47,993 citations for drug-induced liver injury, only 18 (0.037%) were related to elements of ATP/ADO signal transduction. Indeed, this presents an opportunity to encourage basic and clinical groups to continue the biomedical research on liver diseases and cellular purinergic communication in the near future.
An interesting parallelism exists between the liver and kidneys from the perspective of purinergic physiopathology. Both organs are highly dependent on nutrient processing, they are formed by the coordinated function of different cellular components, and the hepatic and renal responses to ATP are proinflammatory, whereas ADO acts as an anti-inflammatory factor [226]. In this context, it has been recognized that the presence of P2X signaling cascades play a role in acute kidney injury [226,227], and that the dysregulation of the P2 receptor in urologic diseases, as well as the participation of purinergic receptors in urinary infections by bacteria [228] and kidney transplantation, also play a role [229].
7. Concluding Remarks
The liver is a complex system. It is formed by a variety of cellular types that harmonize their operation to achieve the dynamic adaptation needed for a healthy hepatic physiology. In this context, efficient communication and coordination between the parenchymal and non-parenchymal cells is imperative to ensure a correct metabolic and protective hepatic response. Among the multiple cellular messengers that exist within the liver, this review focused on how hepatic ATP and ADO signaling during fibrotic, cirrhotic, and cancerous damage affects HSCs, KCs, biliary cells, and LSECs.
Indeed, this approach is necessary to complement the literature on purinergic function in hepatocytes and ultimately achieve an integrated overview of the liver in both healthy and pathological conditions.
Funding
This research was funded by Dirección General de Asuntos del Personal Académico (DGAPA-UNAM-México), #IN205223, to FGVC and #IN207224 to MDM and by the Consejo Nacional de Humanidades, Ciencia y Tecnología (CONAHCYT-México), #CBF2023-2024-200, to FGVC. Esperanza Mata Martínez is a postdoctoral fellow funded by CONAHCYT-México 2023(1); María Guadalupe Ramírez Ledesma is a postdoctoral fellow funded by CONAHCYT-México 2023(1) and the APC was partially funded by Instituto de Neurobiología-UNAM.
Acknowledgments
We are grateful to Jessica Gonzalez Norris for proofreading the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Taub, R. Liver Regeneration: From Myth to Mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R.; Matz-Soja, M. Liver Zonation: Novel Aspects of Its Regulation and Its Impact on Homeostasis. World J. Gastroenterol. 2014, 20, 8491–8504. [Google Scholar] [CrossRef]
- Jungermann, K.; Katz, N. Functional Specialization of Different Hepatocyte Populations. Physiol. Rev. 1989, 69, 708–764. [Google Scholar] [CrossRef] [PubMed]
- Ghafoory, S.; Stengl, C.; Kopany, S.; Mayadag, M.; Mechtel, N.; Murphy, B.; Schattschneider, S.; Wilhelmi, N.; Wölfl, S. Oxygen Gradient Induced in Microfluidic Chips Can Be Used as a Model for Liver Zonation. Cells 2022, 11, 3734. [Google Scholar] [CrossRef]
- Ma, R.; Martínez-Ramírez, A.S.; Borders, T.L.; Gao, F.; Sosa-Pineda, B. Metabolic and Non-Metabolic Liver Zonation Is Established Non-Synchronously and Requires Sinusoidal Wnts. eLife 2020, 9, e46206. [Google Scholar] [CrossRef]
- Michalopoulos, G.K.; Bhushan, B. Liver Regeneration: Biological and Pathological Mechanisms and Implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Yagi, S.; Hirata, M.; Miyachi, Y.; Uemoto, S. Liver Regeneration after Hepatectomy and Partial Liver Transplantation. Int. J. Mol. Sci. 2020, 21, 8414. [Google Scholar] [CrossRef]
- Shaker, M.E. The Contribution of Sterile Inflammation to the Fatty Liver Disease and the Potential Therapies. Biomed. Pharmacother. 2022, 148, 112789. [Google Scholar] [CrossRef]
- Yuen, M.-F.; Chen, D.-S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.-L. Hepatitis B Virus Infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef]
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef]
- Gong, J.; Tu, W.; Liu, J.; Tian, D. Hepatocytes: A Key Role in Liver Inflammation. Front. Immunol. 2022, 13, 1083780. [Google Scholar] [CrossRef]
- Yang Zhou, J. Innate Immunity and Early Liver Inflammation. Front. Immunol. 2023, 14, 1175147. [Google Scholar] [CrossRef]
- Tiegs, G.; Horst, A.K. TNF in the Liver: Targeting a Central Player in Inflammation. Semin. Immunopathol. 2022, 44, 445–459. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver Inflammation and Fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Ju, D. Inflammasome: A Double-Edged Sword in Liver Diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef]
- Yu, C.; Chen, P.; Miao, L.; Di, G. The Role of the NLRP3 Inflammasome and Programmed Cell Death in Acute Liver Injury. Int. J. Mol. Sci. 2023, 24, 3067. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Ferhat, M.; Salamé, E.; Robin, A.; Herbelin, A.; Gombert, J.-M.; Silvain, C.; Barbarin, A. Interleukin-1 Family Cytokines: Keystones in Liver Inflammatory Diseases. Front. Immunol. 2019, 10, 2014. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, B.; Leszczynska, A.; Reca, A.; Booshehri, L.M.; Onyuru, J.; Tan, Z.; Wree, A.; Friess, H.; Hartmann, D.; Papouchado, B.; et al. NLRP3 Activation in Neutrophils Induces Lethal Autoinflammation, Liver Inflammation, and Fibrosis. EMBO Rep. 2022, 23, e54446. [Google Scholar] [CrossRef] [PubMed]
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular Matrix Molecular Remodeling in Human Liver Fibrosis Evolution. PLoS ONE 2016, 11, e0151736. [Google Scholar] [CrossRef]
- Duarte, S.; Baber, J.; Fujii, T.; Coito, A.J. Matrix Metalloproteinases in Liver Injury, Repair and Fibrosis. Matrix Biol. 2015, 44–46, 147–156. [Google Scholar] [CrossRef]
- Elpek, G.Ö. Cellular and Molecular Mechanisms in the Pathogenesis of Liver Fibrosis: An Update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and Cellular Mechanisms of Liver Fibrosis and Its Regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Massey, V.L.; Dolin, C.E.; Poole, L.G.; Hudson, S.V.; Siow, D.L.; Brock, G.N.; Merchant, M.L.; Wilkey, D.W.; Arteel, G.E. The Hepatic “Matrisome” Responds Dynamically to Injury: Characterization of Transitional Changes to the Extracellular Matrix in Mice. Hepatology 2017, 65, 969–982. [Google Scholar] [CrossRef]
- Zhang, M.; Serna-Salas, S.; Damba, T.; Borghesan, M.; Demaria, M.; Moshage, H. Hepatic Stellate Cell Senescence in Liver Fibrosis: Characteristics, Mechanisms and Perspectives. Mech. Ageing Dev. 2021, 199, 111572. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]
- Fallowfield, J.A.; Mizuno, M.; Kendall, T.J.; Constandinou, C.M.; Benyon, R.C.; Duffield, J.S.; Iredale, J.P. Scar-Associated Macrophages Are a Major Source of Hepatic Matrix Metalloproteinase-13 and Facilitate the Resolution of Murine Hepatic Fibrosis. J. Immunol. 2007, 178, 5288–5295. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts Revert to an Inactive Phenotype during Regression of Liver Fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Kweon, Y.O.; Frederick, J.P.; Wang, X.F.; Rippe, R.A.; Brenner, D.A. The Role of Smad3 in Mediating Mouse Hepatic Stellate Cell Activation. Hepatology 2001, 34, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global Burden of Liver Disease: 2023 Update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Liu, Y.; Zhong, J.; Wang, J.; Sun, L.; Yu, L.; Wang, Y.; Li, Q.; Jin, W.; et al. Remodeling Liver Microenvironment by L-Arginine Loaded Hollow Polydopamine Nanoparticles for Liver Cirrhosis Treatment. Biomaterials 2023, 295, 122028. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver Fibrosis and Repair: Immune Regulation of Wound Healing in a Solid Organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Ginès, P.; Krag, A.; Abraldes, J.G.; Solà, E.; Fabrellas, N.; Kamath, P.S. Liver Cirrhosis. Lancet 2021, 398, 1359–1376. [Google Scholar] [CrossRef]
- Trebicka, J.; Macnaughtan, J.; Schnabl, B.; Shawcross, D.L.; Bajaj, J.S. The Microbiota in Cirrhosis and Its Role in Hepatic Decompensation. J. Hepatol. 2021, 75 (Suppl. S1), S67–S81. [Google Scholar] [CrossRef]
- Hasa, E.; Hartmann, P.; Schnabl, B. Liver Cirrhosis and Immune Dysfunction. Int. Immunol. 2022, 34, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Osawa, Y.; Kimura, K. Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. Int. J. Mol. Sci. 2018, 19, 3103. [Google Scholar] [CrossRef]
- Hartke, J.; Johnson, M.; Ghabril, M. The Diagnosis and Treatment of Hepatocellular Carcinoma. Semin. Diagn. Pathol. 2017, 34, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Zajkowska, M.; Mroczko, B. Chemokines in Primary Liver Cancer. Int. J. Mol. Sci. 2022, 23, 8846. [Google Scholar] [CrossRef]
- Plentz, R.R.; Schlegelberger, B.; Flemming, P.; Gebel, M.; Kreipe, H.; Manns, M.P.; Rudolph, K.L.; Wilkens, L. Telomere Shortening Correlates with Increasing Aneuploidy of Chromosome 8 in Human Hepatocellular Carcinoma. Hepatology 2005, 42, 522–526. [Google Scholar] [CrossRef]
- Marrogi, A.J.; Khan, M.A.; van Gijssel, H.E.; Welsh, J.A.; Rahim, H.; Demetris, A.J.; Kowdley, K.V.; Hussain, S.P.; Nair, J.; Bartsch, H.; et al. Oxidative Stress and p53 Mutations in the Carcinogenesis of Iron Overload-Associated Hepatocellular Carcinoma. J. Natl. Cancer Inst. 2001, 93, 1652–1655. [Google Scholar] [CrossRef]
- Díaz-Muñoz, M.; Salín-Pascual, R. Purine Molecules as Hypnogenic Factors Role of Adenosine, ATP, and Caffeine. Cent. Nerv. Syst. Agents Med. Chem. 2010, 10, 259–268. [Google Scholar] [CrossRef]
- Vultaggio-Poma, V.; Falzoni, S.; Salvi, G.; Giuliani, A.L.; Di Virgilio, F. Signalling by Extracellular Nucleotides in Health and Disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2022, 1869, 119237. [Google Scholar] [CrossRef] [PubMed]
- Camici, M.; Garcia-Gil, M.; Tozzi, M.G. The Inside Story of Adenosine. Int. J. Mol. Sci. 2018, 19, 784. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. History of Ectonucleotidases and Their Role in Purinergic Signaling. Biochem. Pharmacol. 2021, 187, 114322. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Carlón, N.; Salgado-García, R.L.; Guerrero-Tortolero, D.A.; Kraffe, E.; Campos-Ramos, R.; Racotta, I.S. Biochemical Composition and Adenylate Energy Charge Shifts in Longfin Yellowtail (Seriola rivoliana) Embryos during Development under Different Temperatures. J. Therm. Biol. 2023, 112, 103470. [Google Scholar] [CrossRef]
- Dale, N. Biological Insights from the Direct Measurement of Purine Release. Biochem. Pharmacol. 2021, 187, 114416. [Google Scholar] [CrossRef]
- O’Grady, S.M.; Kita, H. ATP Functions as a Primary Alarmin in Allergen-Induced Type 2 Immunity. Am. J. Physiol. Cell Physiol. 2023, 325, C1369–C1386. [Google Scholar] [CrossRef]
- Alvarez, C.L.; Troncoso, M.F.; Espelt, M.V. Extracellular ATP and Adenosine in Tumor Microenvironment: Roles in Epithelial-Mesenchymal Transition, Cell Migration, and Invasion. J. Cell. Physiol. 2022, 237, 389–400. [Google Scholar] [CrossRef]
- Mohlin, C.; Säve, S.; Nilsson, M.; Persson, K. Studies of the Extracellular ATP-Adenosine Pathway in Human Urinary Tract Epithelial Cells. Pharmacology 2009, 84, 196–202. [Google Scholar] [CrossRef]
- Moreno, E.; Canet, J.; Gracia, E.; Lluís, C.; Mallol, J.; Canela, E.I.; Cortés, A.; Casadó, V. Molecular Evidence of Adenosine Deaminase Linking Adenosine A Receptor and CD26 Proteins. Front. Pharmacol. 2018, 9, 106. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Burnstock, G. Purinergic and Glutamatergic Receptors on Astroglia. Adv. Neurobiol. 2014, 11, 55–79. [Google Scholar]
- Ase, A.R.; Therrien, É.; Séguéla, P. An Allosteric Inhibitory Site Conserved in the Ectodomain of P2X Receptor Channels. Front. Cell. Neurosci. 2019, 13, 121. [Google Scholar] [CrossRef] [PubMed]
- Von Kügelgen, I. Pharmacological Characterization of P2Y Receptor Subtypes—An Update. Purinergic Signal. 2024, 20, 99–108. [Google Scholar] [CrossRef]
- Effendi, W.I.; Nagano, T.; Kobayashi, K.; Nishimura, Y. Focusing on Adenosine Receptors as a Potential Targeted Therapy in Human Diseases. Cells 2020, 9, 785. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Reklow, R.J.; Alvares, T.S.; Zhang, Y.; Miranda Tapia, A.P.; Biancardi, V.; Katzell, A.K.; Frangos, S.M.; Hansen, M.A.; Toohey, A.W.; Cass, C.E.; et al. The Purinome and the preBötzinger Complex—A Ménage of Unexplored Mechanisms That May Modulate/Shape the Hypoxic Ventilatory Response. Front. Cell. Neurosci. 2019, 13, 365. [Google Scholar] [CrossRef]
- Vaughn, B.P.; Robson, S.C.; Longhi, M.S. Purinergic Signaling in Liver Disease. Dig. Dis. 2014, 32, 516–524. [Google Scholar] [CrossRef]
- Velázquez-Miranda, E.; Díaz-Muñoz, M.; Vázquez-Cuevas, F.G. Purinergic Signaling in Hepatic Disease. Purinergic Signal. 2019, 15, 477–489. [Google Scholar] [CrossRef]
- Jain, S.; Jacobson, K.A. Purinergic Signaling in Liver Pathophysiology. Front. Endocrinol. 2021, 12, 718429. [Google Scholar] [CrossRef]
- Tackett, B.C.; Sun, H.; Mei, Y.; Maynard, J.P.; Cheruvu, S.; Mani, A.; Hernandez-Garcia, A.; Vigneswaran, N.; Karpen, S.J.; Thevananther, S. P2Y2 Purinergic Receptor Activation Is Essential for Efficient Hepatocyte Proliferation in Response to Partial Hepatectomy. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G1073–G1087. [Google Scholar] [CrossRef] [PubMed]
- Besnard, A.; Gautherot, J.; Julien, B.; Tebbi, A.; Garcin, I.; Doignon, I.; Péan, N.; Gonzales, E.; Cassio, D.; Grosse, B.; et al. The P2X4 Purinergic Receptor Impacts Liver Regeneration after Partial Hepatectomy in Mice through the Regulation of Biliary Homeostasis. Hepatology 2016, 64, 941–953. [Google Scholar] [CrossRef]
- Gonzales, E.; Julien, B.; Serrière-Lanneau, V.; Nicou, A.; Doignon, I.; Lagoudakis, L.; Garcin, I.; Azoulay, D.; Duclos-Vallée, J.-C.; Castaing, D.; et al. ATP Release after Partial Hepatectomy Regulates Liver Regeneration in the Rat. J. Hepatol. 2010, 52, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Ohana, G.; Cohen, S.; Rath-Wolfson, L.; Fishman, P. A3 Adenosine Receptor Agonist, CF102, Protects against Hepatic Ischemia/reperfusion Injury Following Partial Hepatectomy. Mol. Med. Rep. 2016, 14, 4335–4341. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Imai, M.; Nowak-Machen, M.; Guckelberger, O.; Enjyoji, K.; Wu, Y.; Khalpey, Z.; Berberat, P.; Munasinghe, J.; Robson, S.C. Liver Damage and Systemic Inflammatory Responses Are Exacerbated by the Genetic Deletion of CD39 in Total Hepatic Ischemia. Purinergic Signal. 2011, 7, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, J.A.; Kruglov, E.A.; Abreu-Lanfranco, O.; Nguyen, T.; Arora, G.; Jain, D. Prevention of Liver Fibrosis by the Purinoceptor Antagonist Pyridoxal-Phosphate-6-Azophenyl-2′,4′-Disulfonate (PPADS). In Vivo 2007, 21, 957–965. [Google Scholar] [PubMed]
- Huang, C.; Yu, W.; Cui, H.; Wang, Y.; Zhang, L.; Han, F.; Huang, T. P2X7 Blockade Attenuates Mouse Liver Fibrosis. Mol. Med. Rep. 2014, 9, 57–62. [Google Scholar] [CrossRef]
- Jiang, M.; Cui, B.-W.; Wu, Y.-L.; Zhang, Y.; Shang, Y.; Liu, J.; Yang, H.-X.; Qiao, C.-Y.; Zhan, Z.-Y.; Ye, H.; et al. P2X7R Orchestrates the Progression of Murine Hepatic Fibrosis by Making a Feedback Loop from Macrophage to Hepatic Stellate Cells. Toxicol. Lett. 2020, 333, 22–32. [Google Scholar] [CrossRef]
- Velázquez-Miranda, E.; Molina-Aguilar, C.; González-Gallardo, A.; Vázquez-Martínez, O.; Díaz-Muñoz, M.; Vázquez-Cuevas, F.G. Increased Purinergic Responses Dependent on P2Y2 Receptors in Hepatocytes from CCl4-Treated Fibrotic Mice. Int. J. Mol. Sci. 2020, 21, 2305. [Google Scholar] [CrossRef]
- Chan, E.S.L.; Montesinos, M.C.; Fernandez, P.; Desai, A.; Delano, D.L.; Yee, H.; Reiss, A.B.; Pillinger, M.H.; Chen, J.-F.; Schwarzschild, M.A.; et al. Adenosine A2A Receptors Play a Role in the Pathogenesis of Hepatic Cirrhosis. Br. J. Pharmacol. 2006, 148, 1144–1155. [Google Scholar] [CrossRef]
- Yang, P.; Chen, P.; Wang, T.; Zhan, Y.; Zhou, M.; Xia, L.; Cheng, R.; Guo, Y.; Zhu, L.; Zhang, J. Loss of A1 Adenosine Receptor Attenuates Alpha-Naphthylisothiocyanate-Induced Cholestatic Liver Injury in Mice. Toxicol. Sci. 2013, 131, 128–138. [Google Scholar] [CrossRef]
- Yang, P.; Han, Z.; Chen, P.; Zhu, L.; Wang, S.; Hua, Z.; Zhang, J. A Contradictory Role of A1 Adenosine Receptor in Carbon Tetrachloride- and Bile Duct Ligation-Induced Liver Fibrosis in Mice. J. Pharmacol. Exp. Ther. 2010, 332, 747–754. [Google Scholar] [CrossRef]
- Chatterjee, S.; Rana, R.; Corbett, J.; Kadiiska, M.B.; Goldstein, J.; Mason, R.P. P2X7 Receptor-NADPH Oxidase Axis Mediates Protein Radical Formation and Kupffer Cell Activation in Carbon Tetrachloride-Mediated Steatohepatitis in Obese Mice. Free Radic. Biol. Med. 2012, 52, 1666–1679. [Google Scholar] [CrossRef]
- Feldbrügge, L.; Jiang, Z.G.; Csizmadia, E.; Mitsuhashi, S.; Tran, S.; Yee, E.U.; Rothweiler, S.; Vaid, K.A.; Sévigny, J.; Schmelzle, M.; et al. Distinct Roles of Ecto-Nucleoside Triphosphate Diphosphohydrolase-2 (NTPDase2) in Liver Regeneration and Fibrosis. Purinergic Signal. 2018, 14, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Amaral, S.S.; Oliveira, A.G.; Marques, P.E.; Quintão, J.L.D.; Pires, D.A.; Resende, R.R.; Sousa, B.R.; Melgaço, J.G.; Pinto, M.A.; Russo, R.C.; et al. Altered Responsiveness to Extracellular ATP Enhances Acetaminophen Hepatotoxicity. Cell Commun. Signal. 2013, 11, 10. [Google Scholar] [CrossRef]
- Abdelaziz, H.A.; Shaker, M.E.; Hamed, M.F.; Gameil, N.M. Repression of Acetaminophen-Induced Hepatotoxicity by a Combination of Celastrol and Brilliant Blue G. Toxicol. Lett. 2017, 275, 6–18. [Google Scholar] [CrossRef]
- Xie, Y.; Williams, C.D.; McGill, M.R.; Lebofsky, M.; Ramachandran, A.; Jaeschke, H. Purinergic Receptor Antagonist A438079 Protects against Acetaminophen-Induced Liver Injury by Inhibiting p450 Isoenzymes, Not by Inflammasome Activation. Toxicol. Sci. 2013, 131, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Hoque, R.; Sohail, M.A.; Salhanick, S.; Malik, A.F.; Ghani, A.; Robson, S.C.; Mehal, W.Z. P2X7 Receptor-Mediated Purinergic Signaling Promotes Liver Injury in Acetaminophen Hepatotoxicity in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1171–G1179. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C.K.; Ganal, S.C.; Hockenjos, B.; Willim, K.; Vieira, R.P.; Grimm, M.; Robaye, B.; Boeynaems, J.M.; Di Virgilio, F.; Pellegatti, P.; et al. Purinergic P2Y2 Receptors Promote Neutrophil Infiltration and Hepatocyte Death in Mice with Acute Liver Injury. Gastroenterology 2012, 143, 1620–1629.e4. [Google Scholar] [CrossRef] [PubMed]
- Le Guilcher, C.; Garcin, I.; Dellis, O.; Cauchois, F.; Tebbi, A.; Doignon, I.; Guettier, C.; Julien, B.; Tordjmann, T. The P2X4 Purinergic Receptor Regulates Hepatic Myofibroblast Activation during Liver Fibrogenesis. J. Hepatol. 2018, 69, 644–653. [Google Scholar] [CrossRef]
- Imarisio, C.; Alchera, E.; Sutti, S.; Valente, G.; Boccafoschi, F.; Albano, E.; Carini, R. Adenosine A2a Receptor Stimulation Prevents Hepatocyte Lipotoxicity and Non-Alcoholic Steatohepatitis (NASH) in Rats. Clin. Sci. 2012, 123, 323–332. [Google Scholar] [CrossRef]
- Das, S.; Seth, R.K.; Kumar, A.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Purinergic Receptor X7 Is a Key Modulator of Metabolic Oxidative Stress-Mediated Autophagy and Inflammation in Experimental Nonalcoholic Steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G950–G963. [Google Scholar] [CrossRef]
- Blasetti Fantauzzi, C.; Menini, S.; Iacobini, C.; Rossi, C.; Santini, E.; Solini, A.; Pugliese, G. Deficiency of the Purinergic Receptor 2X Attenuates Nonalcoholic Steatohepatitis Induced by High-Fat Diet: Possible Role of the NLRP3 Inflammasome. Oxid. Med. Cell. Longev. 2017, 2017, 8962458. [Google Scholar] [CrossRef] [PubMed]
- Baeza-Raja, B.; Goodyear, A.; Liu, X.; Lam, K.; Yamamoto, L.; Li, Y.; Dodson, G.S.; Takeuchi, T.; Kisseleva, T.; Brenner, D.A.; et al. Pharmacological Inhibition of P2RX7 Ameliorates Liver Injury by Reducing Inflammation and Fibrosis. PLoS ONE 2020, 15, e0234038. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-Mediated Dysbiosis Regulates Progression of NAFLD and Obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Dong, Z.; Wei, Y.; Tao, M.; Zhang, L. Activation of the Purinergic Receptor P2X7 Improves Hepatosteatosis by Promoting Lipophagy. FEBS Lett. 2021, 595, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Dusabimana, T.; Park, E.J.; Je, J.; Jeong, K.; Yun, S.P.; Kim, H.J.; Kim, H.; Park, S.W. P2Y2R Deficiency Ameliorates Hepatic Steatosis by Reducing Lipogenesis and Enhancing Fatty Acid β-Oxidation through AMPK and PGC-1α Induction in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2021, 22, 5528. [Google Scholar] [CrossRef]
- Alchera, E.; Rolla, S.; Imarisio, C.; Bardina, V.; Valente, G.; Novelli, F.; Carini, R. Adenosine A2a Receptor Stimulation Blocks Development of Nonalcoholic Steatohepatitis in Mice by Multilevel Inhibition of Signals That Cause Immunolipotoxicity. Transl. Res. 2017, 182, 75–87. [Google Scholar] [CrossRef]
- Cai, Y.; Li, H.; Liu, M.; Pei, Y.; Zheng, J.; Zhou, J.; Luo, X.; Huang, W.; Ma, L.; Yang, Q.; et al. Disruption of Adenosine 2A Receptor Exacerbates NAFLD through Increasing Inflammatory Responses and SREBP1c Activity. Hepatology 2018, 68, 48–61. [Google Scholar] [CrossRef]
- Fishman, P.; Cohen, S.; Itzhak, I.; Amer, J.; Salhab, A.; Barer, F.; Safadi, R. The A3 Adenosine Receptor Agonist, Namodenoson, Ameliorates Non-alcoholic Steatohepatitis in Mice. Int. J. Mol. Med. 2019, 44, 2256–2264. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.X.; Sheng, X.D.; Wang, Y.L.; Wen Lv, X. Blocking P2X4 Purinergic Receptor Attenuates Alcohol-Related Liver Fibrosis by Inhibiting Hepatic Stellate Cell Activation through PI3K/AKT Signaling Pathway. Int. Immunopharmacol. 2022, 113, 109326. [Google Scholar] [CrossRef]
- Xia, G.-Q.; Cai, J.-N.; Wu, X.; Fang, Q.; Zhao, N.; Lv, X.-W. The Mechanism by Which ATP Regulates Alcoholic Steatohepatitis through P2X4 and CD39. Eur. J. Pharmacol. 2022, 916, 174729. [Google Scholar] [CrossRef]
- Yang, P.; Wang, Z.; Zhan, Y.; Wang, T.; Zhou, M.; Xia, L.; Yang, X.; Zhang, J. Endogenous A1 Adenosine Receptor Protects Mice from Acute Ethanol-Induced Hepatotoxicity. Toxicology 2013, 309, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.-W.; Rothweiler, S.; Wei, G.; Ikenaga, N.; Liu, S.B.; Sverdlov, D.Y.; Vaid, K.A.; Longhi, M.S.; Kuang, M.; Robson, S.C.; et al. The Ectonucleotidase ENTPD1/CD39 Limits Biliary Injury and Fibrosis in Mouse Models of Sclerosing Cholangitis. Hepatol. Commun. 2017, 1, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; de Andrade Mello, P.; Figliuolo, V.R.; de Avelar Almeida, T.F.; Santana, P.T.; Oliveira, S.D.S.; Silva, C.L.M.; Feldbrügge, L.; Csizmadia, E.; Minshall, R.D.; et al. CD39 Limits P2X7 Receptor Inflammatory Signaling and Attenuates Sepsis-Induced Liver Injury. J. Hepatol. 2017, 67, 716–726. [Google Scholar] [CrossRef]
- Ni, J.; Zhang, Z.; Luo, X.; Xiao, L.; Wang, N. Plasticizer DBP Activates NLRP3 Inflammasome through the P2X7 Receptor in HepG2 and L02 Cells. J. Biochem. Mol. Toxicol. 2016, 30, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Schulien, I.; Hockenjos, B.; van Marck, V.; Ayata, C.K.; Follo, M.; Thimme, R.; Hasselblatt, P. Extracellular ATP and Purinergic P2Y Receptor Signaling Promote Liver Tumorigenesis in Mice by Exacerbating DNA Damage. Cancer Res. 2020, 80, 699–708. [Google Scholar] [CrossRef]
- Fishman, P.; Stemmer, S.M.; Bareket-Samish, A.; Silverman, M.H.; Kerns, W.D. Targeting the A3 Adenosine Receptor to Treat Hepatocellular Carcinoma: Anti-Cancer and Hepatoprotective Effects. Purinergic Signal. 2023, 19, 513–522. [Google Scholar] [CrossRef]
- Kamm, D.R.; McCommis, K.S. Hepatic Stellate Cells in Physiology and Pathology. J. Physiol. 2022, 600, 1825–1837. [Google Scholar] [CrossRef]
- Giampieri, M.P.; Jezequel, A.M.; Orlandi, F. The Lipocytes in Normal Human Liver. A Quantitative Study. Digestion 1981, 22, 165–169. [Google Scholar] [CrossRef]
- Andrews, T.S.; Atif, J.; Liu, J.C.; Perciani, C.T.; Ma, X.-Z.; Thoeni, C.; Slyper, M.; Eraslan, G.; Segerstolpe, A.; Manuel, J.; et al. Single-Cell, Single-Nucleus, and Spatial RNA Sequencing of the Human Liver Identifies Cholangiocyte and Mesenchymal Heterogeneity. Hepatol. Commun. 2022, 6, 821–840. [Google Scholar] [CrossRef]
- Diamanti, K.; Inda Díaz, J.S.; Raine, A.; Pan, G.; Wadelius, C.; Cavalli, M. Single Nucleus Transcriptomics Data Integration Recapitulates the Major Cell Types in Human Liver. Hepatol. Res. 2021, 51, 233–238. [Google Scholar] [CrossRef]
- Payen, V.L.; Lavergne, A.; Alevra Sarika, N.; Colonval, M.; Karim, L.; Deckers, M.; Najimi, M.; Coppieters, W.; Charloteaux, B.; Sokal, E.M.; et al. Single-Cell RNA Sequencing of Human Liver Reveals Hepatic Stellate Cell Heterogeneity. JHEP Rep. 2021, 3, 100278. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate Tracing Reveals Hepatic Stellate Cells as Dominant Contributors to Liver Fibrosis Independent of Its Aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Xiong, S.; Hazra, S.; Tsukamoto, H. Adipogenic Transcriptional Regulation of Hepatic Stellate Cells. J. Biol. Chem. 2005, 280, 4959–4967. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H. Adipogenic Phenotype of Hepatic Stellate Cells. Alcohol. Clin. Exp. Res. 2005, 29, 132S–133S. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Wu, L.; Zhang, W.; Ma, W.-T.; Yang, G.-Y.; Zhang, J.; Xue, D.-Y.; Chen, B.; Liu, C. Peroxisome Proliferator-Activated Receptor γ Inhibits Hepatic Stellate Cell Activation Regulated by miR-942 in Chronic Hepatitis B Liver Fibrosis. Life Sci. 2020, 253, 117572. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Wang, S.-S.; Tang, X.T.; Lin, M.; Yuan, J.; Peng, Y.J.; Yin, X.; Shang, G.; Ge, G.; Ren, Z.; Zhou, B.O. Perivenous Stellate Cells Are the Main Source of Myofibroblasts and Cancer-Associated Fibroblasts Formed After Chronic Liver Injuries. Hepatology 2021, 74, 1578–1594. [Google Scholar] [CrossRef]
- Yang, W.; He, H.; Wang, T.; Su, N.; Zhang, F.; Jiang, K.; Zhu, J.; Zhang, C.; Niu, K.; Wang, L.; et al. Single-Cell Transcriptomic Analysis Reveals a Hepatic Stellate Cell-Activation Roadmap and Myofibroblast Origin During Liver Fibrosis in Mice. Hepatology 2021, 74, 2774–2790. [Google Scholar] [CrossRef]
- Takemura, S.; Kawada, N.; Hirohashi, K.; Kinoshita, H.; Inoue, M. Nucleotide Receptors in Hepatic Stellate Cells of the Rat. FEBS Lett. 1994, 354, 53–56. [Google Scholar] [CrossRef]
- Dranoff, J.A.; Ogawa, M.; Kruglov, E.A.; Gaça, M.D.A.; Sévigny, J.; Robson, S.C.; Wells, R.G. Expression of P2Y Nucleotide Receptors and Ectonucleotidases in Quiescent and Activated Rat Hepatic Stellate Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G417–G424. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, Y.; Wang, S.; Xu, R.; Lv, X. Purinergic P2X7 Receptor Mediates Acetaldehyde-Induced Hepatic Stellate Cells Activation via PKC-Dependent GSK3β Pathway. Int. Immunopharmacol. 2017, 43, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Filliol, A.; Affo, S.; Nair, A.; Hernandez, C.; Sun, Q.; Hamberger, F.; Brundu, F.; Chen, Y.; Ravichandra, A.; et al. The Purinergic P2Y14 Receptor Links Hepatocyte Death to Hepatic Stellate Cell Activation and Fibrogenesis in the Liver. Sci. Transl. Med. 2022, 14, eabe5795. [Google Scholar] [CrossRef]
- Shuai, C.; Xia, G.-Q.; Yuan, F.; Wang, S.; Lv, X.-W. CD39-Mediated ATP-Adenosine Signalling Promotes Hepatic Stellate Cell Activation and Alcoholic Liver Disease. Eur. J. Pharmacol. 2021, 905, 174198. [Google Scholar] [CrossRef]
- Andrade, C.M.B.; Wink, M.R.; Margis, R.; Borojevic, R.; Battastini, A.M.O.; Guma, F.C.R. Activity and Expression of Ecto-Nucleotide Pyrophosphate/phosphodiesterases in a Hepatic Stellate Cell Line. Mol. Cell. Biochem. 2009, 325, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Fernandez, P.; Wilder, T.; Yee, H.; Chiriboga, L.; Chan, E.S.L.; Cronstein, B.N. Ecto-5′-Nucleotidase (CD73)-Mediated Extracellular Adenosine Production Plays a Critical Role in Hepatic Fibrosis. Nucleosides Nucleotides Nucleic Acids 2008, 27, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Fausther, M.; Sheung, N.; Saiman, Y.; Bansal, M.B.; Dranoff, J.A. Activated Hepatic Stellate Cells Upregulate Transcription of Ecto-5′-nucleotidase/CD73 via Specific SP1 and SMAD Promoter Elements. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G904–G914. [Google Scholar] [CrossRef]
- Jia, W.-Q.; Zhou, T.-C.; Dai, J.-W.; Liu, Z.-N.; Zhang, Y.-F.; Zang, D.-D.; Lv, X.-W. CD73 Regulates Hepatic Stellate Cells Activation and Proliferation through Wnt/β-Catenin Signaling Pathway. Eur. J. Pharmacol. 2021, 890, 173667. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, B.; Liu, X.; Wu, X.; Du, J.; Xia, G.; Cai, J.; Zhu, H.; Sheng, X.; Zhang, M.; et al. CD73/NT5E-Mediated Ubiquitination of AURKA Regulates Alcohol-Related Liver Fibrosis via Modulating Hepatic Stellate Cell Senescence. Int. J. Biol. Sci. 2023, 19, 950–966. [Google Scholar] [CrossRef]
- Yang, L.; Gao, Z.-W.; Wu, X.-N.; Liu, C.; Zhang, J.; Zhang, H.-Z.; Dong, K. CD73 Blockade Alleviated Hepatic Fibrosis via Inhibiting Hepatic Stellate Cells Proliferation and Activation. Curr. Mol. Pharmacol. 2023, 17, e220323214863. [Google Scholar] [CrossRef]
- Wu, X.; Liu, X.-Q.; Liu, Z.-N.; Xia, G.-Q.; Zhu, H.; Zhang, M.; Wu, B.-M.; Lv, X.-W. CD73 Aggravates Alcohol-Related Liver Fibrosis by Promoting Autophagy Mediated Activation of Hepatic Stellate Cells through AMPK/AKT/mTOR Signaling Pathway. Int. Immunopharmacol. 2022, 113, 109229. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.Z.; Hakim, W.; Kruglov, E.A.; Watanabe, A.; Watkins, W.; Dranoff, J.A.; Mehal, W.Z. Adenosine Inhibits Cytosolic Calcium Signals and Chemotaxis in Hepatic Stellate Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G395–G401. [Google Scholar] [CrossRef][Green Version]
- Che, J.; Chan, E.S.L.; Cronstein, B.N. Adenosine A2A Receptor Occupancy Stimulates Collagen Expression by Hepatic Stellate Cells via Pathways Involving Protein Kinase A, Src, and Extracellular Signal-Regulated Kinases 1/2 Signaling Cascade or p38 Mitogen-Activated Protein Kinase Signaling Pathway. Mol. Pharmacol. 2007, 72, 1626–1636. [Google Scholar] [PubMed]
- Block, E.T.; Cronstein, B.N. Interferon-Gamma Inhibits Adenosine A2A Receptor Function in Hepatic Stellate Cells by STAT1-Mediated Repression of Adenylyl Cyclase. Int. J. Infereron. Cytokine Mediat. Res. 2010, 2010, 113–126. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sohail, M.A.; Hashmi, A.Z.; Hakim, W.; Watanabe, A.; Zipprich, A.; Groszmann, R.J.; Dranoff, J.A.; Torok, N.J.; Mehal, W.Z. Adenosine Induces Loss of Actin Stress Fibers and Inhibits Contraction in Hepatic Stellate Cells via Rho Inhibition. Hepatology 2009, 49, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.K.; Mehal, W.Z. Activation of Adenosine Receptor A2A Increases HSC Proliferation and Inhibits Death and Senescence by down-Regulation of p53 and Rb. Front. Pharmacol. 2014, 5, 69. [Google Scholar] [CrossRef]
- Wang, H.; Guan, W.; Yang, W.; Wang, Q.; Zhao, H.; Yang, F.; Lv, X.; Li, J. Caffeine Inhibits the Activation of Hepatic Stellate Cells Induced by Acetaldehyde via Adenosine A2A Receptor Mediated by the cAMP/PKA/SRC/ERK1/2/P38 MAPK Signal Pathway. PLoS ONE 2014, 9, e92482. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Saito, S.-Y.; Nishiyama, R.; Nakamura, M.; Todoroki, K.; Toyo’oka, T.; Ishikawa, T. Caffeine Suppresses the Activation of Hepatic Stellate Cells cAMP-Independently by Antagonizing Adenosine Receptors. Biol. Pharm. Bull. 2017, 40, 658–664. [Google Scholar] [CrossRef]
- Velasco-Loyden, G.; Pérez-Carreón, J.I.; Agüero, J.F.C.; Romero, P.C.; Vidrio-Gómez, S.; Martínez-Pérez, L.; Yáñez-Maldonado, L.; Hernández-Muñoz, R.; Macías-Silva, M.; de Sánchez, V.C. Prevention of In Vitro Hepatic Stellate Cells Activation by the Adenosine Derivative Compound IFC305. Biochem. Pharmacol. 2010, 80, 1690–1699. [Google Scholar] [CrossRef]
- Wang, Q.; Dai, X.; Yang, W.; Wang, H.; Zhao, H.; Yang, F.; Yang, Y.; Li, J.; Lv, X. Caffeine Protects against Alcohol-Induced Liver Fibrosis by Dampening the cAMP/PKA/CREB Pathway in Rat Hepatic Stellate Cells. Int. Immunopharmacol. 2015, 25, 340–352. [Google Scholar] [CrossRef]
- James Phillips, M. The Liver: An Atlas and Text of Ultrastructural Pathology; Raven Press: New York, NY, USA, 1987; ISBN 9780608058740. [Google Scholar]
- Liaskou, E.; Wilson, D.V.; Oo, Y.H. Innate Immune Cells in Liver Inflammation. Mediat. Inflamm. 2012, 2012, 949157. [Google Scholar] [CrossRef] [PubMed]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer Cells in Host Defense and Liver Disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef]
- Naito, M.; Hasegawa, G.; Takahashi, K. Development, Differentiation, and Maturation of Kupffer Cells. Microsc. Res. Tech. 1997, 39, 350–364. [Google Scholar] [CrossRef]
- Bouwens, L.; Baekeland, M.; De Zanger, R.; Wisse, E. Quantitation, Tissue Distribution and Proliferation Kinetics of Kupffer Cells in Normal Rat Liver. Hepatology 1986, 6, 718–722. [Google Scholar] [CrossRef]
- Kolios, G.; Valatas, V.; Kouroumalis, E. Role of Kupffer Cells in the Pathogenesis of Liver Disease. World J. Gastroenterol. 2006, 12, 7413–7420. [Google Scholar] [CrossRef]
- Klaver, D.; Thurnher, M. Control of Macrophage Inflammation by P2Y Purinergic Receptors. Cells 2021, 10, 1098. [Google Scholar] [CrossRef]
- Del Rey, A.; Renigunta, V.; Dalpke, A.H.; Leipziger, J.; Matos, J.E.; Robaye, B.; Zuzarte, M.; Kavelaars, A.; Hanley, P.J. Knock-out Mice Reveal the Contributions of P2Y and P2X Receptors to Nucleotide-Induced Ca2+ Signaling in Macrophages. J. Biol. Chem. 2006, 281, 35147–35155. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal to Promote Phagocytic Clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef]
- Stober, C.B.; Lammas, D.A.; Li, C.M.; Kumararatne, D.S.; Lightman, S.L.; McArdle, C.A. ATP-Mediated Killing of Mycobacterium Bovis Bacille Calmette-Guérin within Human Macrophages Is Calcium Dependent and Associated with the Acidification of Mycobacteria-Containing Phagosomes. J. Immunol. 2001, 166, 6276–6286. [Google Scholar] [CrossRef]
- De la Rosa, G.; Gómez, A.I.; Baños, M.C.; Pelegrín, P. Signaling Through Purinergic Receptor P2Y Enhances Macrophage IL-1β Production. Int. J. Mol. Sci. 2020, 21, 4686. [Google Scholar] [CrossRef]
- Ren, W.; Rubini, P.; Tang, Y.; Engel, T.; Illes, P. Inherent P2X7 Receptors Regulate Macrophage Functions during Inflammatory Diseases. Int. J. Mol. Sci. 2021, 23, 232. [Google Scholar] [CrossRef] [PubMed]
- Gudipaty, L.; Humphreys, B.D.; Buell, G.; Dubyak, G.R. Regulation of P2X7 Nucleotide Receptor Function in Human Monocytes by Extracellular Ions and Receptor Density. Am. J. Physiol. Cell Physiol. 2001, 280, C943–C953. [Google Scholar] [CrossRef] [PubMed]
- Buisman, H.P.; Steinberg, T.H.; Fischbarg, J.; Silverstein, S.C.; Vogelzang, S.A.; Ince, C.; Ypey, D.L.; Leijh, P.C. Extracellular ATP Induces a Large Nonselective Conductance in Macrophage Plasma Membranes. Proc. Natl. Acad. Sci. USA 1988, 85, 7988–7992. [Google Scholar] [CrossRef] [PubMed]
- Nurkhametova, D.; Siniavin, A.; Streltsova, M.; Kudryavtsev, D.; Kudryavtsev, I.; Giniatullina, R.; Tsetlin, V.; Malm, T.; Giniatullin, R. Does Cholinergic Stimulation Affect the P2X7 Receptor-Mediated Dye Uptake in Mast Cells and Macrophages? Front. Cell. Neurosci. 2020, 14, 548376. [Google Scholar] [CrossRef]
- Bockstiegel, J.; Engelhardt, J.; Weindl, G. P2X7 Receptor Activation Leads to NLRP3-Independent IL-1β Release by Human Macrophages. Cell Commun. Signal. 2023, 21, 335. [Google Scholar] [CrossRef]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1 Beta Secretion Stimulated by P2X7 Receptors Is Dependent on Inflammasome Activation and Correlated with Exosome Release in Murine Macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef]
- Sun, S.; Gong, D.; Liu, R.; Wang, R.; Chen, D.; Yuan, T.; Wang, S.; Xing, C.; Lv, Y.; Du, G.; et al. Puerarin Inhibits NLRP3-Caspase-1-GSDMD-Mediated Pyroptosis via P2X7 Receptor in Cardiomyocytes and Macrophages. Int. J. Mol. Sci. 2023, 24, 13169. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Xiang, Z.; Lv, J.; Jiang, P.; Chen, C.; Jiang, B.; Burnstock, G. Expression of P2X Receptors on Immune Cells in the Rat Liver during Postnatal Development. Histochem. Cell Biol. 2006, 126, 453–463. [Google Scholar] [CrossRef]
- Pei, H.; He, Z.; Du, R.; Han, C.; Sheng, Y.; Wang, J.; Zhou, X.; Li, W.; Cao, C.; Sheng, J.; et al. Imidacloprid Activates Kupffer Cells Pyroptosis to Induce Liver Injury in Mice via P2X7. Int. Immunopharmacol. 2023, 119, 110179. [Google Scholar] [CrossRef]
- Toki, Y.; Takenouchi, T.; Harada, H.; Tanuma, S.-I.; Kitani, H.; Kojima, S.; Tsukimoto, M. Extracellular ATP Induces P2X7 Receptor Activation in Mouse Kupffer Cells, Leading to Release of IL-1β, HMGB1, and PGE2, Decreased MHC Class I Expression and Necrotic Cell Death. Biochem. Biophys. Res. Commun. 2015, 458, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Negishi, Y.; Tsukimoto, M.; Takenouchi, T.; Kitani, H.; Takeda, K. Purinergic Signaling via P2X7 Receptor Mediates IL-1β Production in Kupffer Cells Exposed to Silica Nanoparticle. Toxicology 2014, 321, 13–20. [Google Scholar] [CrossRef]
- Yuan, F.; Cai, J.-N.; Dai, M.; Lv, X. Inhibition of P2Y Receptor Expression in Kupffer Cells Alleviates Alcoholic Steatohepatitis in Mice. Int. Immunopharmacol. 2022, 109, 108909. [Google Scholar] [CrossRef]
- Duarte, F.V.; Amorim, J.A.; Varela, A.T.; Teodoro, J.S.; Gomes, A.P.; Cunha, R.A.; Palmeira, C.M.; Rolo, A.P. Adenosine Receptors: Regulatory Players in the Preservation of Mitochondrial Function Induced by Ischemic Preconditioning of Rat Liver. Purinergic Signal. 2017, 13, 179–190. [Google Scholar] [CrossRef]
- Ben-Ari, Z.; Pappo, O.; Sulkes, J.; Cheporko, Y.; Vidne, B.A.; Hochhauser, E. Effect of Adenosine A2A Receptor Agonist (CGS) on Ischemia/reperfusion Injury in Isolated Rat Liver. Apoptosis 2005, 10, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Reinstein, L.J.; Lichtman, S.N.; Currin, R.T.; Wang, J.; Thurman, R.G.; Lemasters, J.J. Suppression of Lipopolysaccharide-Stimulated Release of Tumor Necrosis Factor by Adenosine: Evidence for A2 Receptors on Rat Kupffer Cells. Hepatology 1994, 19, 1445–1452. [Google Scholar] [PubMed]
- Zhao, N.; Xia, G.; Cai, J.; Li, Z.; Lv, X.-W. Adenosine Receptor A2B Mediates Alcoholic Hepatitis by Regulating cAMP Levels and the NF-KB Pathway. Toxicol. Lett. 2022, 359, 84–95. [Google Scholar] [CrossRef]
- Rothweiler, S.; Feldbrügge, L.; Jiang, Z.G.; Csizmadia, E.; Longhi, M.S.; Vaid, K.; Enjyoji, K.; Popov, Y.V.; Robson, S.C. Selective Deletion of ENTPD1/CD39 in Macrophages Exacerbates Biliary Fibrosis in a Mouse Model of Sclerosing Cholangitis. Purinergic Signal. 2019, 15, 375–385. [Google Scholar] [CrossRef]
- Gao, J.; Zuo, B.; He, Y. Liver Sinusoidal Endothelial Cells as Potential Drivers of Liver Fibrosis (Review). Mol. Med. Rep. 2024, 29, 40. [Google Scholar] [CrossRef]
- DeLeve, L.D. Liver Sinusoidal Endothelial Cells in Hepatic Fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrød, B. The Scavenger Endothelial Cell: A New Player in Homeostasis and Immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, L.P.; Mates, J.M.; Cheplowitz, A.M.; Avila, C.L.; Zimmerer, J.M.; Yao, Z.; Maiseyeu, A.; Rajaram, M.V.S.; Robinson, J.M.; Anderson, C.L. Scavenger Receptor B1, the HDL Receptor, Is Expressed Abundantly in Liver Sinusoidal Endothelial Cells. Sci. Rep. 2016, 6, 20646. [Google Scholar] [CrossRef]
- Van Berkel, T.J.; De Rijke, Y.B.; Kruijt, J.K. Different Fate in Vivo of Oxidatively Modified Low Density Lipoprotein and Acetylated Low Density Lipoprotein in Rats. Recognition by Various Scavenger Receptors on Kupffer and Endothelial Liver Cells. J. Biol. Chem. 1991, 266, 2282–2289. [Google Scholar] [CrossRef]
- Pandey, E.; Nour, A.S.; Harris, E.N. Prominent Receptors of Liver Sinusoidal Endothelial Cells in Liver Homeostasis and Disease. Front. Physiol. 2020, 11, 873. [Google Scholar] [CrossRef] [PubMed]
- Boeynaems, J.M.; Pearson, J.D. P2 Purinoceptors on Vascular Endothelial Cells: Physiological Significance and Transduction Mechanisms. Trends Pharmacol. Sci. 1990, 11, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Motte, S.; Communi, D.; Pirotton, S.; Boeynaems, J.M. Involvement of Multiple Receptors in the Actions of Extracellular ATP: The Example of Vascular Endothelial Cells. Int. J. Biochem. Cell Biol. 1995, 27, 1–7. [Google Scholar] [CrossRef]
- Hashimoto, N.; Watanabe, T.; Shiratori, Y.; Ikeda, Y.; Kato, H.; Han, K.; Yamada, H.; Toda, G.; Kurokawa, K. Prostanoid Secretion by Rat Hepatic Sinusoidal Endothelial Cells and Its Regulation by Exogenous Adenosine Triphosphate. Hepatology 1995, 21, 1713–1718. [Google Scholar]
- Beldi, G.; Wu, Y.; Sun, X.; Imai, M.; Enjyoji, K.; Csizmadia, E.; Candinas, D.; Erb, L.; Robson, S.C. Regulated Catalysis of Extracellular Nucleotides by Vascular CD39/ENTPD1 Is Required for Liver Regeneration. Gastroenterology 2008, 135, 1751–1760. [Google Scholar] [CrossRef][Green Version]
- Feng, L.; Sun, X.; Csizmadia, E.; Han, L.; Bian, S.; Murakami, T.; Wang, X.; Robson, S.C.; Wu, Y. Vascular CD39/ENTPD1 Directly Promotes Tumor Cell Growth by Scavenging Extracellular Adenosine Triphosphate. Neoplasia 2011, 13, 206–216. [Google Scholar] [CrossRef]
- Mandili, G.; Alchera, E.; Merlin, S.; Imarisio, C.; Chandrashekar, B.R.; Riganti, C.; Bianchi, A.; Novelli, F.; Follenzi, A.; Carini, R. Mouse Hepatocytes and LSEC Proteome Reveal Novel Mechanisms of Ischemia/reperfusion Damage and Protection by A2aR Stimulation. J. Hepatol. 2015, 62, 573–580. [Google Scholar] [CrossRef]
- Kaczmarek, E.; Koziak, K.; Sévigny, J.; Siegel, J.B.; Anrather, J.; Beaudoin, A.R.; Bach, F.H.; Robson, S.C. Identification and Characterization of CD39/vascular ATP Diphosphohydrolase. J. Biol. Chem. 1996, 271, 33116–33122. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, C.; Sundberg, C.; Sévigny, J.; Enjyoji, K.; Hoshi, T.; Csizmadia, E.; Robson, S. Disordered Cellular Migration and Angiogenesis in cd39-Null Mice. Circulation 2001, 104, 3109–3115. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte Pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Björkström, N.K. Immunobiology of the Biliary Tract System. J. Hepatol. 2022, 77, 1657–1669. [Google Scholar] [CrossRef]
- Wang, W.; Chen, D.; Wang, J.; Wen, L. Cellular Homeostasis and Repair in the Biliary Tree. Semin. Liver Dis. 2022, 42, 271–282. [Google Scholar] [CrossRef]
- Lanzoni, G.; Cardinale, V.; Carpino, G. The Hepatic, Biliary, and Pancreatic Network of Stem/progenitor Cell Niches in Humans: A New Reference Frame for Disease and Regeneration. Hepatology 2016, 64, 277–286. [Google Scholar] [CrossRef]
- Alpini, G.; McGill, J.M.; Larusso, N.F. The Pathobiology of Biliary Epithelia. Hepatology 2002, 35, 1256–1268. [Google Scholar] [CrossRef]
- Marzioni, M.; Glaser, S.S.; Francis, H.; Phinizy, J.L.; LeSage, G.; Alpini, G. Functional Heterogeneity of Cholangiocytes. Semin. Liver Dis. 2002, 22, 227–240. [Google Scholar] [CrossRef]
- Alpini, G.; Roberts, S.; Kuntz, S.M.; Ueno, Y.; Gubba, S.; Podila, P.V.; LeSage, G.; LaRusso, N.F. Morphological, Molecular, and Functional Heterogeneity of Cholangiocytes from Normal Rat Liver. Gastroenterology 1996, 110, 1636–1643. [Google Scholar] [CrossRef]
- Yoo, K.-S.; Lim, W.T.; Choi, H.S. Biology of Cholangiocytes: From Bench to Bedside. Gut Liver 2016, 10, 687–698. [Google Scholar] [CrossRef]
- Mancinelli, R.; Franchitto, A.; Gaudio, E.; Onori, P.; Glaser, S.; Francis, H.; Venter, J.; Demorrow, S.; Carpino, G.; Kopriva, S.; et al. After Damage of Large Bile Ducts by Gamma-Aminobutyric Acid, Small Ducts Replenish the Biliary Tree by Amplification of Calcium-Dependent Signaling and de Novo Acquisition of Large Cholangiocyte Phenotypes. Am. J. Pathol. 2010, 176, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Gouw, A.S.H.; Clouston, A.D.; Theise, N.D. Ductular Reactions in Human Liver: Diversity at the Interface. Hepatology 2011, 54, 1853–1863. [Google Scholar] [CrossRef]
- Maroni, L.; Ninfole, E.; Pinto, C.; Benedetti, A.; Marzioni, M. Gut-Liver Axis and Inflammasome Activation in Cholangiocyte Pathophysiology. Cells 2020, 9, 736. [Google Scholar] [CrossRef] [PubMed]
- Giordano, D.M.; Pinto, C.; Maroni, L.; Benedetti, A.; Marzioni, M. Inflammation and the Gut-Liver Axis in the Pathophysiology of Cholangiopathies. Int. J. Mol. Sci. 2018, 19, 3003. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gao, W.-K.; Shu, Y.-Y.; Ye, J. Mechanisms of Ductular Reaction in Non-Alcoholic Steatohepatitis. World J. Gastroenterol. 2022, 28, 2088–2099. [Google Scholar] [CrossRef]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology 2019, 69, 420–430. [Google Scholar] [CrossRef]
- Boyer, J.L.; Soroka, C.J. Bile Formation and Secretion: An Update. J. Hepatol. 2021, 75, 190–201. [Google Scholar] [CrossRef]
- Pinzani, M.; Luong, T.V. Pathogenesis of Biliary Fibrosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Fabris, L.; Spirli, C.; Cadamuro, M.; Fiorotto, R.; Strazzabosco, M. Emerging Concepts in Biliary Repair and Fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G102–G116. [Google Scholar] [CrossRef]
- Carey, E.J.; Ali, A.H.; Lindor, K.D. Primary Biliary Cirrhosis. Lancet 2015, 386, 1565–1575. [Google Scholar] [CrossRef]
- Salter, K.D.; Fitz, J.G.; Roman, R.M. Domain-Specific Purinergic Signaling in Polarized Rat Cholangiocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 278, G492–G500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Roman, R.; Lidofsky, S.D.; Fitz, J.G. Autocrine Signaling through ATP Release Represents a Novel Mechanism for Cell Volume Regulation. Proc. Natl. Acad. Sci. USA 1996, 93, 12020–12025. [Google Scholar] [CrossRef]
- Taylor, A.L.; Kudlow, B.A.; Marrs, K.L.; Gruenert, D.C.; Guggino, W.B.; Schwiebert, E.M. Bioluminescence Detection of ATP Release Mechanisms in Epithelia. Am. J. Physiol. 1998, 275, C1391–C1406. [Google Scholar] [CrossRef]
- Roman, R.M.; Wang, Y.; Lidofsky, S.D.; Feranchak, A.P.; Lomri, N.; Scharschmidt, B.F.; Fitz, J.G. Hepatocellular ATP-Binding Cassette Protein Expression Enhances ATP Release and Autocrine Regulation of Cell Volume. J. Biol. Chem. 1997, 272, 21970–21976. [Google Scholar] [CrossRef] [PubMed]
- Scoazec, J.Y.; Bringuier, A.F.; Medina, J.F.; Martínez-Ansó, E.; Veissiere, D.; Feldmann, G.; Housset, C. The Plasma Membrane Polarity of Human Biliary Epithelial Cells: In Situ Immunohistochemical Analysis and Functional Implications. J. Hepatol. 1997, 26, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Pasyk, E.A.; Foskett, J.K. Cystic Fibrosis Transmembrane Conductance Regulator-Associated ATP and Adenosine 3′-Phosphate 5’-Phosphosulfate Channels in Endoplasmic Reticulum and Plasma Membranes. J. Biol. Chem. 1997, 272, 7746–7751. [Google Scholar] [CrossRef]
- Krause, U.; Rider, M.H.; Hue, L. Protein Kinase Signaling Pathway Triggered by Cell Swelling and Involved in the Activation of Glycogen Synthase and Acetyl-CoA Carboxylase in Isolated Rat Hepatocytes. J. Biol. Chem. 1996, 271, 16668–16673. [Google Scholar] [CrossRef]
- Feranchak, A.P.; Roman, R.M.; Schwiebert, E.M.; Fitz, J.G. Phosphatidylinositol 3-Kinase Contributes to Cell Volume Regulation through Effects on ATP Release. J. Biol. Chem. 1998, 273, 14906–14911. [Google Scholar] [CrossRef]
- Feranchak, A.P.; Roman, R.M.; Doctor, R.B.; Salter, K.D.; Toker, A.; Fitz, J.G. The Lipid Products of Phosphoinositide 3-Kinase Contribute to Regulation of Cholangiocyte ATP and Chloride Transport. J. Biol. Chem. 1999, 274, 30979–30986. [Google Scholar] [CrossRef]
- Schlosser, S.F.; Burgstahler, A.D.; Nathanson, M.H. Isolated Rat Hepatocytes Can Signal to Other Hepatocytes and Bile Duct Cells by Release of Nucleotides. Proc. Natl. Acad. Sci. USA 1996, 93, 9948–9953. [Google Scholar] [CrossRef]
- Roman, R.M.; Feranchak, A.P.; Salter, K.D.; Wang, Y.; Fitz, J.G. Endogenous ATP Release Regulates Cl− Secretion in Cultured Human and Rat Biliary Epithelial Cells. Am. J. Physiol. 1999, 276, G1391–G1400. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.M.; Fitz, J.G. Emerging Roles of Purinergic Signaling in Gastrointestinal Epithelial Secretion and Hepatobiliary Function. Gastroenterology 1999, 116, 964–979. [Google Scholar] [CrossRef] [PubMed]
- Gatof, D.; Kilic, G.; Fitz, J.G. Vesicular Exocytosis Contributes to Volume-Sensitive ATP Release in Biliary Cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G538–G546. [Google Scholar] [CrossRef]
- Schlenker, T.; Romac, J.M.; Sharara, A.I.; Roman, R.M.; Kim, S.J.; LaRusso, N.; Liddle, R.A.; Fitz, J.G. Regulation of Biliary Secretion through Apical Purinergic Receptors in Cultured Rat Cholangiocytes. Am. J. Physiol. 1997, 273, G1108–G1117. [Google Scholar] [CrossRef]
- Dranoff, J.A.; Masyuk, A.I.; Kruglov, E.A.; LaRusso, N.F.; Nathanson, M.H. Polarized Expression and Function of P2Y ATP Receptors in Rat Bile Duct Epithelia. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1059–G1067. [Google Scholar] [CrossRef] [PubMed]
- Woo, K.; Sathe, M.; Kresge, C.; Esser, V.; Ueno, Y.; Venter, J.; Glaser, S.S.; Alpini, G.; Feranchak, A.P. Adenosine Triphosphate Release and Purinergic (P2) Receptor-Mediated Secretion in Small and Large Mouse Cholangiocytes. Hepatology 2010, 52, 1819–1828. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Masyuk, T.V.; LaRusso, N.F. Cholangiocyte Primary Cilia in Liver Health and Disease. Dev. Dyn. 2008, 237, 2007–2012. [Google Scholar] [CrossRef]
- Masyuk, A.I.; Gradilone, S.A.; Banales, J.M.; Huang, B.Q.; Masyuk, T.V.; Lee, S.-O.; Splinter, P.L.; Stroope, A.J.; Larusso, N.F. Cholangiocyte Primary Cilia Are Chemosensory Organelles That Detect Biliary Nucleotides via P2Y12 Purinergic Receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G725–G734. [Google Scholar] [CrossRef]
- Doctor, R.B.; Matzakos, T.; McWilliams, R.; Johnson, S.; Feranchak, A.P.; Fitz, J.G. Purinergic Regulation of Cholangiocyte Secretion: Identification of a Novel Role for P2X Receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G779–G786. [Google Scholar] [CrossRef]
- Dutta, A.K.; Woo, K.; Khimji, A.-K.; Kresge, C.; Feranchak, A.P. Mechanosensitive Cl− Secretion in Biliary Epithelium Mediated through TMEM16A. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G87–G98. [Google Scholar] [CrossRef]
- Godoy, V.; Banales, J.M.; Medina, J.F.; Pastor-Anglada, M. Functional Crosstalk between the Adenosine Transporter CNT3 and Purinergic Receptors in the Biliary Epithelia. J. Hepatol. 2014, 61, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.P.; Lee, J.-S.; Sohn, B.H.; Yu, X.; Lopez-Terrada, D.; Finegold, M.J.; Goss, J.A.; Thevananther, S. P2X3 Purinergic Receptor Overexpression Is Associated with Poor Recurrence-Free Survival in Hepatocellular Carcinoma Patients. Oncotarget 2015, 6, 41162–41179. [Google Scholar] [CrossRef] [PubMed]
- Lertsuwan, J.; Ruchirawat, M. Inhibitory Effects of ATP and Adenosine on Cholangiocarcinoma Cell Proliferation and Motility. Anticancer Res. 2017, 37, 3553–3561. [Google Scholar]
- Burnstock, G. Purinergic Signalling: Therapeutic Developments. Front. Pharmacol. 2017, 8, 661. [Google Scholar]
- Burnstock, G. The Therapeutic Potential of Purinergic Signalling. Biochem. Pharmacol. 2018, 151, 157–165. [Google Scholar] [CrossRef]
- Chiang, D.J.; Roychowdhury, S.; Bush, K.; McMullen, M.R.; Pisano, S.; Niese, K.; Olman, M.A.; Pritchard, M.T.; Nagy, L.E. Adenosine 2A Receptor Antagonist Prevented and Reversed Liver Fibrosis in a Mouse Model of Ethanol-Exacerbated Liver Fibrosis. PLoS ONE 2013, 8, e69114. [Google Scholar] [CrossRef]
- Khalid, M.; Brisson, L.; Tariq, M.; Hao, Y.; Guibon, R.; Fromont, G.; Mortadza, S.A.S.; Mousawi, F.; Manzoor, S.; Roger, S.; et al. Carcinoma-Specific Expression of P2Y11 Receptor and Its Contribution in ATP-Induced Purinergic Signalling and Cell Migration in Human Hepatocellular Carcinoma Cells. Oncotarget 2017, 8, 37278–37290. [Google Scholar] [CrossRef]
- Cohen, S.; Stemmer, S.M.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.A.; Fishman, P. CF102 an A3 Adenosine Receptor Agonist Mediates Anti-Tumor and Anti-Inflammatory Effects in the Liver. J. Cell. Physiol. 2011, 226, 2438–2447. [Google Scholar] [CrossRef]
- Suresh, R.R.; Jain, S.; Chen, Z.; Tosh, D.K.; Ma, Y.; Podszun, M.C.; Rotman, Y.; Salvemini, D.; Jacobson, K.A. Design and In Vivo Activity of A Adenosine Receptor Agonist Prodrugs. Purinergic Signal. 2020, 16, 367–377. [Google Scholar] [CrossRef]
- Saito, T.; Muramatsu, M.; Ishii, Y.; Saigo, Y.; Konuma, T.; Toriniwa, Y.; Miyajima, K.; Ohta, T. Pathophysiological Analysis of the Progression of Hepatic Lesions in STAM Mice. Physiol. Res. 2017, 66, 791–799. [Google Scholar] [CrossRef]
- Stemmer, S.M.; Benjaminov, O.; Medalia, G.; Ciuraru, N.B.; Silverman, M.H.; Bar-Yehuda, S.; Fishman, S.; Harpaz, Z.; Farbstein, M.; Cohen, S.; et al. CF102 for the Treatment of Hepatocellular Carcinoma: A Phase I/II, Open-Label, Dose-Escalation Study. Oncologist 2013, 18, 25–26. [Google Scholar] [CrossRef]
- Pérez-Carreón, J.I.; Martínez-Pérez, L.; Loredo, M.L.; Yañez-Maldonado, L.; Velasco-Loyden, G.; Vidrio-Gómez, S.; Ramírez-Salcedo, J.; Hernández-Luis, F.; Velázquez-Martínez, I.; Suárez-Cuenca, J.A.; et al. An Adenosine Derivative Compound, IFC305, Reverses Fibrosis and Alters Gene Expression in a Pre-Established CCl4-Induced Rat Cirrhosis. Int. J. Biochem. Cell Biol. 2010, 42, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cabeza de Vaca, R.; Domínguez-López, M.; Guerrero-Celis, N.; Rodríguez-Aguilera, J.R.; Chagoya de Sánchez, V. Inflammation Is Regulated by the Adenosine Derivative Molecule, IFC-305, during Reversion of Cirrhosis in a CCl Rat Model. Int. Immunopharmacol. 2018, 54, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Sandoval, G.; Meza-Toledo, S.E.; Aguirre-Alvarado, C.; Zaragoza-Martinéz, F.; Rodríguez-Páez, L.; Nogueda-Torres, B.; Baeza-Ramírez, I.; Wong-Ramírez, C.; Chagoya-de Sánchez, V.; Suárez-Cuenca, J.A.; et al. New drugs development in Mexico. Gac. Med. Mex. 2007, 143, 33–59. [Google Scholar]
- Menzies, R.I.; Tam, F.W.; Unwin, R.J.; Bailey, M.A. Purinergic Signaling in Kidney Disease. Kidney Int. 2017, 91, 315–323. [Google Scholar] [CrossRef]
- Mishra, S.; Shelke, V.; Dagar, N.; Lech, M.; Gaikwad, A.B. Molecular insights into P2X signalling cascades in acute kidney injury. Purinergic Signal. 2024, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, H. The Bacteria and the Host: A Story of Purinergic Signaling in Urinary Tract Infections. Am. J. Physiol. Cell Physiol. 2021, 321, C134–C146. [Google Scholar] [CrossRef]
- Dwyer, K.M. Burnstock Oration—Purinergic Signalling in Kidney Transplantation. Purinergic Signal. 2022, 18, 387–393. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).