
The Current Landscape of Secondary Malignancies after CAR T-Cell Therapies: How Could Malignancies Be Prevented?


Abstract
:1. Introduction: Current CAR T-Cell Treatment Landscape
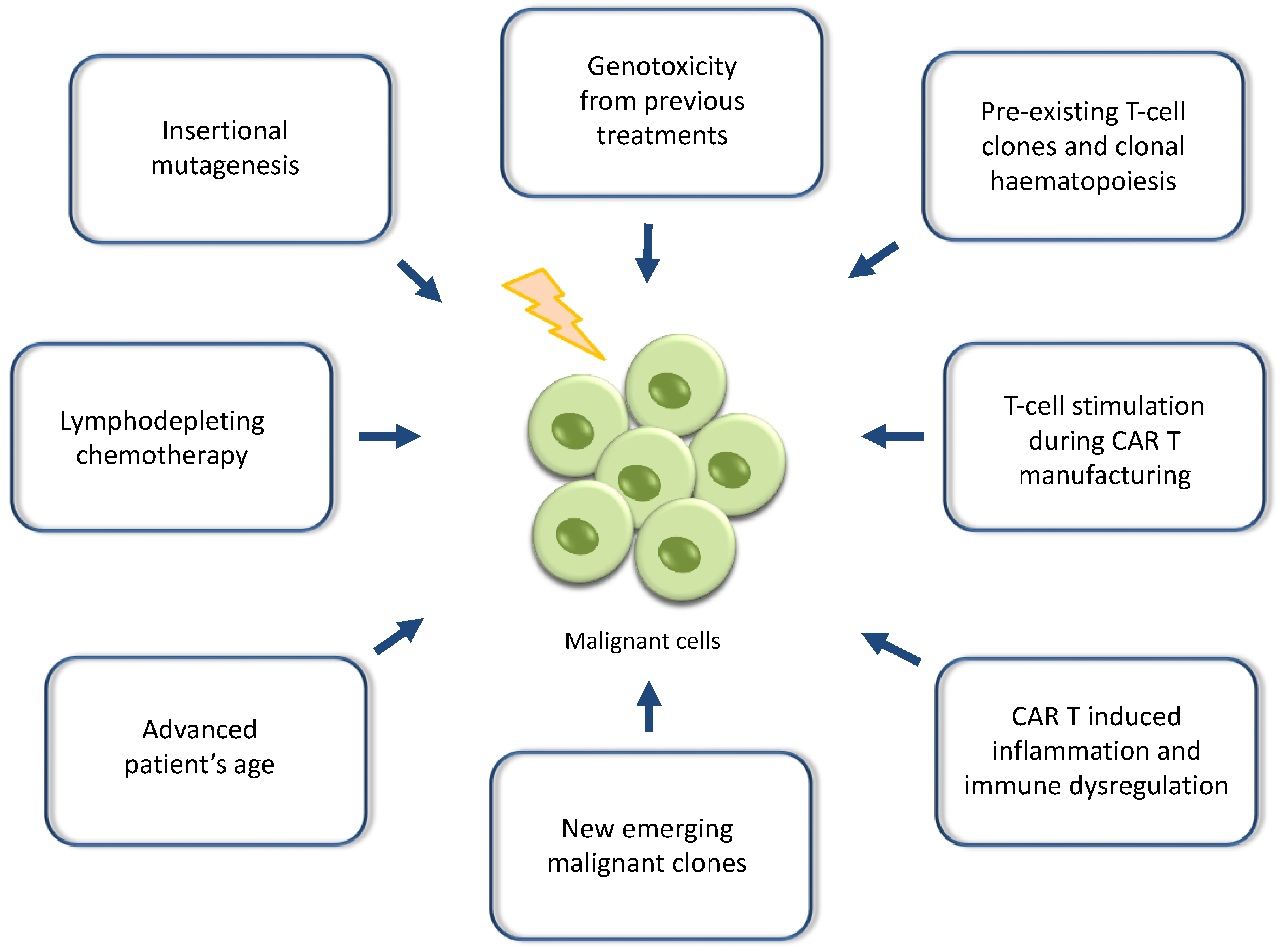
2. Reports of Secondary Malignancies after CAR T-Cell Therapies and Potential Mechanistic Causes
3. Viral Vectors and Genetic Engineering Technologies in Gene Therapy and CAR T-Cell Manufacturing
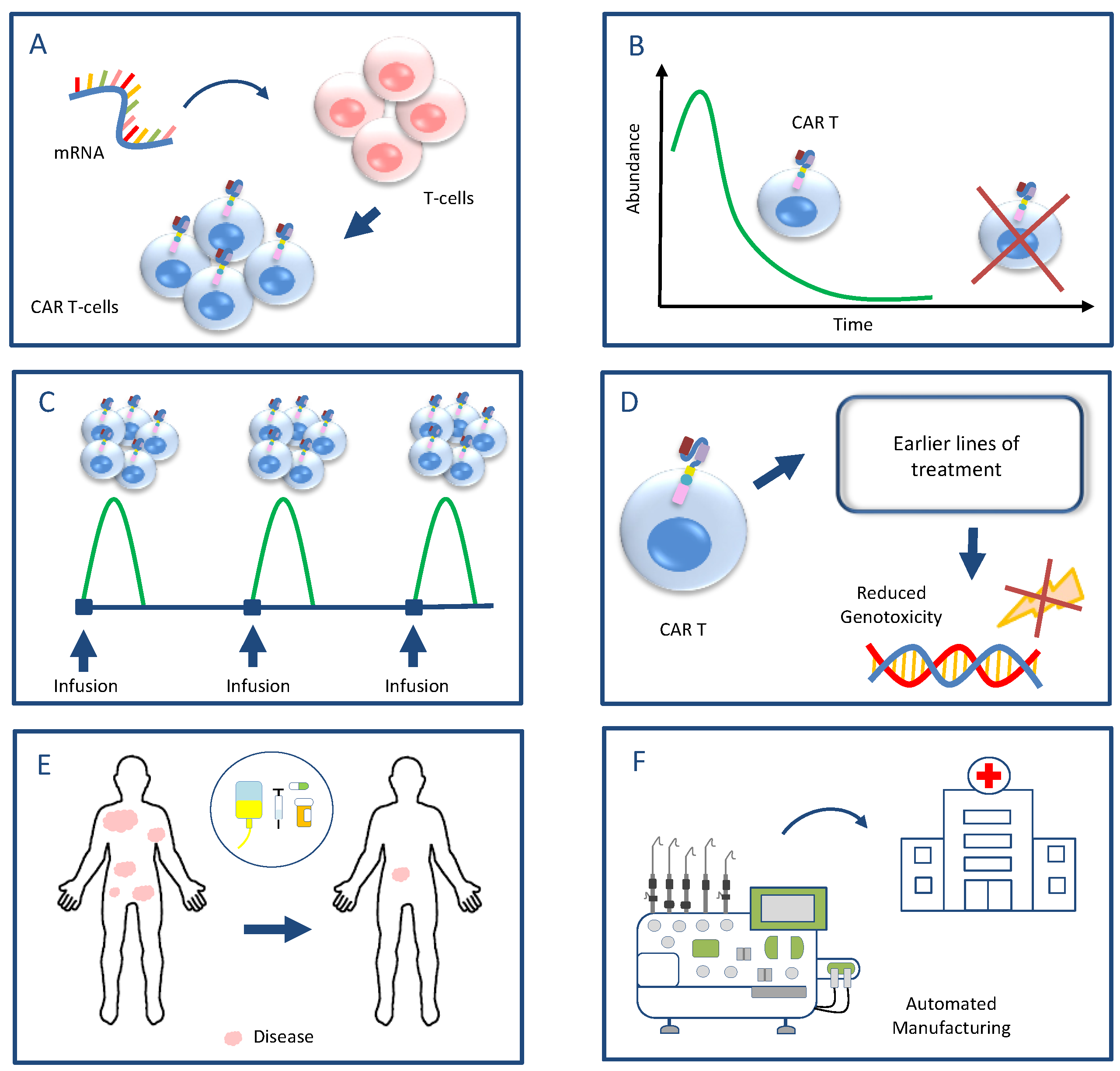
4. A Proposed Strategy for Future Research Aiming to Mitigate the Risk of Secondary Malignancies
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guerra, E.; Di Pietro, R.; Basile, M.; Trerotola, M.; Alberti, S. Cancer-Homing CAR-T Cells and Endogenous Immune Population Dynamics. Int. J. Mol. Sci. 2022, 23, 405. [Google Scholar] [CrossRef] [PubMed]
- Ayala Ceja, M.; Khericha, M.; Harris, C.M.; Puig-Saus, C.; Chen, Y.Y. CAR-T cell manufacturing: Major process parameters and next-generation strategies. J. Exp. Med. 2024, 221, e20230903. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Barua, A.; Huang, L.; Ganguly, S.; Feng, Q.; He, B. From bench to bedside: The history and progress of CAR T cell therapy. Front. Immunol. 2023, 14, 1188049. [Google Scholar] [CrossRef] [PubMed]
- Asmamaw Dejenie, T.; Tiruneh G/Medhin, M.; Dessie Terefe, G.; Tadele Admasu, F.; Wale Tesega, W.; Chekol Abebe, E. Current updates on generations, approvals, and clinical trials of CAR T-cell therapy. Hum. Vaccin. Immunother. 2022, 18, 2114254. [Google Scholar] [CrossRef]
- Korell, F.; Berger, T.R.; Maus, M.V. Understanding CAR T cell-tumor interactions: Paving the way for successful clinical outcomes. Med 2022, 3, 538–564. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Jacobson, C.A.; Ghobadi, A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood 2023, 141, 2307–2315. [Google Scholar] [PubMed]
- Laetsch, T.W.; Maude, S.L.; Rives, S.; Hiramatsu, H.; Bittencourt, H.; Bader, P.; Baruchel, A.; Boyer, M.; De Moerloose, B.; Qayed, M.; et al. Three-Year Update of Tisagenlecleucel in Pediatric and Young Adult Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia in the ELIANA Trial. J. Clin. Oncol. 2023, 41, 1664–1669. [Google Scholar] [CrossRef]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti–B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2023, 41, 1265–1274. [Google Scholar] [CrossRef]
- Approved Cellular and Gene Therapy Products. US Food & Drug Administration. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 26 April 2024).
- U.S. FDA Approves Bristol Myers Squibb’s Breyanzi as the First and Only CAR T Cell Therapy for Adults with Relapsed or Refractory Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL). Available online: https://news.bms.com/news/details/2024/U.S.-FDA-Approves-Bristol-Myers-Squibbs-Breyanzi--as-the-First-and-Only-CAR-T-Cell-Therapy-for-Adults-with-Relapsed-or-Refractory-Chronic-Lymphocytic-Leukemia-CLL-or-Small-Lymphocytic-Lymphoma-SLL/default.aspx (accessed on 3 March 2024).
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukemia remissions with the persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Bouziana, S.; Bouzianas, D. Anti-CD19 CAR-T cells: Digging in the dark side of the golden therapy. Crit. Rev. Oncol. Hematol. 2021, 157, 103096. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer. 2022, 21, 78. [Google Scholar] [CrossRef]
- Young, R.M.; Engel, N.W.; Uslu, U.; Wellhausen, N.; June, C.H. Next-Generation CAR T-cell Therapies. Cancer Discov. 2022, 12, 1625–1633. [Google Scholar] [CrossRef]
- Benjamin, R.; Jain, N.; Maus, M.V.; Boissel, N.; Graham, C.; Jozwik, A.; Yallop, D.; Konopleva, M.; Frigault, M.J.; Teshima, T.; et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): A phase 1, dose-escalation trial. Lancet Haematol. 2022, 9, e833–e843. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.Y.; Jain, M.D.; Nastoupil, L.J.; Tamaresis, J.; Ghobadi, A.; Lin, Y.; Lekakis, L.J.; Reagan, P.M.; Oluwole, O.O.; McGuirk, J.P.; et al. Five year outcomes of patients with large B-Cell lymphoma treated with standard-of-care Axicabtagene Ciloleucel: Results from the US lymphoma CAR-T cell consortium. Blood 2023, 142 (Suppl. S1), 1032. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Current understanding and management of CAR T cell-associated toxicities. Nat. Rev. Clin. Oncol. 2024, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Xiao, Z.; Qin, Z.; Yang, J.; Wen, Y.; Yu, Z.; Li, Y.; Sheppard, N.C.; Fuchs, S.Y.; Xu, X.; et al. Tumor-Derived Small Extracellular Vesicles Inhibit the Efficacy of CAR T Cells against Solid Tumors. Cancer Res. 2023, 83, 2790–2806. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Müller, F.; Mougiakakos, D.; Böltz, S.; Weilhem, A.; Aigner, M.; Völkl, S.; Simon, D.; Kleyer, A.; Munoz, L.; et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat. Med. 2022, 28, 2124–2132. [Google Scholar] [CrossRef]
- Müller, F.; Taubmann, J.; Bucci, L.; Wilhelm, A.; Bergmann, C.; Völkl, S.; Aigner, M.; Rothe, T.; Minopoulou, I.; Tur, C.; et al. CD19 CAR T-Cell Therapy in Autoimmune Disease—A Case Series with Follow-up. N. Engl. J. Med. 2024, 390, 687–700. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Arany, Z.; Baur, J.A.; Epstein, J.A.; June, C.H. CAR T therapy beyond cancer: The evolution of a living drug. Nature 2023, 619, 707–715. [Google Scholar] [CrossRef]
- FDA Investigating Serious Risk of T-cell Malignancy Following BCMA-Directed or CD19-Directed Autologous Chimeric Antigen Receptor (CAR) T cell Immunotherapies. US Food & Drug Administration. Available online: https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-investigating-serious-risk-t-cell-malignancy-following-bcma-directed-or-cd19-directed-autologous (accessed on 28 November 2023).
- Verdun, N.; Marks, P. Secondary Cancers after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2024, 390, 584–586. [Google Scholar] [CrossRef]
- Levine, B.L.; Pasquini, M.C.; Connolly, J.E.; Porter, D.L.; Gustafson, M.P.; Boelens, J.J.; Horwitz, E.M.; Grupp, S.A.; Maus, M.V.; Locke, F.L.; et al. Unanswered questions following reports of secondary malignancies after CAR-T cell therapy. Nat. Med. 2024, 30, 338–341. [Google Scholar] [CrossRef]
- Elsallab, M.; Ellithi, M.; Lunning, M.A.; D’Angelo, C.; Ma, J.; Perales, M.A.; Frigault, M.; Maus, M.V. Second Primary Malignancies After Commercial CAR T Cell Therapy: Analysis of FDA Adverse Events Reporting System (FAERS). Blood 2024, 143, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- FDA Requires Boxed Warning for T cell Malignancies Following Treatment with BCMA-Directed or CD19-Directed Autologous Chimeric Antigen Receptor (CAR) T cell Immunotherapies. US Food & Drug Administration. Available online: https://www.fda.gov/vaccines-blood-biologics/safety-availability-biologics/fda-requires-boxed-warning-t-cell-malignancies-following-treatment-bcma-directed-or-cd19-directed (accessed on 18 April 2024).
- Harrison, S.J.; Nguyen, T.; Rahman, M.; Er, J.; Li, J.; Li, K.; Lendvai, N.; Schecter, J.M.; Banerjee, A.; Roccia, T.; et al. CAR+ T-Cell Lymphoma Post Ciltacabtagene Autoleucel Therapy for Relapsed Refractory Multiple Myeloma. Blood 2023, 142 (Suppl. S1), 6939. [Google Scholar] [CrossRef]
- Heinrich, T.; Rengstl, B.; Muik, A.; Petkova, M.; Schmid, F.; Wistinghausen, R.; Warner, K.; Crispatzu, G.; Hansmann, M.L.; Herling, M.; et al. Mature T-cell lymphomagenesis induced by retroviral insertional activation of Janus kinase 1. Mol. Ther. 2013, 21, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Ghilardi, G.; Fraietta, J.A.; Gerson, J.N.; Van Deerlin, V.M.; Morrissette, J.J.D.; Caponetti, G.C.; Paruzzo, L.; Harris, J.C.; Chong, E.A.; Susanibar Adaniya, S.P.; et al. T-cell Lymphoma and Secondary Primary Malignancy Risk After Commercial CAR T-cell Therapy. Nat. Med. 2024, 30, 984–989. [Google Scholar] [CrossRef]
- Bishop, D.C.; Clancy, L.E.; Simms, R.; Burgess, J.; Mathew, G.; Moezzi, L.; Street, J.A.; Sutrave, G.; Atkins, E.; McGuire, H.M.; et al. Development of CAR T-cell lymphoma in 2 of 10 patients effectively treated with piggyBac-modified CD19 CAR T cells. Blood 2021, 138, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yu, W.J.; Zhou, L.; Yang, M.; Ye, S.; Zhu, J.; Huang, J.; Zhang, Y.; Li, L.; Zhao, J.; et al. C-CAR039, a Novel Anti-CD20/CD19 Bi-Specific CAR T-Cell Therapy Shows Deep and Durable Clinical Benefits in Patients with Relapsed or Refractory (r/r) B-Cell Non-Hodgkin Lymphoma (B-NHL) in Long Term Follow up. Blood 2023, 142 (Suppl. S1), 1025. [Google Scholar] [CrossRef]
- Steffin, D.H.M.; Muhsen, I.N.; Hill, L.C.; Ramos, C.A.; Ahmed, N.; Hegde, M.; Wang, T.; Wu, M.; Gottschalk, S.; Whittle, S.B.; et al. Long-term follow-up for the development of subsequent malignancies in patients treated with genetically modified IECs. Blood 2022, 140, 16–24. [Google Scholar] [CrossRef]
- Hamilton, M.P.; Sugio, T.; Noordenbos, T.; Shi, S.; Bulterys, P.L.; Liu, C.L.; Kang, X.; Olsen, M.N.; Good, Z.; Dahiya, S.; et al. Risk of Second Tumors and T-Cell Lymphoma after CAR T-Cell Therapy. N. Engl. J. Med. 2024, 390, 2047–2060. [Google Scholar] [CrossRef]
- Chihara, D.; Dores, G.M.; Flowers, C.R.; Morton, L.M. The bidirectional increased risk of B-cell lymphoma and T-cell lymphoma. Blood 2021, 138, 785–789. [Google Scholar] [CrossRef]
- Wartewig, T.; Kurgyis, Z.; Keppler, S.; Pechloff, K.; Hameister, E.; Öllinger, R.; Maresch, R.; Buch, T.; Steiger, K.; Winter, C.; et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 2017, 552, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.A.; Ruella, M.; Schuster, S.J. Lymphoma Program Investigators at the University of Pennsylvania. Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. N. Engl. J. Med. 2021, 384, 673–674. [Google Scholar] [CrossRef]
- Cordeiro, A.; Bezerra, E.D.; Hirayama, A.V.; Hill, J.A.; Wu, Q.V.; Voutsinas, J.; Sorror, M.L.; Turtle, C.J.; Maloney, D.G.; Bar, M. Late events after treatment with CD19-targeted chimeric antigen receptor modified T cells. Biol. Blood Marrow Transpl. 2020, 26, 26–33. [Google Scholar] [CrossRef]
- Tward, J.D.; Wendland, M.M.; Shrieve, D.C.; Szabo, A.; Gaffney, D.K. The risk of secondary malignancies over 30 years after the treatment of non-Hodgkin lymphoma. Cancer 2006, 107, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, E.M.; Myers, R.M.; Yates, B.; Annesley, C.; John, S.; Taraseviciute, A.; Steinberg, S.M.; Sheppard, J.; Chung, P.; Chen, L.; et al. Low rate of subsequent malignant neoplasms after CD19 CAR T-cell therapy. Blood Adv. 2022, 6, 5222–5226. [Google Scholar] [CrossRef]
- Strati, P.; Varma, A.; Adkins, S.; Nastoupil, L.J.; Westin, J.; Hagemeister, F.B.; Fowler, N.H.; Lee, H.J.; Fayad, L.E.; Samaniego, F.; et al. Hematopoietic recovery and immune reconstitution after axicabtagene ciloleucel in patients with large b-cell lymphoma. Haematologica 2021, 106, 2667–2672. [Google Scholar] [CrossRef]
- Panagiota, V.; Kerschbaum, J.F.; Penack, O.; Stein, C.M.; Arends, C.M.; Koenecke, C.; Strzelecka, P.M.; Kloos, A.; Wiegand, L.; Lasch, A.; et al. Clinical implications and dynamics of clonal hematopoiesis in anti- CD19 CAR T-cell treated patients. Hemasphere 2023, 7, e957. [Google Scholar] [CrossRef] [PubMed]
- Teipel, R.; Kroschinsky, F.; Kramer, M.; Kretschmann, T.; Egger-Heidrich, K.; Krüger, T.; Ruhnke, L.; Herold, S.; Stasik, S.; Sockel, K.; et al. Prevalence and variation of CHIP in patients with aggressive lymphomas undergoing CD19-directed CAR T-cell treatment. Blood Adv. 2022, 6, 1941–1946. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.P.; Sworder, B.J.; Alig, S.K.; Good, Z.; Boegeholz, J.; Schroers-Martin, J.; Tamaresis, J.; Shahrokh Esfahani, M.; Lu, Y.; Olsen, M.; et al. CAR19 Therapy Drives Expansion of Clonal Hematopoiesis and Associated Cytopenias. Blood 2023, 142 (Suppl. S1), 360. [Google Scholar] [CrossRef]
- Alkhateeb, H.B.; Mohty, R.; Greipp, P.; Bansal, R.; Hathcock, M.; Rosenthal, A.; Murth, H.; Kharfan-Dabaja, M.; Bisneto Villasboas, J.C.; Bennani, N.; et al. Therapy-related myeloid neoplasms following chimeric antigen receptor T-cell therapy for Non-Hodgkin Lymphoma. Blood Cancer J. 2022, 12, 113. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.G.; Sperling, A.S.; Brea, E.J.; Leick, M.B.; Fell, G.G.; Jan, M.; Gohil, S.H.; Tai, Y.T.; Munshi, N.C.; Wu, C.J.; et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. 2021, 5, 2982–2986. [Google Scholar] [CrossRef] [PubMed]
- Ahmadreza, A.; Srikanthan, J.; Khelil, M.B.; Marcos-Kovandzik, L.; Amine-Hneineh, R.; Sabourin-Cousin, M.; Quivoron, C.; Christophe, M.; Baptiste Micol, J.; Zitvogel, L.; et al. CAR T cells reside in the bone marrow and inhibit hematopoiesis. HemaSphere 2024, 8 (Suppl. S1), 2620. [Google Scholar]
- Zhao, A.; Zhao, M.; Qian, W.; Liang, A.; Li, P.; Liu, H. Secondary myeloid neoplasms after CD19 CAR T therapy in patients with refractory/relapsed B-cell lymphoma: Case series and review of literature. Front. Immunol. 2023, 13, 1063986. [Google Scholar] [CrossRef]
- Sacchi, S.; Marcheselli, L.; Bari, A.; Marcheselli, R.; Pozzi, S.; Luminari, S.; Lombardo, M.; Buda, G.; Lazzaro, A.; Gobbi, P.G.; et al. Secondary malignancies after treatment for indolent non-Hodgkin’s lymphoma: A 16-year follow-up study. Haematologica 2008, 93, 398–404. [Google Scholar] [CrossRef]
- Banerjee, R.; Poh, C.; Hirayama, A.V.; Gauthier, J.; Cassaday, R.D.; Shadman, M.; Cowan, A.J.; Till, B.G.; Green, D.J.; Kiem, H.P.; et al. Answering the “Doctor, can CAR-T therapy cause cancer?” question in clinic. Blood Adv. 2024, 8, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.; Alkhateeb, H.; Shah, S.; Bansal, R.; Hathcock, M.; Rosenthal, A.; Kharfan-Dabaja, M.; Kourelis, T.; Patnaik, M.; Chen, D.; et al. Cytopenias, age and CAR-HEMATOTOX score predict the development of post CAR T-cell therapy-related myeloid neoplasms. Hemasphere 2023, 7 (Suppl. S3), e6718317. [Google Scholar] [CrossRef]
- Galli, E.; Rossi, M.; Pansini, I.; Viscovo, M.; Malara, T.; Colangelo, M.; Alma, E.; Hohaus, S.; Sica, S.; Sorà, F.; et al. Predicting therapy-related myeloid neoplasms after CAR-T: Validation of the clonal hematopoiesis risk score (CHRS). HemaSphere 2024, 8 (Suppl. S1), 398. [Google Scholar]
- Levine, B.L.; Miskin, J.; Wonnacott, K.; Keir, C. Global Manufacturing of CAR T Cell Therapy. Mol. Ther. Methods Clin. Dev. 2016, 4, 92–101. [Google Scholar] [CrossRef]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53. [Google Scholar] [CrossRef]
- Howe, S.J.; Mansour, M.R.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.H.; Pike-Overzet, K.; Chatters, S.J.; de Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Krämer, A.; Schwäble, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Göhring, G.; Steinemann, D.; et al. Gene therapy for Wiskott-Aldrich Syndrome—Long-term reconstitution and clinical benefits, but increased risk for leukemogenesis. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G. Adenovirus: The first effective in vivo gene delivery vector. Hum. Gene Ther. 2014, 25, 3–11. [Google Scholar] [CrossRef]
- Cooray, S.; Howe, S.J.; Thrasher, A.J. Retrovirus and lentivirus vector design and methods of cell conditioning. Methods Enzymol. 2012, 507, 29–57. [Google Scholar]
- David, R.M.; Doherty, A.T. Viral Vectors: The Road to Reducing Genotoxicity. T Toxicol. Sci. 2017, 155, 315–325. [Google Scholar] [CrossRef]
- Labbé, R.P.; Vessillier, S.; Rafiq, Q.A. Lentiviral vectors for T cell engineering: Clinical applications, bioprocessing and future perspectives. Viruses 2021, 13, 1528. [Google Scholar] [CrossRef] [PubMed]
- Moiani, A.; Paleari, Y.; Sartori, D.; Mezzadra, R.; Miccio, A.; Cattoglio, C.; Cocchiarella, F.; Lidonnici, M.R.; Ferrari, G.; Mavilio, F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Investig. 2012, 122, 1653–1666. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Ranzani, M.; Volpin, M.; Bartholomae, C.; Duros, C.; Artus, A.; Merella, S.; Benedicenti, F.; Sergi Sergi, L.; Sanvito, F.; et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014, 22, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Schubert, M.L.; Schmitt1, M.; Wang, L.; Ramos, C.A.; Jordan, K.; Müller-Tidow, C.; Dreger, P. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Qin, H.; Yates, B.; Su, L.; Shalabi, H.; Raffeld, M.; Ahlman, M.A.; Stetler-Stevenson, M.; Yuan, C.; Guo, S.; et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv. 2019, 3, 2317–2322. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.O.; Raymond, G.; Pierpont, E.I.; Kemp, S.; McIvor, R.S.; Rayannavar, A.; Miller, B.; Lund, T.C.; Orchard, P.J. Treatment of cerebral adrenoleukodystrophy: Allogeneic transplantation and lentiviral gene therapy. Exp. Opin. Biol. Ther. 2022, 22, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Brunson, A.; Keegan, T.H.M.; Bang, H.; Mahajan, A.; Paulukonis, S.; Wun, T. Increased risk of leukemia among sickle cell disease patients in California. Blood 2017, 130, 1597–1599. [Google Scholar] [CrossRef]
- Goyal, S.; Tisdale, J.; Schmidt, M.; Kanter, J.; Jaroscak, J.; Whitney, D.; Bitter, H.; Gregory, P.D.; Parsons, G.; Foos, M.; et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N. Engl. J. Med. 2022, 386, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Bonner, M.; Pierciey, F.J.; Uchida, N.; Rottman, J.; Demopoulos, L.; Schmidt, M.; Kanter, J.; Walters, M.C.; Thompson, A.A.; et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020, 4, 2058–2063. [Google Scholar] [CrossRef] [PubMed]
- Global Newswire Uniqure Announces Findings from Reported Case of Hepatocellular Carcinoma (HCC) in Hemophilia B Gene Therapy Program. Available online: https://www.globenewswire.com/news-release/2021/03/29/2200653/0/en/uniQure-Announces-Findings-from-Reported-Case-of-Hepatocellular-Carcinoma-HCC-in-Hemophilia-B-Gene-Therapy-Program.html (accessed on 29 March 2021).
- Chandler, R.J.; LaFave, M.C.; Varshney, G.K.; Trivedi, N.S.; Carrillo-Carrasco, N.; Senac, J.S.; Wu, W.; Hoffmann, V.; Elkahloun, A.G.; Burgess, S.M.; et al. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Investig. 2015, 125, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.A.; Merricks, E.P.; Long, C.T.; et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. US Food & Drug Administration. Available online: www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease (accessed on 8 December 2023).
- Liu, X.; Zhang, Y.; Cheng, C.; Cheng, A.W.; Zhang, X.; Li, N.; Xia, C.; Wei, X.; Liu, X.; Wang, H. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017, 27, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, C.A.; Brandes, N.; Bueno, R.; Trinidad, M.; Mazumder, T.; Yu, B.; Hwang, B.; Chang, C.; Liu, J.; Sun, Y.; et al. Mitigation of chromosome loss in clinical CRISPR-Cas9-engineered T cells. Cell 2023, 186, 4567–4582. [Google Scholar] [CrossRef] [PubMed]
- Höijer, I.; Emmanouilidou, A.; Östlund, R.; van Schendel, R.; Bozorgpana, S.; Tijsterman, M.; Feuk, L.; Gyllensten, U.; den Hoed, M.; Ameur, A. CRISPR-Cas9 induces large structural variants at on-target and off-target sites in vivo that segregate across generations. Nat. Commun. 2022, 13, 627. [Google Scholar] [CrossRef]
- Lemmens, M.; Dorsheimer, L.; Zeller, A.; Dietz-Baum, Y. Non-clinical safety assessment of novel drug modalities: Genome safety perspectives on viral-, nuclease- and nucleotide-based gene therapies. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2024, 896, 503767. [Google Scholar] [CrossRef]
- Micklethwaite, K.P.; Gowrishankar, K.; Gloss, B.S.; Li, Z.; Street, J.A.; Moezzi, L.; Mach, M.A.; Sutrave, G.; Clancy, L.E.; Bishop, D.C.; et al. Investigation of product-derived lymphoma following infusion of piggyBac-modified CD19 chimeric antigen receptor T cells. Blood 2021, 138, 1401–1415. [Google Scholar] [CrossRef]
- Murphy, L.A.; Marians, R.C.; Miller, K.; Brenton, M.D.; Mallo, R.L.V.; Kohler, M.E.; Fry, T.J.; Winters, A.C. Digital polymerase chain reaction strategies for accurate and precise detection of vector copy number in CAR T cell products. Cytotherapy 2023, 25, 94–102. [Google Scholar] [CrossRef]
- Yan, N.; Feng, H.; Sun, Y.; Xin, Y.; Zhang, H.; Lu, H.; Zheng, J.; He, C.; Zuo, Z.; Yuan, T.; et al. Cytosine base editors induce off-target mutations and adverse phenotypic effects in transgenic mice. Nat. Commun. 2023, 14, 1784. [Google Scholar] [CrossRef]
- Slesarenko, Y.S.; Lavrov, A.V.; Smirnikhina, S.A. Off-target effects of base editors: What we know and how we can reduce it. Curr. Genet. 2022, 68, 39–48. [Google Scholar] [CrossRef]
- Odak, A.; Yuan, H.; Feucht, J.; Cantu, V.A.; Mansilla-Soto, J.; Kogel, F.; Eyquem, J.; Everett, J.; Bushman, F.D.; Leslie, C.S.; et al. Novel extragenic genomic safe harbors for precise therapeutic T-cell engineering. Blood 2023, 141, 2698–2712. [Google Scholar] [CrossRef]
- Metzloff, A.E.; Padilla, M.S.; Gong, N.; Billingsley, M.M.; Han, X.; Merolle, M.; Mai, D.; Figueroa-Espada, C.G.; Thatte, A.S.; Haley, R.M.; et al. Antigen Presenting Cell Mimetic Lipid Nanoparticles for Rapid mRNA CAR T Cell Cancer Immunotherapy. Adv. Mater. 2024, 36, e2313226. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wu, W.; Zhou, B.; Li, B. Chimeric antigen receptor therapy meets mRNA technology. Trends Biotech. 2024, 42, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Bouzianas, D.; Bouziana, S. A decade of CD4+ chimeric antigen receptor T-cell evolution in two chronic lymphocytic leukemia patients: Were chronic lymphocytic leukemia cells present? Explor. Target. Antitumor Ther. 2023, 4, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Bouzianas, D.; Bouziana, S. First pediatric B-acute lymphoblastic leukemia patient treated with anti-CD19 chimeric antigen receptor T-cell therapy: Long-term remission or early cure? Hum. Vaccines Immunother. 2024, 20, 2321678. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| CAR T-Cell Product | Brand Name | Company | Disease Indications (R/R) | Year Initially Approved | Target Antigen | Costimulatory Domain | Viral Vector |
|---|---|---|---|---|---|---|---|
| Tisagenlecleucel | Kymriah | Novartis | B-ALL (≤25 years) LBCL FL | 2017 | CD19 | 4-1BB | Lentiviral |
| Axicabtagene ciloleucel | Yescarta | Kite | LBCL FL | 2017 | CD19 | CD28 | Retroviral |
| Brexucabtagene autoleucel | Tecartus | Kite | B-ALL (>25 years) MCL | 2020 | CD19 | CD28 | Retroviral |
| Lisocabtagene maraleucel | Breyanzi | Juno/BMS | LBCL MCL FL CLL/SLL (AA) | 2021 | CD19 | 4-1BB | Lentiviral |
| Idecabtagene vicleucel | Abecma | Celgene/BMS | MM | 2021 | BCMA | 4-1BB | Lentiviral |
| Ciltacabtagene autoleucel | Carvykti | Legend/J&J | MM | 2022 | BCMA | 4-1BB | Lentiviral |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouziana, S.; Bouzianas, D. The Current Landscape of Secondary Malignancies after CAR T-Cell Therapies: How Could Malignancies Be Prevented? Int. J. Mol. Sci. 2024, 25, 9518. https://doi.org/10.3390/ijms25179518
Bouziana S, Bouzianas D. The Current Landscape of Secondary Malignancies after CAR T-Cell Therapies: How Could Malignancies Be Prevented? International Journal of Molecular Sciences. 2024; 25(17):9518. https://doi.org/10.3390/ijms25179518
Chicago/Turabian StyleBouziana, Stella, and Dimitrios Bouzianas. 2024. "The Current Landscape of Secondary Malignancies after CAR T-Cell Therapies: How Could Malignancies Be Prevented?" International Journal of Molecular Sciences 25, no. 17: 9518. https://doi.org/10.3390/ijms25179518
APA StyleBouziana, S., & Bouzianas, D. (2024). The Current Landscape of Secondary Malignancies after CAR T-Cell Therapies: How Could Malignancies Be Prevented? International Journal of Molecular Sciences, 25(17), 9518. https://doi.org/10.3390/ijms25179518