
Gastric Cancer and Intestinal Metaplasia: Differential Metabolic Landscapes and New Pathways to Diagnosis

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Patient Characteristics
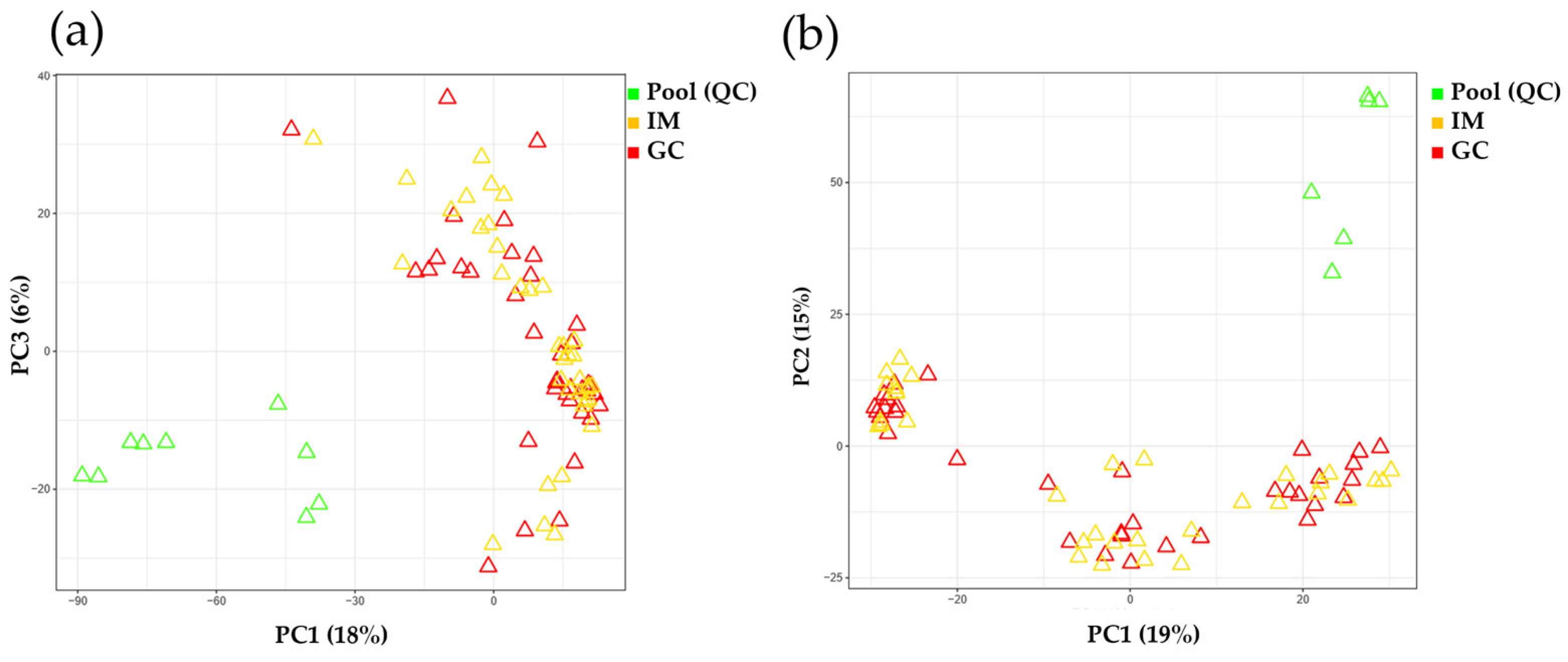
2.2. Analytical Performance Evaluation
2.3. Uni/Multivariate Observation of Metabolic Alterations between IM and GC
2.3.1. Comparison of Aqueous Data
2.3.2. Comparison of Organic Data
2.4. Pathway Enrichment Analysis between IM and GC
2.5. Relative Intensities of Metabolites Significantly Altered by GC
2.6. Alteration of Metabolite Intensities According to GC Stage
3. Discussion
4. Materials and Methods
4.1. Study Design and Patient Enrollment
4.2. Endoscopic Sampling
4.3. Quality Control Samples
4.4. Sample Treatment for Metabolite Extraction
4.5. Analysis of Metabolites by Liquid Chromatography with Tandem Mass Spectrometry
4.6. Alteration of Metabolite Intensities Caused by GC Corrected with Stage
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.S.; Lee, J.; Woo, H.; Shin, D.; Kong, S.H.; Lee, H.J.; Shin, A.; Yang, H.K. National cancer screening program for gastric cancer in Korea: Nationwide treatment benefit and cost. Cancer 2020, 126, 1929–1939. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, N. Review of atrophic gastritis and intestinal metaplasia as a premalignant lesion of gastric cancer. J. Cancer Prev. 2015, 20, 25–40. [Google Scholar] [CrossRef]
- ASGE Standards of Practice Committee; Ben-Menachem, T.; Decker, G.A.; Early, D.S.; Evans, J.; Fanelli, R.D.; Fisher, D.A.; Fisher, L.; Fukami, N.; Hwang, J.H.; et al. Adverse events of upper GI endoscopy. Gastrointest. Endosc. 2012, 76, 707–718. [Google Scholar] [CrossRef]
- Menon, S.; Trudgill, N. How commonly is upper gastrointestinal cancer missed at endoscopy? A meta-analysis. Endosc. Int. Open 2014, 2, E46–E50. [Google Scholar] [CrossRef]
- Kim, S.J.; Choi, C.W. Common Locations of Gastric Cancer: Review of Research from the Endoscopic Submucosal Dissection Era. J. Korean Med. Sci. 2019, 34, e231. [Google Scholar] [CrossRef]
- Emoto, S.; Ishigami, H.; Yamashita, H.; Yamaguchi, H.; Kaisaki, S.; Kitayama, J. Clinical significance of CA125 and CA72-4 in gastric cancer with peritoneal dissemination. Gastric Cancer 2012, 15, 154–161. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Y.; Lu, M.; Shen, L. Predictive value of serum CEA, CA19-9 and CA72.4 in early diagnosis of recurrence after radical resection of gastric cancer. Hepatogastroenterology 2011, 58, 2166–2170. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhou, Y.; Sheng, S.; Qian, S.Y.; Huo, X. Evaluation of Serum CEA, CA19-9, CA72-4, CA125 and Ferritin as Diagnostic Markers and Factors of Clinical Parameters for Colorectal Cancer. Sci. Rep. 2018, 8, 2732. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Zhang, P.; Ma, J.; Xu, J.; Yang, L.; Xu, W.; Que, H.; Chen, M.; Xu, H. Serum biomarker panels for the diagnosis of gastric cancer. Cancer Med. 2019, 8, 1576–1583. [Google Scholar] [CrossRef]
- Cho, J.; Ahn, S.; Son, D.S.; Kim, N.K.; Lee, K.W.; Kim, S.; Lee, J.; Park, S.H.; Park, J.O.; Kang, W.K.; et al. Bridging genomics and phenomics of gastric carcinoma. Int. J. Cancer 2019, 145, 2407–2417. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Choi, S.A.; Na, J.; Pamungkas, A.D.; Jung, K.J.; Jee, S.H.; Park, Y.H. Noninvasive Serum Metabolomic Profiling Reveals Elevated Kynurenine Pathway’s Metabolites in Humans with Prostate Cancer. J. Proteome Res. 2019, 18, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Kos, Z.; Dabbs, D.J. Biomarker assessment and molecular testing for prognostication in breast cancer. Histopathology 2016, 68, 70–85. [Google Scholar] [CrossRef]
- Pandey, R.; Caflisch, L.; Lodi, A.; Brenner, A.J.; Tiziani, S. Metabolomic signature of brain cancer. Mol. Carcinog. 2017, 56, 2355–2371. [Google Scholar] [CrossRef]
- Zeleznik, O.A.; Eliassen, A.H.; Kraft, P.; Poole, E.M.; Rosner, B.A.; Jeanfavre, S.; Deik, A.A.; Bullock, K.; Hitchcock, D.S.; Avila-Pacheco, J.; et al. A Prospective Analysis of Circulating Plasma Metabolites Associated with Ovarian Cancer Risk. Cancer Res. 2020, 80, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Moschetta, M.; Uccello, M.; Kasenda, B.; Mak, G.; McClelland, A.; Boussios, S.; Forster, M.; Arkenau, H.T. Dynamics of Neutrophils-to-Lymphocyte Ratio Predict Outcomes of PD-1/PD-L1 Blockade. BioMed Res. Int. 2017, 2017, 1506824. [Google Scholar] [CrossRef]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef]
- Yoon, H.; Kim, N. Diagnosis and management of high risk group for gastric cancer. Gut Liver 2015, 9, 5–17. [Google Scholar] [CrossRef]
- Xiao, S.; Zhou, L. Gastric cancer: Metabolic and metabolomics perspectives (Review). Int. J. Oncol. 2017, 51, 5–17. [Google Scholar] [CrossRef]
- Hirayama, A.; Kami, K.; Sugimoto, M.; Sugawara, M.; Toki, N.; Onozuka, H.; Kinoshita, T.; Saito, N.; Ochiai, A.; Tomita, M.; et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009, 69, 4918–4925. [Google Scholar] [CrossRef]
- Jung, J.; Jung, Y.; Bang, E.J.; Cho, S.I.; Jang, Y.J.; Kwak, J.M.; Ryu, D.H.; Park, S.; Hwang, G.S. Noninvasive diagnosis and evaluation of curative surgery for gastric cancer by using NMR-based metabolomic profiling. Ann. Surg. Oncol. 2014, 21 (Suppl. S4), S736–S742. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, L.; Liu, H.L.; Wu, X.B.; Wang, H.S.; Liu, Z.H.; Li, Y.; Diao, D.C.; Chen, H.L.; Peng, J.S. Tissue metabolomic fingerprinting reveals metabolic disorders associated with human gastric cancer morbidity. Oncol. Rep. 2011, 26, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhao, J.S.; Li, J.J.; Peng, D.N.; Wang, X.Y.; Chen, T.L.; Qiu, Y.P.; Chen, P.P.; Li, W.J.; Xu, L.Y.; et al. A combined proteomics and metabolomics profiling of gastric cardia cancer reveals characteristic dysregulations in glucose metabolism. Mol. Cell. Proteom. 2010, 9, 2617–2628. [Google Scholar] [CrossRef]
- Aa, J.; Yu, L.; Sun, M.; Liu, L.; Li, M.; Cao, B.; Shi, J.; Xu, J.; Cheng, L.; Zhou, J.; et al. Metabolic features of the tumor microenvironment of gastric cancer and the link to the systemic macroenvironment. Metabolomics 2012, 8, 164–173. [Google Scholar] [CrossRef]
- Venerito, M.; Ford, A.C.; Rokkas, T.; Malfertheiner, P. Review: Prevention and management of gastric cancer. Helicobacter 2020, 25 (Suppl. S1), e12740. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Ji, X.; Ji, J. Prognostic biomarker in advanced gastric cancer. Transl. Gastrointest. Cancer 2016, 5, 16–29. [Google Scholar]
- Necula, L.; Matei, L.; Dragu, D.; Neagu, A.I.; Mambet, C.; Nedeianu, S.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Recent advances in gastric cancer early diagnosis. World J. Gastroenterol. 2019, 25, 2029–2044. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wu, F.; Zhou, Y.; Bao, Z.; Li, H.; Zheng, P.; Zhao, S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell Death Dis. 2019, 10, 918. [Google Scholar] [CrossRef]
- Hassani, B.; Attar, Z.; Firouzabadi, N. The renin-angiotensin-aldosterone system (RAAS) signaling pathways and cancer: Foes versus allies. Cancer Cell Int. 2023, 23, 254. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, J.; Park, R.W.; Shin, S.J.; Kim, J.; Sung, J.D.; Kim, D.J.; Yang, K. Renin-Angiotensin-Aldosterone System Inhibitors and Risk of Cancer: A Population-Based Cohort Study Using a Common Data Model. Diagnostics 2022, 12, 263. [Google Scholar] [CrossRef]
- Busada, J.T.; Ramamoorthy, S.; Cain, D.W.; Xu, X.; Cook, D.N.; Cidlowski, J.A. Endogenous glucocorticoids prevent gastric metaplasia by suppressing spontaneous inflammation. J. Clin. Investig. 2019, 129, 1345–1358. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2,3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Choi, J.M.; Park, W.S.; Song, K.Y.; Lee, H.J.; Jung, B.H. Development of simultaneous analysis of tryptophan metabolites in serum and gastric juice—An investigation towards establishing a biomarker test for gastric cancer diagnosis. Biomed. Chromatogr. 2016, 30, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Chen, G.; Shi, Z.; Yang, J.; Wang, X.; Pan, J.; Zhu, L. Comprehensive multi-omics analysis of tryptophan metabolism-related gene expression signature to predict prognosis in gastric cancer. Front. Pharmacol. 2023, 14, 1267186. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Lamas, B.; Richard, M.L.; Leducq, V.; Pham, H.P.; Michel, M.L.; Da Costa, G.; Bridonneau, C.; Jegou, S.; Hoffmann, T.W.; Natividad, J.M.; et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 2016, 22, 598–605. [Google Scholar] [CrossRef]
- Fu, D.J.; Cui, X.X.; Zhu, T.; Zhang, Y.B.; Hu, Y.Y.; Zhang, L.R.; Wang, S.H.; Zhang, S.Y. Discovery of novel indole derivatives that inhibit NEDDylation and MAPK pathways against gastric cancer MGC803 cells. Bioorganic Chem. 2021, 107, 104634. [Google Scholar] [CrossRef]
- Cao, K.; Lyu, Y.; Chen, J.; He, C.; Lyu, X.; Zhang, Y.; Chen, L.; Jiang, Y.; Xiang, J.; Liu, B.; et al. Prognostic Implication of Plasma Metabolites in Gastric Cancer. Int. J. Mol. Sci. 2023, 24, 12774. [Google Scholar] [CrossRef]
- Cao, F.Y.; Wang, C.H.; Li, X.; Ma, M.Z.; Tao, G.C.; Yang, C.; Li, K.; He, X.B.; Tong, S.L.; Zhao, Q.C.; et al. Guanylate binding protein 5 accelerates gastric cancer progression via the JAK1-STAT1/GBP5/CXCL8 positive feedback loop. Am. J. Cancer Res. 2023, 13, 1310–1328. [Google Scholar] [PubMed]
- Gao, J.; Pan, H.; Zhu, Z.; Yu, T.; Huang, B.; Zhou, Y. Guanine nucleotide-binding protein subunit beta-4 promotes gastric cancer progression via activating Erk1/2. Acta Biochim. Biophys. Sin. 2020, 52, 975–987. [Google Scholar] [CrossRef]
- Allegrini, S.; Garcia-Gil, M.; Pesi, R.; Camici, M.; Tozzi, M.G. The Good, the Bad and the New about Uric Acid in Cancer. Cancers 2022, 14, 4959. [Google Scholar] [CrossRef]
- Miyagi, Y.; Higashiyama, M.; Gochi, A.; Akaike, M.; Ishikawa, T.; Miura, T.; Saruki, N.; Bando, E.; Kimura, H.; Imamura, F.; et al. Plasma free amino acid profiling of five types of cancer patients and its application for early detection. PLoS ONE 2011, 6, e24143. [Google Scholar] [CrossRef]
- Bednarz-Misa, I.; Fleszar, M.G.; Fortuna, P.; Lewandowski, L.; Mierzchala-Pasierb, M.; Diakowska, D.; Krzystek-Korpacka, M. Altered L-Arginine Metabolic Pathways in Gastric Cancer: Potential Therapeutic Targets and Biomarkers. Biomolecules 2021, 11, 1086. [Google Scholar] [CrossRef]
- Wallimann, T.; Tokarska-Schlattner, M.; Schlattner, U. The creatine kinase system and pleiotropic effects of creatine. Amino Acids 2011, 40, 1271–1296. [Google Scholar] [CrossRef]
- Kazak, L.; Cohen, P. Creatine metabolism: Energy homeostasis, immunity and cancer biology. Nat. Rev. Endocrinol. 2020, 16, 421–436. [Google Scholar] [CrossRef] [PubMed]
- Tatsuki, N.; Fumiaki, S.; Takashi, S.; Yoko, O.; Satoshi, Y.; Yuichiro, O.; Hideaki, O.; Kimihiko, F.; Hideaki, S. Urinary N1, N12-Diacetylspermine Level in the Patients with Various Cancer; A Pilot Study in Seven Types of Cancer. Toho J. Med. 2023, 9, 29–35. [Google Scholar]
- Parker, A.L.; Toulabi, L.; Oike, T.; Kanke, Y.; Patel, D.; Tada, T.; Taylor, S.; Beck, J.A.; Bowman, E.; Reyzer, M.L.; et al. Creatine riboside is a cancer cell-derived metabolite associated with arginine auxotrophy. J. Clin. Investig. 2022, 132, e157410. [Google Scholar] [CrossRef] [PubMed]
- Sari, I.N.; Setiawan, T.; Kim, K.S.; Wijaya, Y.T.; Cho, K.W.; Kwon, H.Y. Metabolism and function of polyamines in cancer progression. Cancer Lett. 2021, 519, 91–104. [Google Scholar] [CrossRef]
- Allemang, A.; Lester, C.; Roth, T.; Pfuhler, S.; Peuschel, H.; Kosemund, K.; Mahony, C.; Bergeland, T.; O’Keeffe, L. Assessing the genotoxicity and carcinogenicity of 2-chloroethanol through structure activity relationships and in vitro testing approaches. Food Chem. Toxicol. 2022, 168, 113290. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef]
- Vorkas, P.A.; Isaac, G.; Anwar, M.A.; Davies, A.H.; Want, E.J.; Nicholson, J.K.; Holmes, E. Untargeted UPLC-MS profiling pipeline to expand tissue metabolome coverage: Application to cardiovascular disease. Anal. Chem. 2015, 87, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Park, Y.; Johnson, J.M.; Jones, D.P. apLCMS—Adaptive processing of high-resolution LC/MS data. Bioinformatics 2009, 25, 1930–1936. [Google Scholar] [CrossRef] [PubMed]
- Uppal, K.; Soltow, Q.A.; Strobel, F.H.; Pittard, W.S.; Gernert, K.M.; Yu, T.; Jones, D.P. xMSanalyzer: Automated pipeline for improved feature detection and downstream analysis of large-scale, non-targeted metabolomics data. BMC Bioinform. 2013, 14, 15. [Google Scholar] [CrossRef]
- Uppal, K.; Soltow, Q.A.; Promislow, D.E.; Wachtman, L.M.; Quyyumi, A.A.; Jones, D.P. MetabNet: An R Package for Metabolic Association Analysis of High-Resolution Metabolomics Data. Front. Bioeng. Biotechnol. 2015, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Uppal, K.; Walker, D.I.; Liu, K.; Li, S.; Go, Y.M.; Jones, D.P. Computational Metabolomics: A Framework for the Million Metabolome. Chem. Res. Toxicol. 2016, 29, 1956–1975. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Fujibuchi, W.; Kanehisa, M. Computation with the KEGG pathway database. Biosystems 1998, 47, 119–128. [Google Scholar] [CrossRef]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total (n = 37) |
|---|---|
| Age, median | 62.5 ± 13.9 |
| Sex, n (%) | |
| Male | 23 (62.2) |
| Female | 14 (37.8) |
| Location, n (%) | |
| Upper | 5 (13.5) |
| Middle | 11 (29.7) |
| Lower | 21 (56.8) |
| Stage, n (%) | |
| I | 15 (40.5) |
| II | 7 (18.9) |
| III | 4 (10.8) |
| IV | 11 (29.7) |
| Pathologic differentiation | |
| Poorly | 22 (59.5) |
| Moderately | 13 (35.1) |
| Not applicable | 2 (5.4) |
| Lauren classification | |
| Intestinal | 17 (45.9) |
| Diffuse | 12 (32.4) |
| Mixed | 8 (21.6) |
| Current Helicobacter pylori infection | 26 (70.3) |
| Family history of gastric cancer | 5 (13.5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.J.; Choi, H.S.; Kim, H.; Lee, J.M.; Kim, S.H.; Yoon, J.H.; Keum, B.; Kim, H.J.; Chun, H.J.; Park, Y.H. Gastric Cancer and Intestinal Metaplasia: Differential Metabolic Landscapes and New Pathways to Diagnosis. Int. J. Mol. Sci. 2024, 25, 9509. https://doi.org/10.3390/ijms25179509
Choi SJ, Choi HS, Kim H, Lee JM, Kim SH, Yoon JH, Keum B, Kim HJ, Chun HJ, Park YH. Gastric Cancer and Intestinal Metaplasia: Differential Metabolic Landscapes and New Pathways to Diagnosis. International Journal of Molecular Sciences. 2024; 25(17):9509. https://doi.org/10.3390/ijms25179509
Chicago/Turabian StyleChoi, Seong Ji, Hyuk Soon Choi, Hyunil Kim, Jae Min Lee, Seung Han Kim, Jai Hoon Yoon, Bora Keum, Hyo Jung Kim, Hoon Jai Chun, and Youngja H. Park. 2024. "Gastric Cancer and Intestinal Metaplasia: Differential Metabolic Landscapes and New Pathways to Diagnosis" International Journal of Molecular Sciences 25, no. 17: 9509. https://doi.org/10.3390/ijms25179509