
Navigating the Complement Pathway to Optimize PNH Treatment with Pegcetacoplan and Other Currently Approved Complement Inhibitors


Abstract
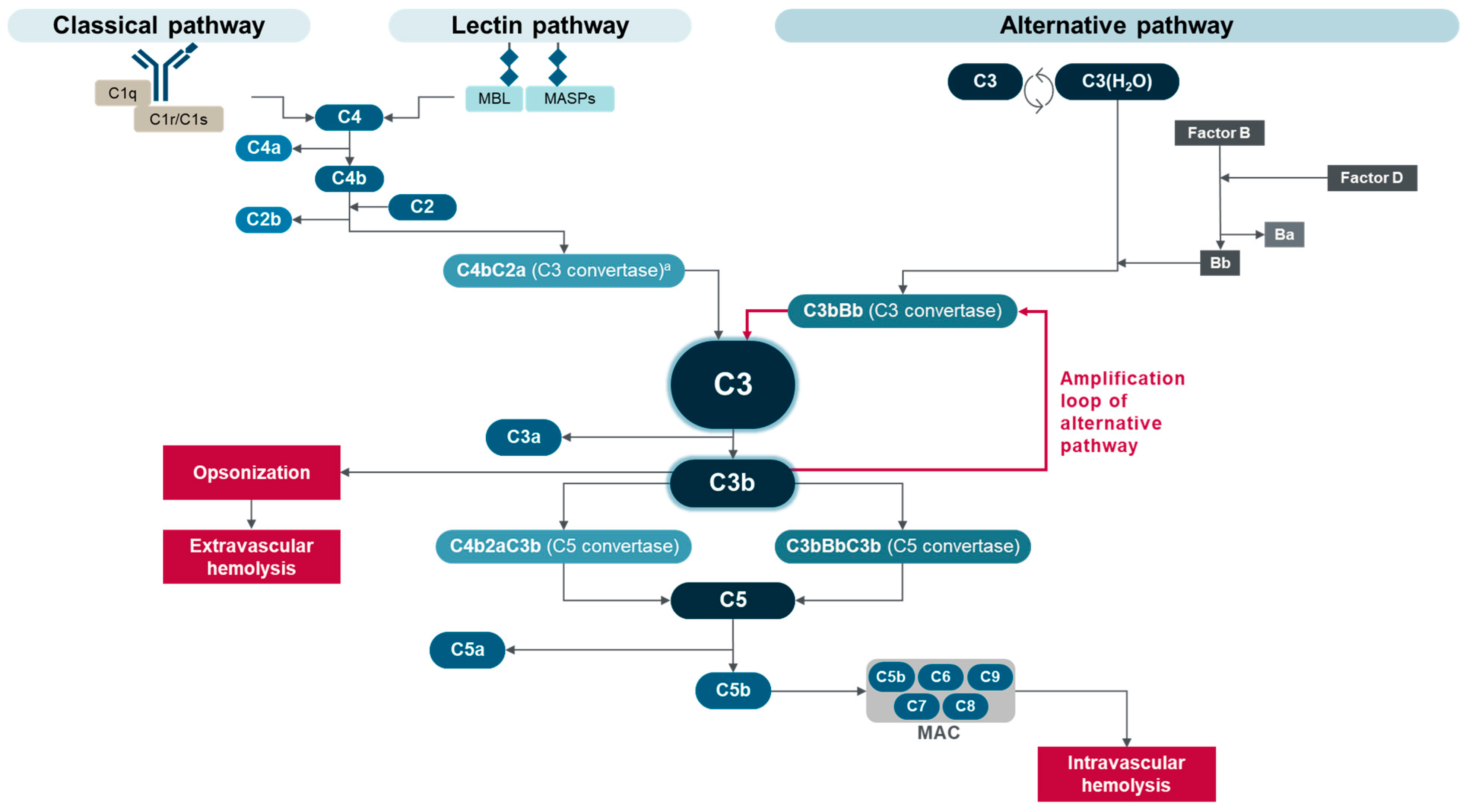
:1. Paroxysmal Nocturnal Hemoglobinuria
2. Eculizumab, Ravulizumab, and Crovalimab, Terminal Complement Inhibitors for PNH
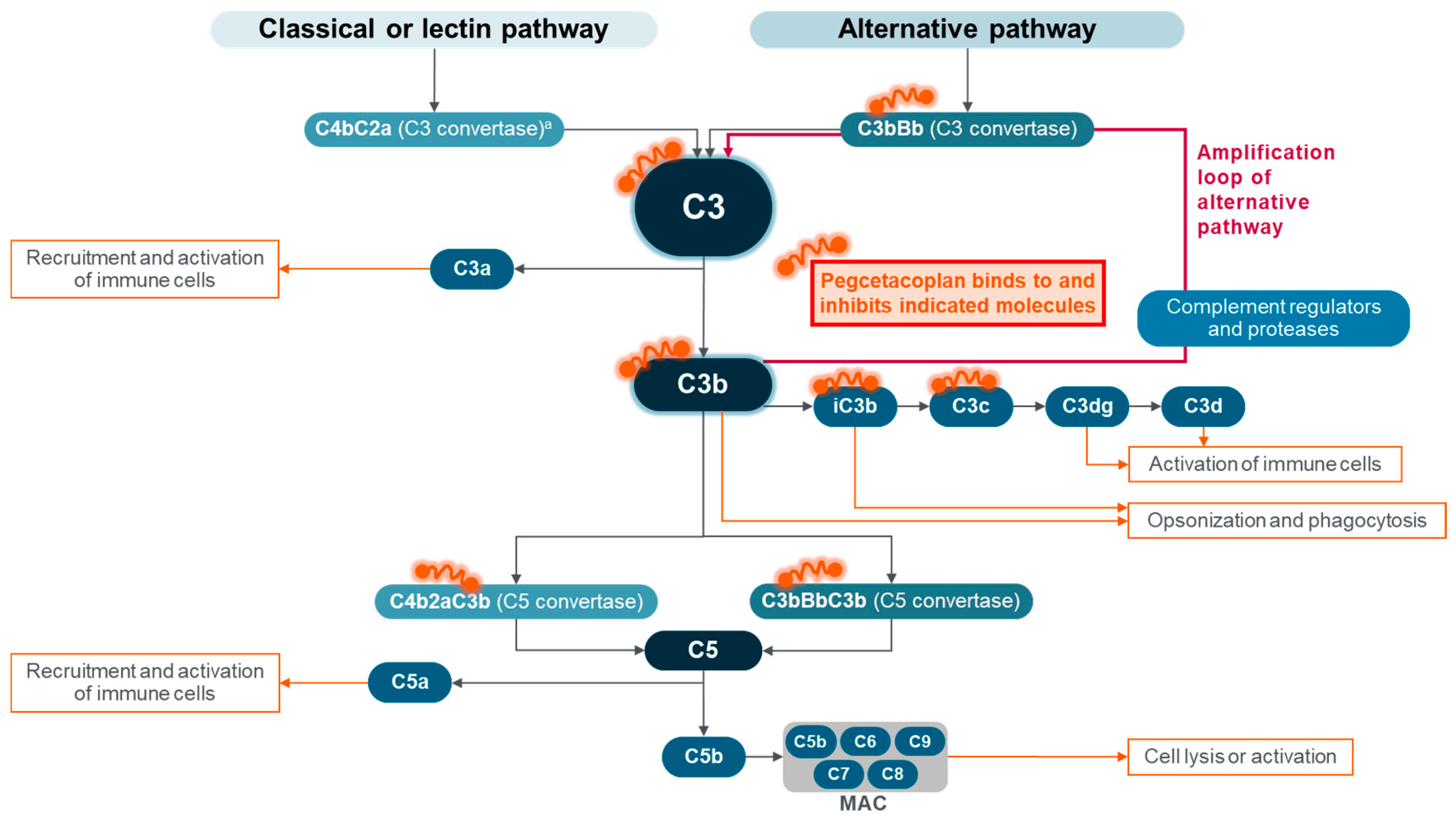
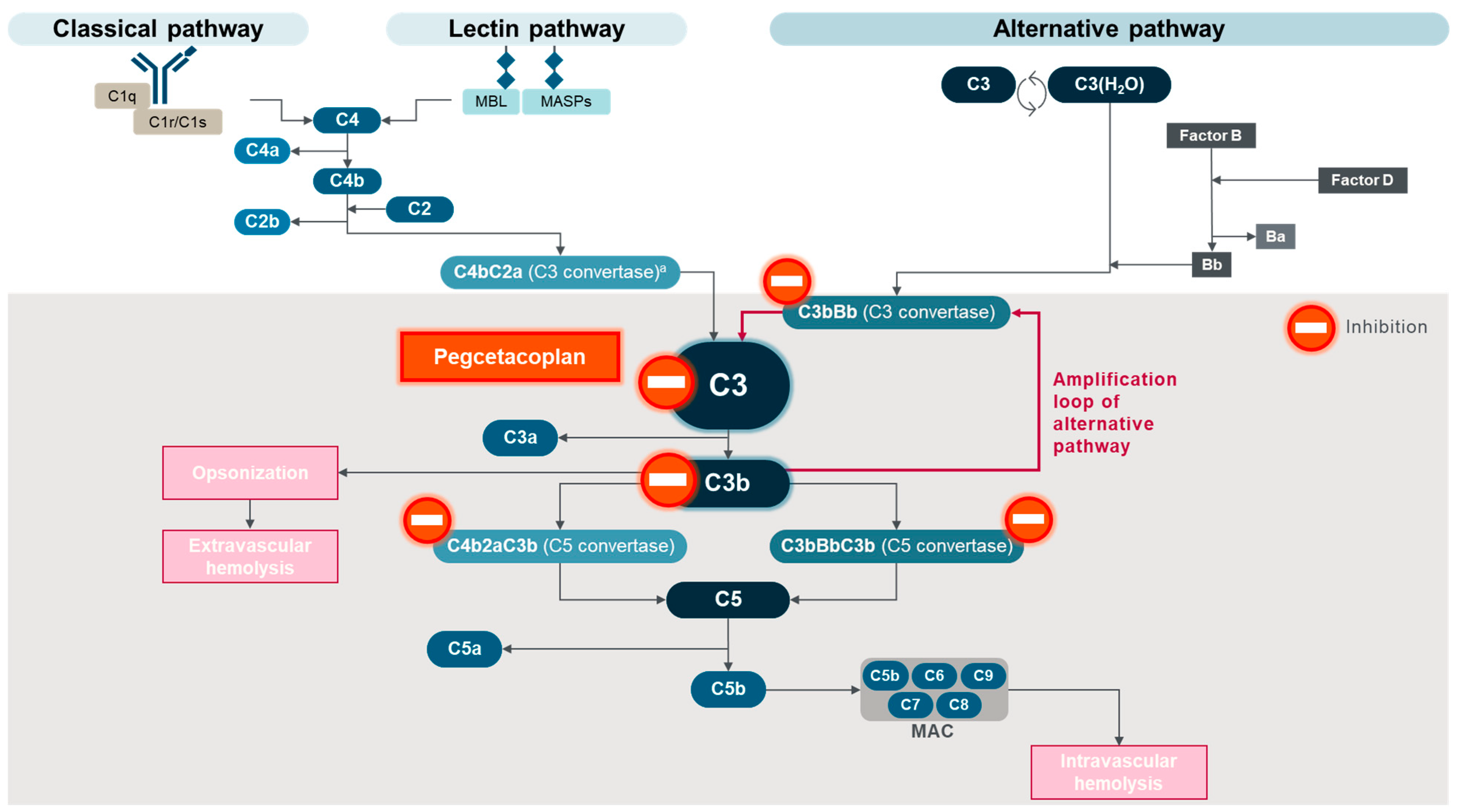
3. Pegcetacoplan, the First C3 Complement Inhibitor for PNH
4. Iptacopan and Danicopan, New Proximal Complement Inhibitors for PNH
5. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, A.; DeZern, A.E.; Kinoshita, T.; Brodsky, R.A. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Primers 2017, 3, 17028. [Google Scholar] [CrossRef]
- Jalbert, J.J.; Chaudhari, U.; Zhang, H.; Weyne, J.; Shammo, J.M. Epidemiology of PNH and real-world treatment patterns following an incident PNH diagnosis in the US. Blood 2019, 134, 3407. [Google Scholar] [CrossRef]
- Brodsky, R.A. How I treat paroxysmal nocturnal hemoglobinuria. Blood 2021, 137, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.J.; Painter, D.; Dickinson, A.J.; Griffin, M.; Munir, T.; Arnold, L.; Payne, D.; Pike, A.; Muus, P.; Hill, A.; et al. The incidence and prevalence of patients with paroxysmal nocturnal haemoglobinuria and aplastic anaemia PNH syndrome: A retrospective analysis of the UK’s population-based haematological malignancy research network 2004–2018. Eur. J. Haematol. 2021, 107, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Versino, F.; Fattizzo, B. Complement inhibition in paroxysmal nocturnal hemoglobinuria: From biology to therapy. Int. J. Lab. Hematol. 2024, 46, 43–54. [Google Scholar] [CrossRef]
- Mahoney, J.F.; Urakaze, M.; Hall, S.; DeGasperi, R.; Chang, H.M.; Sugiyama, E.; Warren, C.D.; Borowitz, M.; Nicholson-Weller, A.; Rosse, W.F.; et al. Defective glycosylphosphatidylinositol anchor synthesis in paroxysmal nocturnal hemoglobinuria granulocytes. Blood 1992, 79, 1400–1403. [Google Scholar] [CrossRef]
- Parker, C.; Omine, M.; Richards, S.; Nishimura, J.; Bessler, M.; Ware, R.; Hillmen, P.; Luzzatto, L.; Young, N.; Kinoshita, T.; et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005, 106, 3699–3709. [Google Scholar] [CrossRef]
- Hillmen, P.; Lewis, S.M.; Bessler, M.; Luzzatto, L.; Dacie, J.V. Natural history of paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 1995, 333, 1253–1258. [Google Scholar] [CrossRef]
- Schrezenmeier, H.; Röth, A.; Araten, D.J.; Kanakura, Y.; Larratt, L.; Shammo, J.M.; Wilson, A.; Shayan, G.; Maciejewski, J.P. Baseline clinical characteristics and disease burden in patients with paroxysmal nocturnal hemoglobinuria (PNH): Updated analysis from the International PNH Registry. Ann. Hematol. 2020, 99, 1505–1514. [Google Scholar] [CrossRef]
- Parker, C.J. Update on the diagnosis and management of paroxysmal nocturnal hemoglobinuria. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 208–216. [Google Scholar] [CrossRef]
- Babushok, D.V. When does a PNH clone have clinical significance? Hematology 2021, 2021, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Bessler, M.; Mason, P.J.; Hillmen, P.; Miyata, T.; Yamada, N.; Takeda, J.; Luzzatto, L.; Kinoshita, T. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994, 13, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Bat, T.; Abdelhamid, O.N.; Balasubramanian, S.K.; Mai, A.; Radivoyevitch, T.; Clemente, M.; Maciejewski, J.P. The evolution of paroxysmal nocturnal haemoglobinuria depends on intensity of immunosuppressive therapy. Br. J. Haematol. 2018, 182, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Marotta, S.; Ricci, P.; Marano, L.; Frieri, C.; Cacace, F.; Sica, M.; Kulasekararaj, A.; Calado, R.T.; Scheinberg, P.; et al. Anti-complement treatment for paroxysmal nocturnal hemoglobinuria: Time for proximal complement inhibition? A position paper from the SAAWP of the EBMT. Front. Immunol. 2019, 10, 1157. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Matsuda, I.; Utsunomiya, N.; Samori, M.; Hirata, S.; Okada, M.; Hirota, S.; Higasa, S.; Yoshihara, S. High prevalence of PNH-phenotype cells in patients who received CD19-targeted CAR T-cell therapy. Hemasphere 2021, 5, e628. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef]
- Holers, V.M. Complement and its receptors: New insights into human disease. Annu. Rev. Immunol. 2014, 32, 433–459. [Google Scholar] [CrossRef]
- Kolev, M.; Barbour, T.; Baver, S.; Francois, C.; Deschatelets, P. With complements: C3 inhibition in the clinic. Immunol. Rev. 2023, 313, 358–375. [Google Scholar] [CrossRef]
- West, E.E.; Woodruff, T.; Fremeaux-Bacchi, V.; Kemper, C. Complement in human disease: Approved and up-and-coming therapeutics. Lancet 2024, 403, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Arbore, G.; Kemper, C.; Kolev, M. Intracellular complement—The complosome—In immune cell regulation. Mol. Immunol. 2017, 89, 2–9. [Google Scholar] [CrossRef]
- Goldberg, R.A.; Kitchens, J.W.; Lad, E.; Zarbin, M.A. The evolving treatment paradigm in geographic atrophy. A panel of experts translates clinical trial data to real-world practice. Retin. Today. 2024. Available online: https://retinatoday.com/articles/2024-may-june-supplement2/the-evolving-treatment-paradigm-in-geographic-atrophy (accessed on 28 August 2024).
- Pouw, R.B.; Ricklin, D. Tipping the balance: Intricate roles of the complement system in disease and therapy. Semin. Immunopathol. 2021, 43, 757–771. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Holt, M.; Vidler, J.; Arnold, L.M.; Large, J.; Forrest, B.; Barnfield, C.; Pike, A.; Griffin, M.; Munir, T.; et al. Treatment outcomes of complement protein C5 inhibition in 509 UK patients with paroxysmal nocturnal hemoglobinuria. Blood 2024, 143, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Höchsmann, B.; Szer, J.; Kulasekararaj, A.; de Guibert, S.; Röth, A.; Weitz, I.C.; Armstrong, E.; Risitano, A.M.; Patriquin, C.J.; et al. Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2015, 373, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Mei, L. A case report of pegcetacoplan use for a pregnant woman with paroxysmal nocturnal hemoglobinuria. Res. Pract. Thromb. Haemost. 2024, 8, 102435. [Google Scholar] [CrossRef]
- Patriquin, C.J.; Kiss, T.; Caplan, S.; Chin-Yee, I.; Grewal, K.; Grossman, J.; Larratt, L.; Marceau, D.; Nevill, T.; Sutherland, D.R.; et al. How we treat paroxysmal nocturnal hemoglobinuria: A consensus statement of the Canadian PNH Network and review of the national registry. Eur. J. Haematol. 2019, 102, 36–52. [Google Scholar] [CrossRef]
- Terriou, L.; Lee, J.W.; Forsyth, C.; Griffin, M.; Szer, J.; Röth, A.; Gustovic, P.; Metzger, J.; Patel, A.S.; Patriquin, C.J. Long-term effectiveness of eculizumab: Data from the International PNH Registry. Eur. J. Haematol. 2023, 111, 796–804. [Google Scholar] [CrossRef]
- Kulasekararaj, A.G.; Kuter, D.J.; Griffin, M.; Weitz, I.C.; Röth, A. Biomarkers and laboratory assessments for monitoring the treatment of patients with paroxysmal nocturnal hemoglobinuria: Differences between terminal and proximal complement inhibition. Blood Rev. 2023, 59, 101041. [Google Scholar] [CrossRef]
- Schrezenmeier, H.; Muus, P.; Socié, G.; Szer, J.; Urbano-Ispizua, A.; Maciejewski, J.P.; Brodsky, R.A.; Bessler, M.; Kanakura, Y.; Rosse, W.; et al. Baseline characteristics and disease burden in patients in the International Paroxysmal Nocturnal Hemoglobinuria Registry. Haematologica 2014, 99, 922–929. [Google Scholar] [CrossRef]
- Hill, A.; Rother, R.P.; Arnold, L.; Kelly, R.; Cullen, M.J.; Richards, S.J.; Hillmen, P. Eculizumab prevents intravascular hemolysis in patients with paroxysmal nocturnal hemoglobinuria and unmasks low-level extravascular hemolysis occurring through C3 opsonization. Haematologica 2010, 95, 567–573. [Google Scholar] [CrossRef]
- Heier, J.S.; Lad, E.M.; Holz, F.G.; Rosenfeld, P.J.; Guymer, R.H.; Boyer, D.; Grossi, F.; Baumal, C.R.; Korobelnik, J.F.; Slakter, J.S.; et al. Pegcetacoplan for the treatment of geographic atrophy secondary to age-related macular degeneration (OAKS and DERBY): Two multicentre, randomised, double-masked, sham-controlled, phase 3 trials. Lancet 2023, 402, 1434–1448. [Google Scholar] [CrossRef]
- Dixon, B.P.; Greenbaum, L.A.; Huang, L.; Rajan, S.; Ke, C.; Zhang, Y.; Li, L. Clinical safety and efficacy of pegcetacoplan in a phase 2 study of patients with C3 glomerulopathy and other complement-mediated glomerular diseases. Kidney Int. Rep. 2023, 8, 2284–2293. [Google Scholar] [CrossRef]
- Noris, M.; Daina, E.; Remuzzi, G. Membranoproliferative glomerulonephritis: No longer the same disease and may need very different treatment. Nephrol. Dial. Transplant. 2023, 38, 283–290. [Google Scholar] [CrossRef]
- Soliris (Eculizumab) US Prescribing Information. June 2024. Available online: https://alexion.com/Documents/Soliris_USPI.pdf (accessed on 26 August 2024).
- European Medicines Agency. Soliris (Eculizumab). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/soliris#authorisation-details (accessed on 26 August 2024).
- Ultomiris (Ravulizumab). US Prescribing Information. June 2024. Available online: https://alexion.com/Documents/ultomiris_uspi (accessed on 26 August 2024).
- European Medicines Agency. Ultomiris (Ravulizumab). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ultomiris#product-details (accessed on 26 August 2024).
- Piasky (Crovalimab-Akkz). US Prescribing Information. June 2024. Available online: https://www.gene.com/download/pdf/piasky_prescribing.pdf (accessed on 26 August 2024).
- Empaveli (Pegcetacoplan) US Prescribing Information. February 2024. Available online: https://pi.apellis.com/files/PI_Empaveli.pdf (accessed on 26 August 2024).
- European Medicines Agency. Aspaveli (Pegcetacoplan). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/aspaveli#authorisation-details (accessed on 26 August 2024).
- Fabhalta (Iptacopan). US Prescribing Information. August 2024. Available online: https://www.novartis.com/us-en/sites/novartis_us/files/fabhalta.pdf (accessed on 26 August 2024).
- European Medicines Agency. Fabhalta (Iptacopan). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/fabhalta#product-details (accessed on 26 August 2024).
- Voydeya (Danicopan). US Prescribing Information. March 2024. Available online: https://alexion.com/Documents/voydeya_uspi (accessed on 26 August 2024).
- European Medicines Agency. Voydeya (Danicopan). Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/voydeya#product-details (accessed on 26 August 2024).
- Hillmen, P.; Young, N.S.; Schubert, J.; Brodsky, R.A.; Socié, G.; Muus, P.; Röth, A.; Szer, J.; Elebute, M.O.; Nakamura, R.; et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2006, 355, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Peffault de Latour, R. How we(‘ll) treat paroxysmal nocturnal haemoglobinuria: Diving into the future. Br. J. Haematol. 2022, 196, 288–303. [Google Scholar] [CrossRef]
- Hillmen, P.; Hall, C.; Marsh, J.C.; Elebute, M.; Bombara, M.P.; Petro, B.E.; Cullen, M.J.; Richards, S.J.; Rollins, S.A.; Mojcik, C.F.; et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2004, 350, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Kulasekararaj, A.G.; Hill, A.; Rottinghaus, S.T.; Langemeijer, S.; Wells, R.; Gonzalez-Fernandez, F.A.; Gaya, A.; Lee, J.W.; Gutierrez, E.O.; Piatek, C.I.; et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor-experienced adult patients with PNH: The 302 study. Blood 2019, 133, 540–549. [Google Scholar] [CrossRef]
- Lee, J.W.; Sicre de Fontbrune, F.; Wong Lee Lee, L.; Pessoa, V.; Gualandro, S.; Füreder, W.; Ptushkin, V.; Rottinghaus, S.T.; Volles, L.; Shafner, L.; et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: The 301 study. Blood 2019, 133, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Röth, A.; He, G.; Tong, H.; Lin, Z.; Wang, X.; Chai-Adisaksopha, C.; Lee, J.H.; Brodsky, A.; Hantaweepant, C.; Dumagay, T.E.; et al. Phase 3 randomized COMMODORE 2 trial: Crovalimab versus eculizumab in patients with paroxysmal nocturnal hemoglobinuria naive to complement inhibition. Am. J. Hematol. 2024, 99, 1768–1777. [Google Scholar] [CrossRef]
- Scheinberg, P.; Clé, D.V.; Kim, J.S.; Nur, E.; Yenerel, M.N.; Barcellini, W.; Bonito, D.; Giai, V.; Hus, M.; Lee, Y.; et al. Phase 3 randomized COMMODORE 1 trial: Crovalimab versus eculizumab in complement inhibitor-experienced patients with paroxysmal nocturnal hemoglobinuria. Am. J. Hematol. 2024, 99, 1757–1767. [Google Scholar] [CrossRef]
- Brodsky, R.A.; Young, N.S.; Antonioli, E.; Risitano, A.M.; Schrezenmeier, H.; Schubert, J.; Gaya, A.; Coyle, L.; de Castro, C.; Fu, C.L.; et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood 2008, 111, 1840–1847. [Google Scholar] [CrossRef]
- Hillmen, P.; Muus, P.; Röth, A.; Elebute, M.O.; Risitano, A.M.; Schrezenmeier, H.; Szer, J.; Browne, P.; Maciejewski, J.P.; Schubert, J.; et al. Long-term safety and efficacy of sustained eculizumab treatment in patients with paroxysmal nocturnal haemoglobinuria. Br. J. Haematol. 2013, 162, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Risitano, A.M.; Notaro, R.; Marando, L.; Serio, B.; Ranaldi, D.; Seneca, E.; Ricci, P.; Alfinito, F.; Camera, A.; Gianfaldoni, G.; et al. Complement fraction 3 binding on erythrocytes as additional mechanism of disease in paroxysmal nocturnal hemoglobinuria patients treated by eculizumab. Blood 2009, 113, 4094–4100. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Schmidt, C.Q.; Koutsogiannaki, S.; Ricci, P.; Risitano, A.M.; Lambris, J.D.; Ricklin, D. Complement C3dg-mediated erythrophagocytosis: Implications for paroxysmal nocturnal hemoglobinuria. Blood 2015, 126, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Harder, M.J.; Kuhn, N.; Schrezenmeier, H.; Höchsmann, B.; von Zabern, I.; Weinstock, C.; Simmet, T.; Ricklin, D.; Lambris, J.D.; Skerra, A.; et al. Incomplete inhibition by eculizumab: Mechanistic evidence for residual C5 activity during strong complement activation. Blood 2017, 129, 970–980. [Google Scholar] [CrossRef]
- Gerber, G.F.; Brodsky, R.A. Pegcetacoplan for paroxysmal nocturnal hemoglobinuria. Blood 2022, 139, 3361–3365. [Google Scholar] [CrossRef]
- Nakayama, H.; Usuki, K.; Echizen, H.; Ogawa, R.; Orii, T. Eculizumab dosing intervals longer than 17 days may be associated with greater risk of breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria. Biol. Pharm. Bull. 2016, 39, 285–288. [Google Scholar] [CrossRef]
- Notaro, R.; Luzzatto, L. Breakthrough hemolysis in PNH with proximal or terminal complement inhibition. N. Engl. J. Med. 2022, 387, 160–166. [Google Scholar] [CrossRef]
- Brodsky, R.A.; Peffault de Latour, R.; Rottinghaus, S.T.; Röth, A.; Risitano, A.M.; Weitz, I.C.; Hillmen, P.; Maciejewski, J.P.; Szer, J.; Lee, J.W.; et al. Characterization of breakthrough hemolysis events observed in the phase 3 randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica 2021, 106, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Peffault de Latour, R.; Fremeaux-Bacchi, V.; Porcher, R.; Xhaard, A.; Rosain, J.; Castaneda, D.C.; Vieira-Martins, P.; Roncelin, S.; Rodriguez-Otero, P.; Plessier, A.; et al. Assessing complement blockade in patients with paroxysmal nocturnal hemoglobinuria receiving eculizumab. Blood 2015, 125, 775–783. [Google Scholar] [CrossRef]
- Fattizzo, B.; Metafuni, E.; Beggiato, E.; Giai, V.; Oliva, E.; Ricchiuti, A.; Bortolotti, M.; Raso, S.; Carlino, D.; Memoli, M.; et al. Breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria treated with complement inhibitors: A multicenter international study. HemaSphere 2024, 8, 1445–1446. [Google Scholar]
- Risitano, A.M.; Ricklin, D.; Huang, Y.; Reis, E.S.; Chen, H.; Ricci, P.; Lin, Z.; Pascariello, C.; Raia, M.; Sica, M.; et al. Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood 2014, 123, 2094–2101, Correction in: Blood 2017, 129, 2205. [Google Scholar] [CrossRef]
- Sahu, A.; Kay, B.K.; Lambris, J.D. Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J. Immunol. 1996, 157, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Lambris, J.D. Compstatin: A complement inhibitor on its way to clinical application. Adv. Exp. Med. Biol. 2008, 632, 273–292. [Google Scholar] [CrossRef] [PubMed]
- de Castro, C.; Grossi, F.; Weitz, I.C.; Maciejewski, J.; Sharma, V.; Roman, E.; Brodsky, R.A.; Tan, L.; Di Casoli, C.; El Mehdi, D.; et al. C3 inhibition with pegcetacoplan in subjects with paroxysmal nocturnal hemoglobinuria treated with eculizumab. Am. J. Hematol. 2020, 95, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.; Xue, X.; Smieško, M.; van Son, H.; Wagner, B.; Berger, N.; Sfyroera, G.; Gros, P.; Lambris, J.D.; Ricklin, D. Insight into mode-of-action and structural determinants of the compstatin family of clinical complement inhibitors. Nat. Commun. 2022, 13, 5519. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Yancopoulou, D.; Kokkinos, P.; Huber-Lang, M.; Hajishengallis, G.; Biglarnia, A.R.; Lupu, F.; Nilsson, B.; Risitano, A.M.; Ricklin, D.; et al. Compstatin: A C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur. J. Clin. Investig. 2015, 45, 423–440. [Google Scholar] [CrossRef]
- Peffault de Latour, R.; Szer, J.; Weitz, I.; Röth, A.; Höchsmann, B.; Panse, J.; Usuki, K.; Griffin, M.; Kiladijan, J.-J.; de Castro, C.M.; et al. Forty-eight week efficacy and safety of pegcetacoplan in adult patients with paroxysmal nocturnal hemoglobinuria and suboptimal response to prior eculizumab treatment. HemaSphere 2021, 5, 42–43. [Google Scholar]
- Fakhouri, F.; Bomback, A.; Kavanagh, D.; Remuzzi, G.; Sunder-Plassmann, G.; Kanellis, J.; Daina, E.; Walker, P.; Wang, Z.; Ahmad, Z. Pegcetacoplan maintains early improvements seen in post-transplant recurrent C3G and IC-MPGN: 1-year results from the phase II NOBLE trial. Nephrol. Dial. Transplant. 2024, 39, i727–i728. [Google Scholar]
- Wong, R.S.M.; Pullon, H.W.H.; Amine, I.; Bogdanovic, A.; Deschatelets, P.; Francois, C.G.; Ignatova, K.; Issaragrisil, S.; Niparuck, P.; Numbenjapon, T.; et al. Inhibition of C3 with pegcetacoplan results in normalization of hemolysis markers in paroxysmal nocturnal hemoglobinuria. Ann. Hematol. 2022, 101, 1971–1986. [Google Scholar] [CrossRef]
- Hillmen, P.; Szer, J.; Weitz, I.; Röth, A.; Höchsmann, B.; Panse, J.; Usuki, K.; Griffin, M.; Kiladjian, J.J.; de Castro, C.; et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2021, 384, 1028–1037, Correction in: N. Engl. J. Med. 2024, 390, 1060. [Google Scholar] [CrossRef]
- de Latour, R.P.; Szer, J.; Weitz, I.C.; Röth, A.; Höchsmann, B.; Panse, J.; Usuki, K.; Griffin, M.; Kiladjian, J.J.; de Castro, C.M.; et al. Pegcetacoplan versus eculizumab in patients with paroxysmal nocturnal haemoglobinuria (PEGASUS): 48-week follow-up of a randomised, open-label, phase 3, active-comparator, controlled trial. Lancet Haematol. 2022, 9, e648–e659. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.M.; Navarro-Cabrera, J.R.; Comia, N.S.; Goh, Y.T.; Idrobo, H.; Kongkabpan, D.; Gómez-Almaguer, D.; Al-Adhami, M.; Ajayi, T.; Alvarenga, P.; et al. Pegcetacoplan controls hemolysis in complement inhibitor-naive patients with paroxysmal nocturnal hemoglobinuria. Blood Adv. 2023, 7, 2468–2478. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.; Andreoni, J.; Densen, P. Complement deficiency states and meningococcal disease. Immunol. Res. 1993, 12, 295–311. [Google Scholar] [CrossRef]
- Socié, G.; Caby-Tosi, M.P.; Marantz, J.L.; Cole, A.; Bedrosian, C.L.; Gasteyger, C.; Mujeebuddin, A.; Hillmen, P.; Vande Walle, J.; Haller, H. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br. J. Haematol. 2019, 185, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.B.; Ma, M.; Hsu, N.C.; Carroll, M.C. Local synthesis of C3 within the splenic lymphoid compartment can reconstitute the impaired immune response in C3-deficient mice. J. Immunol. 1998, 160, 2619–2625. [Google Scholar] [CrossRef]
- Verschoor, A.; Brockman, M.A.; Gadjeva, M.; Knipe, D.M.; Carroll, M.C. Myeloid C3 determines induction of humoral responses to peripheral herpes simplex virus infection. J. Immunol. 2003, 171, 5363–5371. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.L.; Heurich, M.; Rodriguez de Cordoba, S.; Morgan, B.P. The complotype: Dictating risk for inflammation and infection. Trends Immunol. 2012, 33, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Panse, J.; Kelly, R.J.; Nishimori, H.; Horneff, R.; Hillmen, P.; Uchendu, U.; Zhang, D.; Lallier, S.; Gerber, G. Thrombosis and meningococcal infection rates in pegcetacoplan patients with paroxysmal nocturnal hemoglobinuria in the post-marketing setting. HemaSphere 2024, 8, 1471–1472. [Google Scholar]
- Griffin, M.; Kelly, R.J.; Panse, J.; de Castro, C.; Szer, J.; Horneff, R.; Tan, L.; Yeh, M.; Peffault de Latour, R. Management of acute breakthrough hemolysis with intensive pegcetacoplan dosing in patients with PNH. Blood Adv. 2024, 8, 1776–1786. [Google Scholar] [CrossRef]
- Peffault de Latour, R.; de Castro, C.; Mulherin, B.; Patriquin, C.J.; Selvaratnam, V.; Kelly, R.J.; Griffin, M.; Surova, E.; Szamosi, J.; Uchendu, U.; et al. Characterization of clinically significant breakthrough hemolysis in patients with paroxysmal nocturnal hemoglobinuria treated with pegcetacoplan. HemaSphere 2024, 8, 1433–1434. [Google Scholar]
- Hill, A.; Kelly, R.J.; Hillmen, P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood 2013, 121, 4985–5105. [Google Scholar] [CrossRef]
- Kelly, R.J.; Nishimori, H.; Horneff, R.; Hillmen, P.; Al-Adhami, M.; Lallier, S.; Gerber, G.F. Thrombosis and meningococcal infection rates in pegcetacoplan-treated patients with paroxysmal nocturnal hemoglobinuria in the clinical trial and postmarketing settings. Res. Pract. Thromb. Haemost. 2024, 8, 102416. [Google Scholar] [CrossRef]
- Caravaca-Fontán, F.; Lucientes, L.; Cavero, T.; Praga, M. Update on C3 glomerulopathy: A complement-mediated disease. Nephron 2020, 144, 272–280. [Google Scholar] [CrossRef]
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Mac Sweeney, A.; Liao, S.M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef] [PubMed]
- Peffault de Latour, R.; Röth, A.; Kulasekararaj, A.G.; Han, B.; Scheinberg, P.; Maciejewski, J.P.; Ueda, Y.; de Castro, C.M.; Di Bona, E.; Fu, R.; et al. Oral iptacopan monotherapy in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2024, 390, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Harboe, M.; Ulvund, G.; Vien, L.; Fung, M.; Mollnes, T.E. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin. Exp. Immunol. 2004, 138, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Harboe, M.; Garred, P.; Karlstrøm, E.; Lindstad, J.K.; Stahl, G.L.; Mollnes, T.E. The down-stream effects of mannan-induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol. Immunol. 2009, 47, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Schmouder, R.; Junge, G.; Milojevic, J.; Nidamarthy, P.K.; Kumatycki, K. Alternative complement pathway pharmacodynamics of iptacopan. Kidney Int. Rep. 2023, 8, S268–S269. [Google Scholar]
- Risitano, A.M.; Kulasekararaj, A.G.; Lee, J.W.; Maciejewski, J.P.; Notaro, R.; Brodsky, R.; Huang, M.; Geffner, M.; Browett, P. Danicopan: An oral complement factor D inhibitor for paroxysmal nocturnal hemoglobinuria. Haematologica 2021, 106, 3188–3197. [Google Scholar] [CrossRef]
- Lee, J.W.; Griffin, M.; Kim, J.S.; Lee Lee, L.W.; Piatek, C.; Nishimura, J.I.; Carrillo Infante, C.; Jain, D.; Liu, P.; Filippov, G.; et al. Addition of danicopan to ravulizumab or eculizumab in patients with paroxysmal nocturnal haemoglobinuria and clinically significant extravascular haemolysis (ALPHA): A double-blind, randomised, phase 3 trial. Lancet Haematol. 2023, 10, e955–e965. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Levels in Untreated Patients with PNH | Physiological Cause |
|---|---|---|
| LDH or LDH ratio (LDH/ULN) (marker of hemolysis) | High often ≥ 1.5 × ULN (up to 10 × ULN) | Increased due to hemolysis of PNH red blood cells (mostly through IVH) |
| Hemoglobin (marker of anemia) | Low (≤10 g/dL) | Decreased due to hemolysis of PNH red blood cells (through IVH and EVH) or bone marrow failure |
| ARC (marker of bone marrow compensation) a | High often > ULN (up to 1.5 × ULN) | Increased due to bone marrow compensation for hemolyzed PNH red blood cells |
| PNH red blood cell clone size | Variable (usually > 5%) | Increased due to PIGA gene somatic mutation and subsequent loss of GPI-linked proteins (e.g., CD59 for red blood cells) |
| PNH granulocyte clone size (diagnostic marker of PNH) | Variable (usually ≥ 10%) |
| Inhibition Target | Drug Name | Approval Date | Indication | Administration Regimen | Molecule | MOA |
|---|---|---|---|---|---|---|
| C5 | Eculizumab [34,35] | 03/2007 | Adults (≥18 years) with PNH | IV infusion every 2 weeks | C5 mAB | Inhibits C5 cleavage to C5a and C5b |
| C5 | Ravulizumab [36,37] | 12/2018 | Adults and children (≥1 month) with PNH | IV infusions every 4 or 8 weeks based on body weight | C5 hmAB | Inhibits C5 cleavage to C5a and C5b |
| C5 | Crovalimab [38] | 06/2024 | Adults and children (≥13 years) with PNH and body weight ≥ 40 kg | SC injection every 4 weeks | C5 hmAB | Inhibits C5 cleavage to C5a and C5b |
| C3 and C3b | Pegcetacoplan [39,40] | 03/2021 | Adults with PNH | SC infusion twice weekly | Pegylated compstatin | Regulates C3 cleavage and generation of downstream effectors |
| Factor B | Iptacopan [41,42] | 12/2023 | Adults with PNH | Orally twice daily | Small peptide | Regulates AP-dependent C3 cleavage and AP amplification loop |
| Factor D | Danicopan [43,44] | 03/2024 | Add-on to ravulizumab or eculizumab for EVH in adults with PNH | Orally 3 times daily | Small peptide | Prevents factor B cleavage to Ba and Bb, regulating C3 cleavage and AP amplification loop |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hillmen, P.; Horneff, R.; Yeh, M.; Kolev, M.; Deschatelets, P. Navigating the Complement Pathway to Optimize PNH Treatment with Pegcetacoplan and Other Currently Approved Complement Inhibitors. Int. J. Mol. Sci. 2024, 25, 9477. https://doi.org/10.3390/ijms25179477
Hillmen P, Horneff R, Yeh M, Kolev M, Deschatelets P. Navigating the Complement Pathway to Optimize PNH Treatment with Pegcetacoplan and Other Currently Approved Complement Inhibitors. International Journal of Molecular Sciences. 2024; 25(17):9477. https://doi.org/10.3390/ijms25179477
Chicago/Turabian StyleHillmen, Peter, Regina Horneff, Michael Yeh, Martin Kolev, and Pascal Deschatelets. 2024. "Navigating the Complement Pathway to Optimize PNH Treatment with Pegcetacoplan and Other Currently Approved Complement Inhibitors" International Journal of Molecular Sciences 25, no. 17: 9477. https://doi.org/10.3390/ijms25179477
APA StyleHillmen, P., Horneff, R., Yeh, M., Kolev, M., & Deschatelets, P. (2024). Navigating the Complement Pathway to Optimize PNH Treatment with Pegcetacoplan and Other Currently Approved Complement Inhibitors. International Journal of Molecular Sciences, 25(17), 9477. https://doi.org/10.3390/ijms25179477