
Single-Cell RNA-Sequencing: Opening New Horizons for Breast Cancer Research


Abstract
:1. Introduction
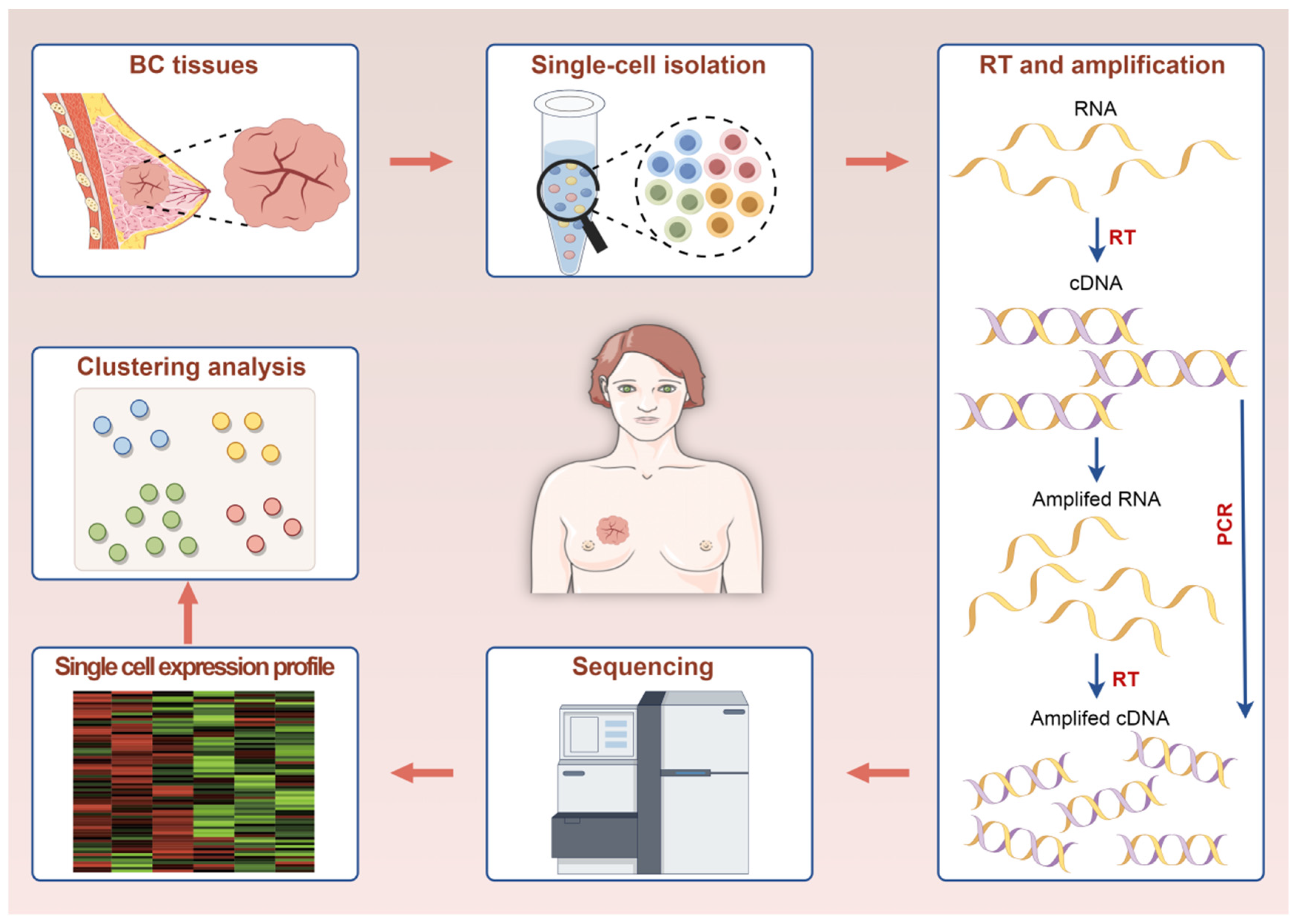
2. The Functional Principle and Workflow of scRNA-Seq
2.1. Single-Cell Isolation
2.2. Reverse Transcription, Amplification, and Sequencing
2.2.1. PCR after polyA Tailing
2.2.2. Template-Switching-Based PCR
2.2.3. In Vitro Transcription (IVT)
2.3. Data Analysis
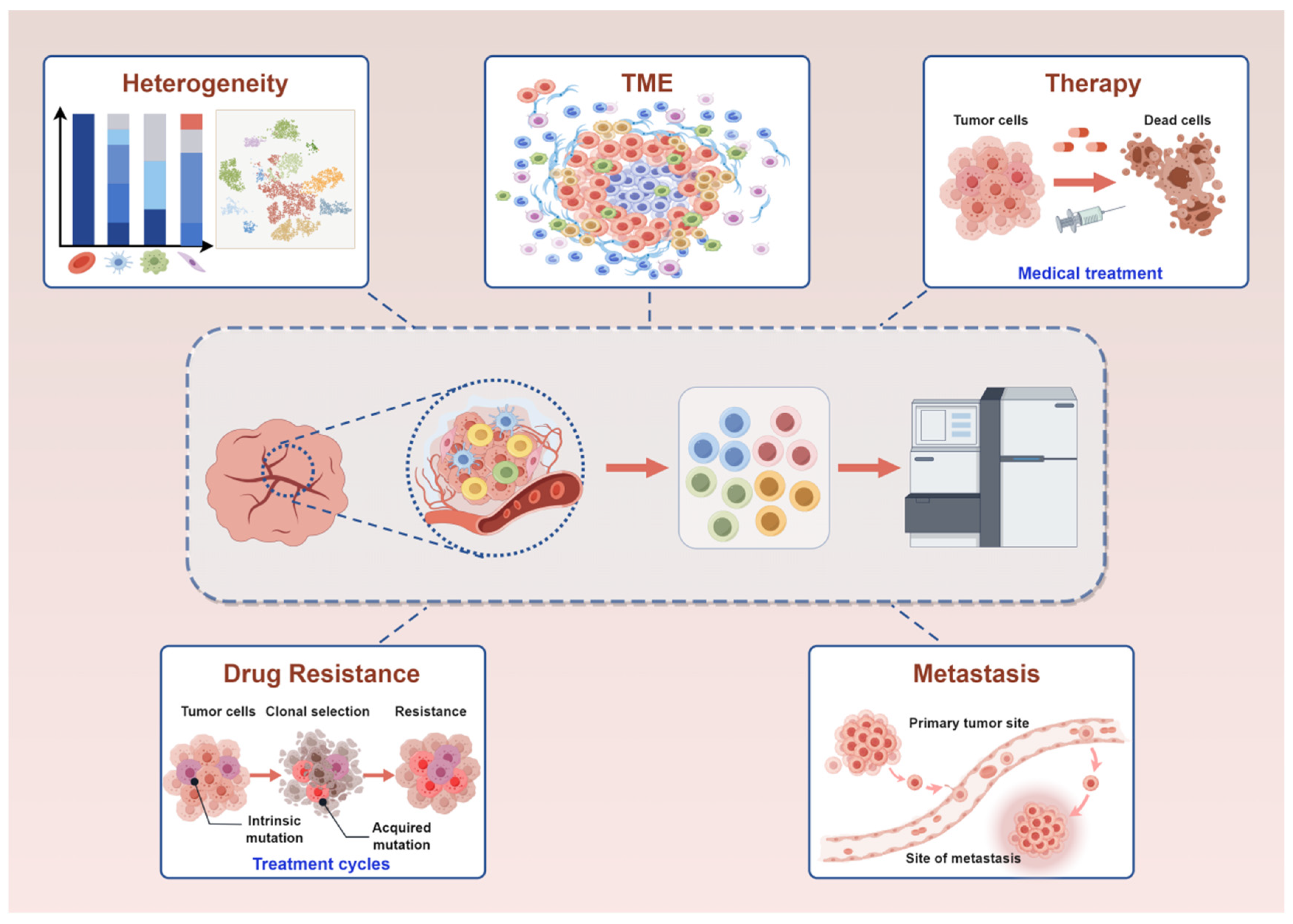
3. Application of scRNA-Seq in Breast Cancer
3.1. Application of scRNA-Seq in Exploring the Heterogeneity of Breast Cancer
3.2. Application of scRNA-Seq in TME of Breast Cancer
3.3. Application of scRNA-Seq in Therapy of Breast Cancer
3.4. Application of scRNA-Seq in Drug Resistance of Breast Cancer
3.5. Application of scRNA-Seq in Metastasis of Breast Cancer
4. Potential Future Directions of scRNA-Seq in Breast Cancer Research
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jiang, G.; Tu, J.; Zhou, L.; Dong, M.; Fan, J.; Chang, Z.; Zhang, L.; Bian, X.; Liu, S. Single-cell transcriptomics reveal the heterogeneity and dynamic of cancer stem-like cells during breast tumor progression. Cell Death Dis. 2021, 12, 979. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, L.; Gathani, T. Understanding breast cancer as a global health concern. Br. J. Radiol. 2022, 95, 20211033. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef]
- Tuasha, N.; Petros, B. Heterogeneity of Tumors in Breast Cancer: Implications and Prospects for Prognosis and Therapeutics. Scientifica 2020, 2020, 4736091. [Google Scholar] [CrossRef]
- Luond, F.; Tiede, S.; Christofori, G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Br. J. Cancer 2021, 125, 164–175. [Google Scholar] [CrossRef]
- Thakur, S.; Haider, S.; Natrajan, R. Implications of tumour heterogeneity on cancer evolution and therapy resistance: Lessons from breast cancer. J. Pathol. 2023, 260, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Deepak, K.G.K.; Vempati, R.; Nagaraju, G.P.; Dasari, V.R.; Nagini, S.; Rao, D.N.; Malla, R.R. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacol. Res. 2020, 153, 104683. [Google Scholar] [CrossRef]
- Manohar, S.M.; Shah, P.; Nair, A. Flow cytometry: Principles, applications and recent advances. Bioanalysis 2021, 13, 181–198. [Google Scholar] [CrossRef]
- Liu, C.C.; Steen, C.B.; Newman, A.M. Computational approaches for characterizing the tumor immune microenvironment. Immunology 2019, 158, 70–84. [Google Scholar] [CrossRef]
- Fincham, R.E.A.; Bashiri, H.; Lau, M.C.; Yeong, J. Editorial: Multiplex Immunohistochemistry/Immunofluorescence Technique: The Potential and Promise for Clinical Application. Front. Mol. Biosci. 2022, 9, 831383. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Nomura, S. Review of Single-Cell RNA Sequencing in the Heart. Int. J. Mol. Sci. 2020, 21, 8345. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mashock, M.; Tong, Z.; Mu, X.; Chen, H.; Zhou, X.; Zhang, H.; Zhao, G.; Liu, B.; Li, X. Changing Technologies of RNA Sequencing and Their Applications in Clinical Oncology. Front. Oncol. 2020, 10, 447. [Google Scholar] [CrossRef]
- Kulkarni, A.; Anderson, A.G.; Merullo, D.P.; Konopka, G. Beyond bulk: A review of single cell transcriptomics methodologies and applications. Curr. Opin. Biotechnol. 2019, 58, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Eberwine, J.; Yeh, H.; Miyashiro, K.; Cao, Y.; Nair, S.; Finnell, R.; Zettel, M.; Coleman, P. Analysis of gene expression in single live neurons. Proc. Natl. Acad. Sci. USA 1992, 89, 3010–3014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, C.; Tian, Y.; Yang, X. Single-Cell RNA Sequencing in Lung Cancer: Revealing Phenotype Shaping of Stromal Cells in the Microenvironment. Front. Immunol. 2021, 12, 802080. [Google Scholar] [CrossRef]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Pensold, D.; Zimmer-Bensch, G. Methods for Single-Cell Isolation and Preparation. In Single-Cell Sequencing and Methylation: Methods and Clinical Applications; Yu, B., Zhang, J., Zeng, Y., Li, L., Wang, X., Eds.; Springer: Singapore, 2020; pp. 7–27. [Google Scholar]
- Foley, J.W.; Zhu, C.; Jolivet, P.; Zhu, S.X.; Lu, P.; Meaney, M.J.; West, R.B. Gene expression profiling of single cells from archival tissue with laser-capture microdissection and Smart-3SEQ. Genome Res. 2019, 29, 1816–1825. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Bravo, B.; Sancho-Bru, P. Laser capture microdissection: Techniques and applications in liver diseases. Hepatol. Int. 2019, 13, 138–147. [Google Scholar] [CrossRef]
- Rao, B.H.; Soucek, P.; Hlavac, V. Laser Capture Microdissection: A Gear for Pancreatic Cancer Research. Int. J. Mol. Sci. 2022, 23, 14566. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk, A.A.; Kim, J.K.; Svensson, V.; Marioni, J.C.; Teichmann, S.A. The technology and biology of single-cell RNA sequencing. Mol. Cell 2015, 58, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, J.; Wu, L.; Guo, J.; Song, Y.; Tian, T.; Wang, W.; Zhu, Z.; Yang, C. Microfluidic Single-Cell Omics Analysis. Small 2020, 16, e1903905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gu, M.L. Single-cell sequencing and its application in breast cancer. Yi Chuan 2020, 42, 250–268. [Google Scholar]
- Tung, P.Y.; Blischak, J.D.; Hsiao, C.J.; Knowles, D.A.; Burnett, J.E.; Pritchard, J.K.; Gilad, Y. Batch effects and the effective design of single-cell gene expression studies. Sci. Rep. 2017, 7, 39921. [Google Scholar] [CrossRef]
- El-Hajjar, L.; Ali, A.F.; Nasr, R.A. Guide to Flow Cytometry: Components, Basic Principles, Experimental Design, and Cancer Research Applications. Curr. Protoc. 2023, 3, e721. [Google Scholar] [CrossRef]
- Ellsworth, D.L.; Blackburn, H.L.; Shriver, C.D.; Rabizadeh, S.; Soon-Shiong, P.; Ellsworth, R.E. Single-cell sequencing and tumorigenesis: Improved understanding of tumor evolution and metastasis. Clin. Transl. Med. 2017, 6, 15. [Google Scholar] [CrossRef]
- Sasagawa, Y.; Nikaido, I.; Hayashi, T.; Danno, H.; Uno, K.D.; Imai, T.; Ueda, H.R. Quartz-Seq: A highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013, 14, R31. [Google Scholar] [CrossRef]
- Sasagawa, Y.; Danno, H.; Takada, H.; Ebisawa, M.; Tanaka, K.; Hayashi, T.; Kurisaki, A.; Nikaido, I. Quartz-Seq2: A high-throughput single-cell RNA-sequencing method that effectively uses limited sequence reads. Genome Biol. 2018, 19, 29. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, X.; Wu, X.; Guo, H.; Hu, Y.; Tang, F.; Huang, Y. Single-cell RNA-seq transcriptome analysis of linear and circular RNAs in mouse preimplantation embryos. Genome Biol. 2015, 16, 148. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.; Zong, C. Single-Cell RNA-Seq by Multiple Annealing and Tailing-Based Quantitative Single-Cell RNA-Seq (MATQ-Seq). Methods Mol. Biol. 2019, 1979, 57–71. [Google Scholar] [PubMed]
- Sheng, K.; Cao, W.; Niu, Y.; Deng, Q.; Zong, C. Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat. Methods 2017, 14, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Picelli, S.; Björklund, Å.K.; Faridani, O.R.; Sagasser, S.; Winberg, G.; Sandberg, R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods 2013, 10, 1096–1098. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Björklund, Å.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Ramsköld, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef]
- Hagemann-Jensen, M.; Ziegenhain, C.; Chen, P.; Ramsköld, D.; Hendriks, G.J.; Larsson, A.J.; Faridani, O.R.; Sandberg, R. Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat. Biotechnol. 2020, 38, 708–714. [Google Scholar] [CrossRef]
- Hahaut, V.; Pavlinic, D.; Carbone, W.; Schuierer, S.; Balmer, P.; Quinodoz, M.; Renner, M.; Roma, G.; Cowan, C.S.; Picelli, S. Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat. Biotechnol. 2023, 40, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.N. Single-Cell Tagged Reverse Transcription (STRT-Seq). Methods Mol. Biol. 2019, 1979, 133–153. [Google Scholar] [PubMed]
- Hochgerner, H.; Lönnerberg, P.; Hodge, R.; Mikes, J.; Heskol, A.; Hubschle, H.; Lin, P.; Picelli, S.; La Manno, G.; Ratz, M.; et al. STRT-seq-2i: Dual-index 5′ single cell and nucleus RNA-seq on an addressable microwell array. Sci. Rep. 2017, 7, 16327. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Hu, C.; Li, H.; Li, X.; Fu, Q.; Czajkowsky, D.M.; Guo, Y.; Shao, Z. Significant improvement in data quality with simplified SCRB-seq. Acta Biochim. Biophys. Sin. 2020, 52, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Bageritz, J.; Raddi, G. Single-Cell RNA Sequencing with Drop-Seq. Methods Mol. Biol. 2019, 1979, 73–85. [Google Scholar] [PubMed]
- Shapiro, E.; Biezuner, T.; Linnarsson, S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013, 14, 618–630. [Google Scholar] [CrossRef]
- Picelli, S. Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol. 2017, 14, 637–650. [Google Scholar] [CrossRef]
- Dal Molin, A.; Di Camillo, B. How to design a single-cell RNA-sequencing experiment: Pitfalls, challenges and perspectives. Brief. Bioinform. 2019, 20, 1384–1394. [Google Scholar] [CrossRef]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef]
- Grun, D.; van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-cell RNA-Seq by multiplexed linear amplification. Cell. Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef]
- Hashimshony, T.; Senderovich, N.; Avital, G.; Klochendler, A.; De Leeuw, Y.; Anavy, L.; Gennert, D.; Li, S.; Livak, K.J.; Rozenblatt-Rosen, O.; et al. CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016, 17, 77. [Google Scholar] [CrossRef]
- Yanai, I.; Hashimshony, T. CEL-Seq2-Single-Cell RNA Sequencing by Multiplexed Linear Amplification. Methods Mol. Biol. 2019, 1979, 45–56. [Google Scholar]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Kenigsberg, E.; Jaitin, D.A.; David, E.; Paul, F.; Tanay, A.; Amit, I. MARS-seq2.0: An experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat. Protoc. 2019, 14, 1841–1862. [Google Scholar] [CrossRef]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef]
- Zappia, L.; Phipson, B.; Oshlack, A. Exploring the single-cell RNA-seq analysis landscape with the scRNA-tools database. PLoS Comput. Biol. 2018, 14, e1006245. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Campbell, K.R.; Lun, A.T.; Wills, Q.F. Scater: Pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics 2017, 33, 1179–1186. [Google Scholar] [CrossRef]
- Wolf, F.A.; Angerer, P.; Theis, F.J. SCANPY: Large-scale single-cell gene expression data analysis. Genome Biol. 2018, 19, 15. [Google Scholar] [CrossRef]
- Slovin, S.; Carissimo, A.; Panariello, F.; Grimaldi, A.; Bouché, V.; Gambardella, G.; Cacchiarelli, D. Single-Cell RNA Sequencing Analysis: A Step-by-Step Overview. Methods Mol. Biol. 2021, 2284, 343–365. [Google Scholar] [PubMed]
- Su, M.; Pan, T.; Chen, Q.Z.; Zhou, W.W.; Gong, Y.; Xu, G.; Yan, H.Y.; Li, S.; Shi, Q.Z.; Zhang, Y.; et al. Data analysis guidelines for single-cell RNA-seq in biomedical studies and clinical applications. Mil. Med. Res. 2022, 9, 68. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Sharon, N.; Bar-Joseph, Z. Temporal modelling using single-cell transcriptomics. Nat. Rev. Genet. 2022, 23, 355–368. [Google Scholar] [CrossRef]
- Cheng, C.; Chen, W.; Jin, H.; Chen, X. A Review of Single-Cell RNA-Seq Annotation, Integration, and Cell-Cell Communication. Cells 2023, 12, 1970. [Google Scholar] [CrossRef] [PubMed]
- Muciño-Olmos, E.A.; Vázquez-Jiménez, A.; Avila-Ponce de León, U.; Matadamas-Guzman, M.; Maldonado, V.; López-Santaella, T.; Hernández-Hernández, A.; Resendis-Antonio, O. Unveiling functional heterogeneity in breast cancer multicellular tumor spheroids through single-cell RNA-seq. Sci. Rep. 2020, 10, 12728. [Google Scholar] [CrossRef]
- Gray, G.K.; Li, C.M.C.; Rosenbluth, J.M.; Selfors, L.M.; Girnius, N.; Lin, J.R.; Schackmann, R.C.; Goh, W.L.; Moore, K.; Shapiro, H.K.; et al. A human breast atlas integrating single-cell proteomics and transcriptomics. Dev. Cell 2022, 57, 1400–1420. [Google Scholar] [CrossRef]
- Kumar, T.; Nee, K.; Wei, R.; He, S.; Nguyen, Q.H.; Bai, S.; Blake, K.; Pein, M.; Gong, Y.; Sei, E. A spatially resolved single-cell genomic atlas of the adult human breast. Nature 2023, 620, 181–191. [Google Scholar] [CrossRef]
- Liu, S.Q.; Gao, Z.J.; Wu, J.; Zheng, H.M.; Li, B.; Sun, S.; Meng, X.Y.; Wu, Q. Single-cell and spatially resolved analysis uncovers cell heterogeneity of breast cancer. J. Hematol. Oncol. 2022, 15, 19. [Google Scholar] [CrossRef]
- Wu, S.Z.; Al-Eryani, G.; Roden, D.L.; Junankar, S.; Harvey, K.; Andersson, A.; Thennavan, A.; Wang, C.; Torpy, J.R.; Bartonicek, N.; et al. A single-cell and spatially resolved atlas of human breast cancers. Nat. Genet. 2021, 53, 1334–1347. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, X. Classification of triple-negative breast cancer based on pathway enrichment levels. Med. Oncol. 2023, 40, 157. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.E.; Yang, P.; Chen, S.; Wei, G.; Yuan, L.; Yang, Z.; Gong, L.; He, L.; Yang, L.; Peng, S.; et al. Clinical and biological heterogeneities in triple-negative breast cancer reveals a non-negligible role of HER2-low. Breast Cancer Res. 2023, 25, 34. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Liu, W.; Yan, M.; Ren, Y.; Qian, C.; Fu, Y.; Wang, H.; Li, Z. Unveiling heterogeneity and prognostic markers in ductal breast cancer through single-cell RNA-seq. Cancer Cell Int. 2024, 24, 266. [Google Scholar] [CrossRef] [PubMed]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Weinberg, R.A. Cancer cell of origin: Spotlight on luminal progenitors. Cell Stem Cell 2010, 7, 271–272. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308. [Google Scholar] [CrossRef]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef]
- Ding, S.; Qiao, N.; Zhu, Q.; Tong, Y.; Wang, S.; Chen, X.; Tian, Q.; Xiao, Y.; Shen, K. Single-cell atlas reveals a distinct immune profile fostered by T cell-B cell crosstalk in triple negative breast cancer. Cancer Commun. 2023, 43, 661–684. [Google Scholar] [CrossRef]
- Rebuffet, L.; Melsen, J.E.; Escalière, B.; Basurto-Lozada, D.; Bhandoola, A.; Björkström, N.K.; Bryceson, Y.T.; Castriconi, R.; Cichocki, F.; Colonna, M.; et al. High-dimensional single-cell analysis of human natural killer cell heterogeneity. Nat. Immunol. 2024, 25, 1474–1488. [Google Scholar] [CrossRef]
- Mao, J.; Liu, L.L.; Shen, Q.; Cen, M. Integrating single-cell transcriptomics and machine learning to predict breast cancer prognosis: A study based on natural killer cell-related genes. J. Cell. Mol. Med. 2024, 28, e18549. [Google Scholar] [CrossRef]
- Xu, M.; Zhang, T.; Xia, R.; Wei, Y.; Wei, X. Targeting the tumor stroma for cancer therapy. Mol. Cancer 2022, 21, 208. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yang, C.; Peng, A.; Sun, T.; Ji, X.; Mi, J.; Wei, L.; Shen, S.; Feng, Q. Pan-cancer spatially resolved single-cell analysis reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment. Mol. Cancer 2023, 22, 170. [Google Scholar] [CrossRef]
- Cords, L.; Tietscher, S.; Anzeneder, T.; Langwieder, C.; Rees, M.; de Souza, N.; Bodenmiller, B. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. Nat. Commun. 2023, 14, 4294. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Quan, C.; Wang, Y.; Wu, Z.; Yuan, P.; Xie, N. scRNA-seq characterizing the heterogeneity of fibroblasts in breast cancer reveals a novel subtype SFRP4(+) CAF that inhibits migration and predicts prognosis. Front. Oncol. 2024, 14, 1348299. [Google Scholar] [CrossRef] [PubMed]
- Croizer, H.; Mhaidly, R.; Kieffer, Y.; Gentric, G.; Djerroudi, L.; Leclere, R.; Pelon, F.; Robley, C.; Bohec, M.; Meng, A.; et al. Deciphering the spatial landscape and plasticity of immunosuppressive fibroblasts in breast cancer. Nat. Commun. 2024, 15, 2806. [Google Scholar] [CrossRef] [PubMed]
- Houthuijzen, J.M.; De Bruijn, R.; Van Der Burg, E.; Drenth, A.P.; Wientjens, E.; Filipovic, T.; Bullock, E.; Brambillasca, C.S.; Pulver, E.M.; Nieuwland, M.; et al. CD26-negative and CD26-positive tissue-resident fibroblasts contribute to functionally distinct CAF subpopulations in breast cancer. Nat. Commun. 2023, 14, 183. [Google Scholar] [CrossRef]
- Gambardella, G.; Viscido, G.; Tumaini, B.; Isacchi, A.; Bosotti, R.; Di Bernardo, D. A single-cell analysis of breast cancer cell lines to study tumour heterogeneity and drug response. Nat. Commun. 2022, 13, 1714. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, J.L.; Ang, L.; Li, M.C.; Zhao, M.; Wang, Y.; Wu, Q. Proposing a novel molecular subtyping scheme for predicting distant recurrence-free survival in breast cancer post-neoadjuvant chemotherapy with close correlation to metabolism and senescence. Front. Endocrinol. 2023, 14, 1265520. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, R.; Guo, J.; Pan, L.; Yao, Y.; Gao, J. Integrated single-cell and bulk RNA sequencing analysis identifies a neoadjuvant chemotherapy-related gene signature for predicting survival and therapy in breast cancer. BMC Med. Genom. 2023, 16, 300. [Google Scholar] [CrossRef]
- Mei, J.; Cai, Y.; Chen, L.; Wu, Y.; Liu, J.; Qian, Z.; Jiang, Y.; Zhang, P.; Xia, T.; Pan, X.; et al. The heterogeneity of tumour immune microenvironment revealing the CRABP2/CD69 signature discriminates distinct clinical outcomes in breast cancer. Br. J. Cancer 2023, 129, 1645–1657. [Google Scholar] [CrossRef]
- Deng, J.; Thennavan, A.; Shah, S.; Bagdatlioglu, E.; Klar, N.; Heguy, A.; Marier, C.; Meyn, P.; Zhang, Y.; Labbe, K.; et al. Serial single-cell profiling analysis of metastatic TNBC during Nab-paclitaxel and pembrolizumab treatment. Breast Cancer Res. Treat. 2021, 185, 85–94. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Mo, H.; Hu, X.; Gao, R.; Zhao, Y.; Liu, B.; Niu, L.; Sun, X.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Brady, S.W.; McQuerry, J.A.; Qiao, Y.; Piccolo, S.R.; Shrestha, G.; Jenkins, D.F.; Layer, R.M.; Pedersen, B.S.; Miller, R.H.; Esch, A.; et al. Combating subclonal evolution of resistant cancer phenotypes. Nat. Commun. 2017, 8, 1231. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Li, J.; Wang, C.; Lou, Z.; Gao, S.; Zhao, L.; Wang, S.; Chaulagain, A.; Zhang, M.; Li, X.; et al. Single cell RNA sequencing for breast cancer: Present and future. Cell Death Discov. 2021, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Shaath, H.; Vishnubalaji, R.; Elango, R.; Khattak, S.; Alajez, N.M. Single-cell long noncoding RNA (lncRNA) transcriptome implicates MALAT1 in triple-negative breast cancer (TNBC) resistance to neoadjuvant chemotherapy. Cell Death Discov. 2021, 7, 23. [Google Scholar] [CrossRef]
- Prieto-Vila, M.; Usuba, W.; Takahashi, R.U.; Shimomura, I.; Sasaki, H.; Ochiya, T.; Yamamoto, Y. Single-Cell Analysis Reveals a Preexisting Drug-Resistant Subpopulation in the Luminal Breast Cancer Subtype. Cancer Res. 2019, 79, 4412–4425. [Google Scholar] [CrossRef]
- Gao, Y.; Li, X.; Zeng, C.; Liu, C.; Hao, Q.; Li, W.; Zhang, K.; Zhang, W.; Wang, S.; Zhao, H.; et al. CD63(+) Cancer-Associated Fibroblasts Confer Tamoxifen Resistance to Breast Cancer Cells through Exosomal miR-22. Adv. Sci. 2020, 7, 2002518. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Zhang, X.; Lu, X.; Ma, X.; Guo, X.; Shi, C.; Ren, X.; Ma, X.; He, Y.; Gao, Y.; et al. PDPN positive CAFs contribute to HER2 positive breast cancer resistance to trastuzumab by inhibiting antibody-dependent NK cell-mediated cytotoxicity. Drug Resist. Updates 2023, 68, 100947. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, N.; Wang, S.; Yang, X.; Wang, X.; Xu, B. Concurrent mutations associated with trastuzumab-resistance revealed by single cell sequencing. Breast Cancer Res. Treat. 2021, 187, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Niu, M.; Wu, Y.; Ge, H.; Jiao, D.; Zhu, S.; Zhang, J.; Yan, Y.; Zhou, P.; Chu, Q.; et al. Combination of oral STING agonist MSA-2 and anti-TGF-beta/PD-L1 bispecific antibody YM101: A novel immune cocktail therapy for non-inflamed tumors. J. Hematol. Oncol. 2022, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wang, R.; Xie, H.; Hu, L.; Wang, C.; Xu, J.; Zhu, C.; Liu, Y.; Gao, F.; Li, X.; et al. Single-cell RNA sequencing reveals cell heterogeneity and transcriptome profile of breast cancer lymph node metastasis. Oncogenesis 2021, 10, 66. [Google Scholar] [CrossRef]
- Xu, K.; Zhang, W.; Wang, C.; Hu, L.; Wang, R.; Wang, C.; Tang, L.; Zhou, G.; Zou, B.; Xie, H.; et al. Integrative analyses of scRNA-seq and scATAC-seq reveal CXCL14 as a key regulator of lymph node metastasis in breast cancer. Hum. Mol. Genet. 2021, 30, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Ge, J.Y.; Chen, Y.F.; Liu, T.; Chen, L.; Liu, C.C.; Ma, D.; Chen, Y.Y.; Cai, Y.W.; Xu, Y.Y.; et al. Combined Single-Cell and Spatial Transcriptomics Reveal the Metabolic Evolvement of Breast Cancer during Early Dissemination. Adv. Sci. 2023, 10, e2205395. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz-Krzywiec, P.; Martín-Pérez, R.; Ehling, M.; García-Caballero, M.; Pinioti, S.; Pretto, S.; Kroes, R.; Aldeni, C.; Di Matteo, M.; Prenen, H.; et al. Podoplanin-Expressing Macrophages Promote Lymphangiogenesis and Lymphoinvasion in Breast Cancer. Cell Metab. 2019, 30, 917–936. [Google Scholar] [CrossRef]
- Shen, J.; Ma, H.; Chen, Y.; Shen, J. ScRNA-seq reveals the correlation between M2 phenotype of tumor-associated macrophages and lymph node metastasis of breast cancer. Oncol. Res. 2023, 31, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Sanjaya, A.; Ratnawati, H.; Adhika, O.A.; Rahmatilah, F.R. The heterogeneity of breast cancer metastasis: A bioinformatics analysis utilizing single-cell RNA sequencing data. Breast Cancer Res. Treat. 2024. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, L.; Wang, Z.; Gu, D.; Zhu, M.; Cai, Y.; Li, L.; Tang, J.; Huang, B.; Bosco, B.; et al. Single-cell transcriptome analysis indicates fatty acid metabolism-mediated metastasis and immunosuppression in male breast cancer. Nat. Commun. 2023, 14, 5590. [Google Scholar] [CrossRef]
- Gulati, G.S.; D’Silva, J.P.; Liu, Y.; Wang, L.; Newman, A.M. Profiling cell identity and tissue architecture with single-cell and spatial transcriptomics. Nat. Rev. Mol. Cell Biol. 2024. [Google Scholar] [CrossRef]
- Stein, C.M.; Weiskirchen, R.; Damm, F.; Strzelecka, P.M. Single-cell omics: Overview, analysis, and application in biomedical science. J. Cell. Biochem. 2021, 122, 1571–1578. [Google Scholar] [CrossRef]
- Srinivasan, S.; Leshchyk, A.; Johnson, N.T.; Korkin, D. A hybrid deep clustering approach for robust cell type profiling using single-cell RNA-seq data. RNA 2020, 26, 1303–1319. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, Y.; Zou, Q. Inferring gene regulatory network from single-cell transcriptomes with graph autoencoder model. PLoS Genet. 2023, 19, e1010942. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Method | RNA-Capture | Transcript Coverage | UMI | Amplification Technology |
|---|---|---|---|---|
| Tang | polyA | full length | No | PCR after polyA tailing |
| Quartz-seq | polyA | full length | No | |
| Quartz-seq2 | polyA | full length | Yes | |
| SUPeR-seq | polyA | full length | No | |
| MATQ-seq | polyA | full length | Yes | |
| SMART-seq | polyA | full length | No | Template-switching-based PCR |
| SMART-seq2 | polyA | full length | No | |
| SMART-seq3 | polyA | full length | Yes | |
| FLASH-seq | polyA | full length | Yes | |
| STRT-seq | polyA | 5′ tag | Yes | |
| STRT-seq-2i | polyA | 5′ tag | Yes | |
| SCRB-seq | polyA | 3′ tag | Yes | |
| Drop-seq | polyA | 3′ tag | Yes | |
| CEL-seq | polyA | 3′ tag | Yes | In vitro transcription(IVT) |
| CEL-seq2 | polyA | 3′ tag | Yes | |
| MARS-seq | polyA | 3′ tag | Yes | |
| MARS-seq2.0 | polyA | 3′ tag | Yes | |
| In Drops | polyA | 3′ tag | Yes |
| Applications | Category | Study | Clinical Significance | References |
|---|---|---|---|---|
| Heterogeneity | Heterogeneity within normal breast tissues |
|
| [67,68] |
| Heterogeneity within breast tumors |
| Offering insights into the refined classification and tailored therapies for breast cancer. | [69,70,71,72] | |
| Heterogeneity between breast cancer malignant cells and reference normal epithelial cells | Revealing evolution mimicry during the specification of breast cancer subtype. | Revealing the origin of tumor cells and providing a foundation for accurate prognostic and therapeutic stratification of breast cancer. | [73] | |
| Heterogeneity among breast cancer cell lines | Investigation of the functional relationship among different cell subtypes in breast cancer cell lines and how this interdependence contributes to tumor development. | Highlighting the systemic nature of cancer and task stratification of cell populations to maintain tumor hallmarks. | [66] | |
| Heterogeneity in gene expression within each tumor | Revealing the phenotypes and biology underlying the genetic evolution and clinical behavior of TNBC. | Highlighting the connection between the functional heterogeneity of TNBC and genomic evolution, and revealing the biological principles that lead to the poor prognosis of TNBC. | [74] | |
| TME | Tumor immune microenvironment(T cells, B cells, macrophages, NK cells) |
|
| [76,77,78,79,80,81] |
| Tumor interstitial microenvironment (CAFs) |
|
| [83,84,85,86,87] | |
| Therapy | Drug sensitivity | Predicting drug sensitivity | Guiding personalized drug treatment for patients. | [88] |
| Predictive markers for NAT | Screening for biomarkers associated with the prognostic response to NAT. | Enabling the identification of subgroups of breast cancer patients who are likely to benefit from NAT. | [89,90,91] | |
| Chemotherapy combined with immunotherapy | Analyses on the changes in the immune microenvironment and immune cell dynamics of breast cancer resulting from chemotherapy combined with immunotherapy. | Highlighting the role and concerns of specific immune cells in combined therapy, which could potentially provide important clues for individualized treatment. | [92,93] | |
| Drug resistance | Drug resistance of TNBC |
|
| [96,97] |
| Drug resistance of luminal breast cancer |
|
| [98,99] | |
| Drug resistance of HER2-positive breast cancer |
|
| [100,101] | |
| Drug resistance of non-inflammatory breast cancer | The role of combined application of MSA-2 and YM101 in immune therapy resistance of non-inflammatory tumors. | Providing a new treatment strategy for non-inflammatory tumors. | [102] | |
| Metastasis | Lymph node metastasis in female breast cancer |
|
| [103,104,105,106,107,108] |
| Metastasis in male breast cancer | Metastatic characteristics of male breast cancer. | Providing a new perspective for the research and treatment of male breast cancer. | [109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, L.; Rao, J.; Yuan, J.; Xie, T.; Yan, H. Single-Cell RNA-Sequencing: Opening New Horizons for Breast Cancer Research. Int. J. Mol. Sci. 2024, 25, 9482. https://doi.org/10.3390/ijms25179482
Xiang L, Rao J, Yuan J, Xie T, Yan H. Single-Cell RNA-Sequencing: Opening New Horizons for Breast Cancer Research. International Journal of Molecular Sciences. 2024; 25(17):9482. https://doi.org/10.3390/ijms25179482
Chicago/Turabian StyleXiang, Lingyan, Jie Rao, Jingping Yuan, Ting Xie, and Honglin Yan. 2024. "Single-Cell RNA-Sequencing: Opening New Horizons for Breast Cancer Research" International Journal of Molecular Sciences 25, no. 17: 9482. https://doi.org/10.3390/ijms25179482