
Multi-Omics Reveals the Role of Arachidonic Acid Metabolism in the Gut–Follicle Axis for the Antral Follicular Development of Holstein Cows


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
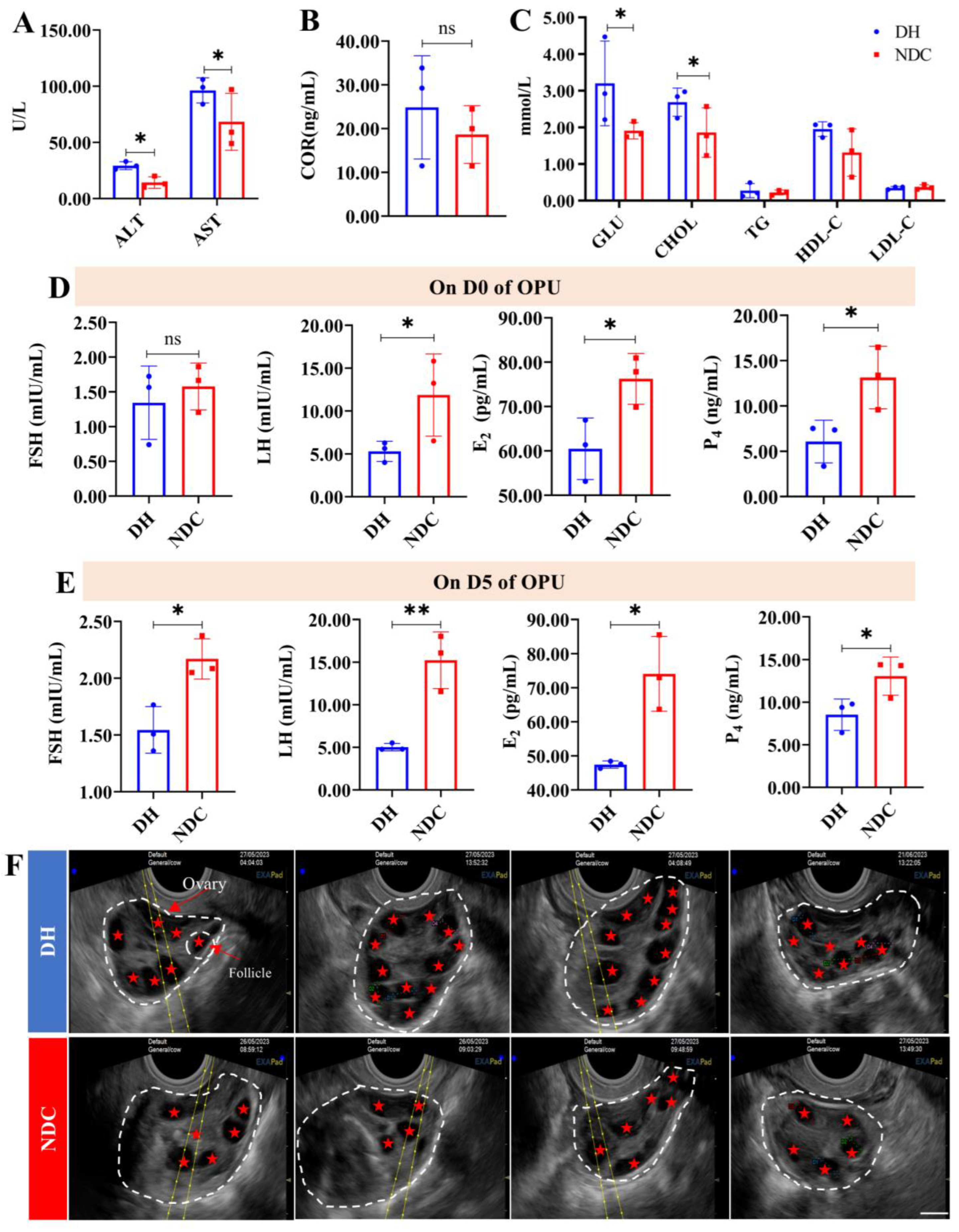
2.1. Comparison of the Efficiency of In Vitro Embryo Production between the DH and NDC Groups
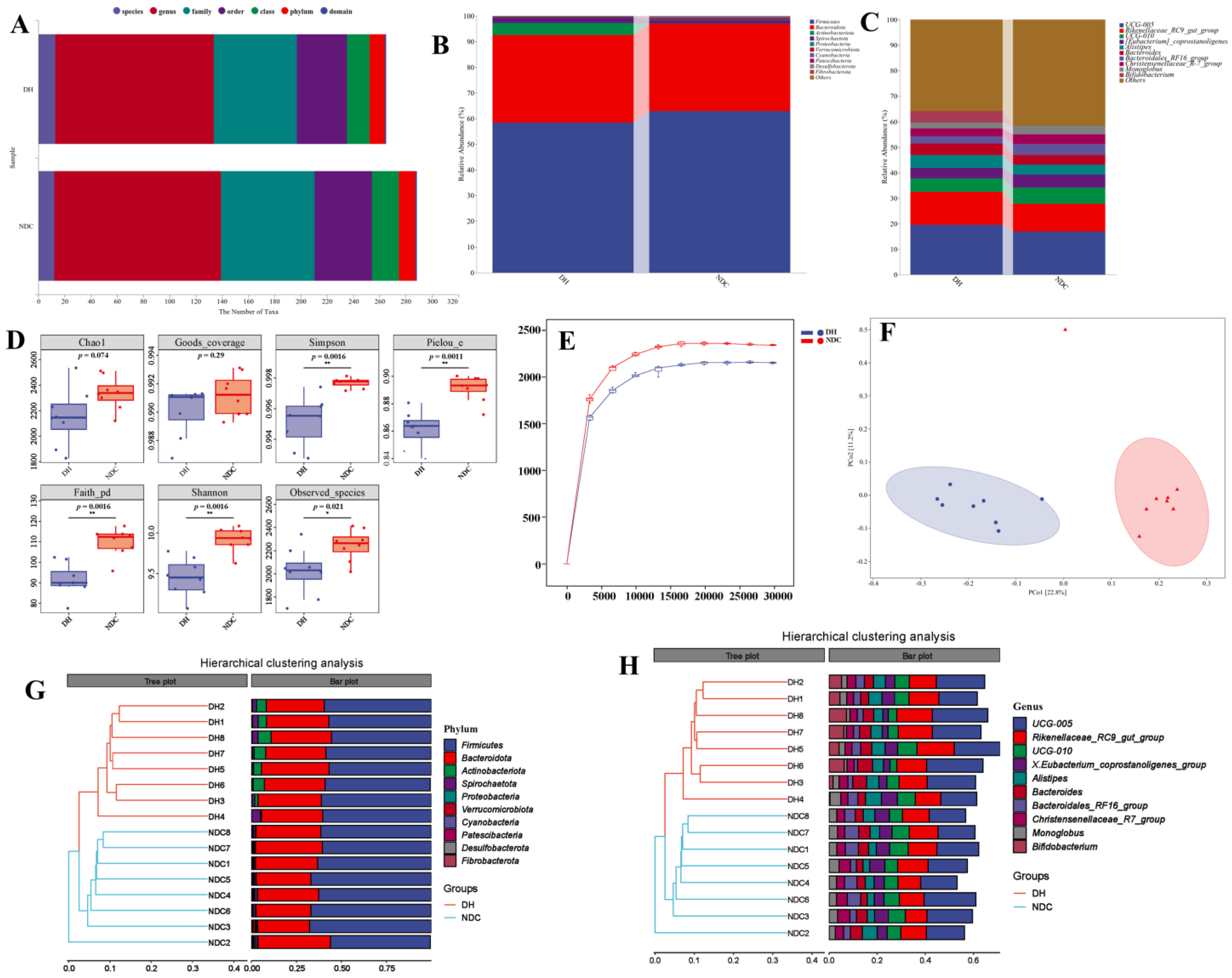
2.2. The Difference in the Gut Microbiota Abundance between the DH and NDC Groups
2.3. Correlation Analysis Reveals the Relationship between the Gut Microbiota and Its Metabolites
2.4. Correlation Analysis Reveals the Relationship between Serum Metabolic Profiles and Gut Microbes
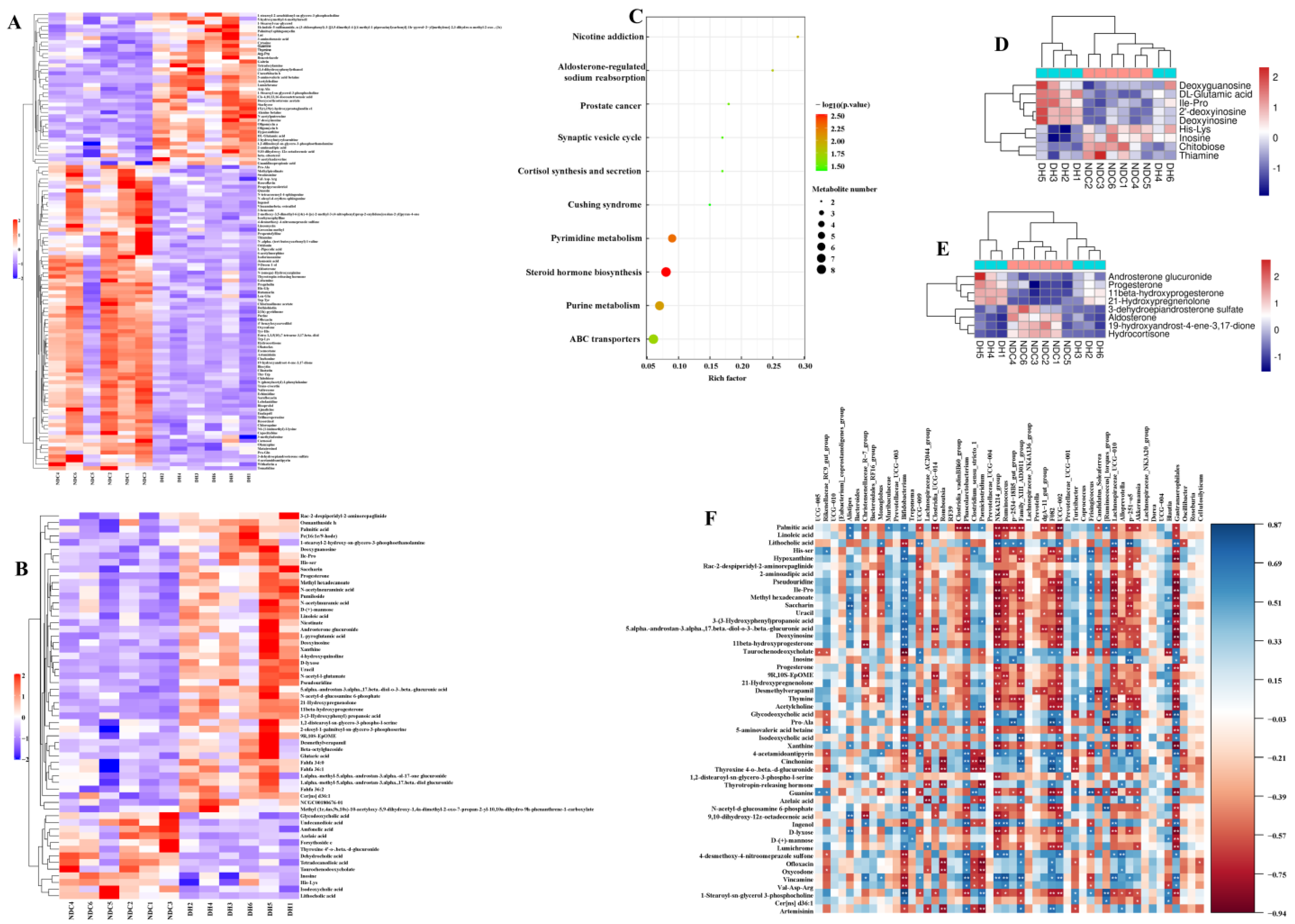
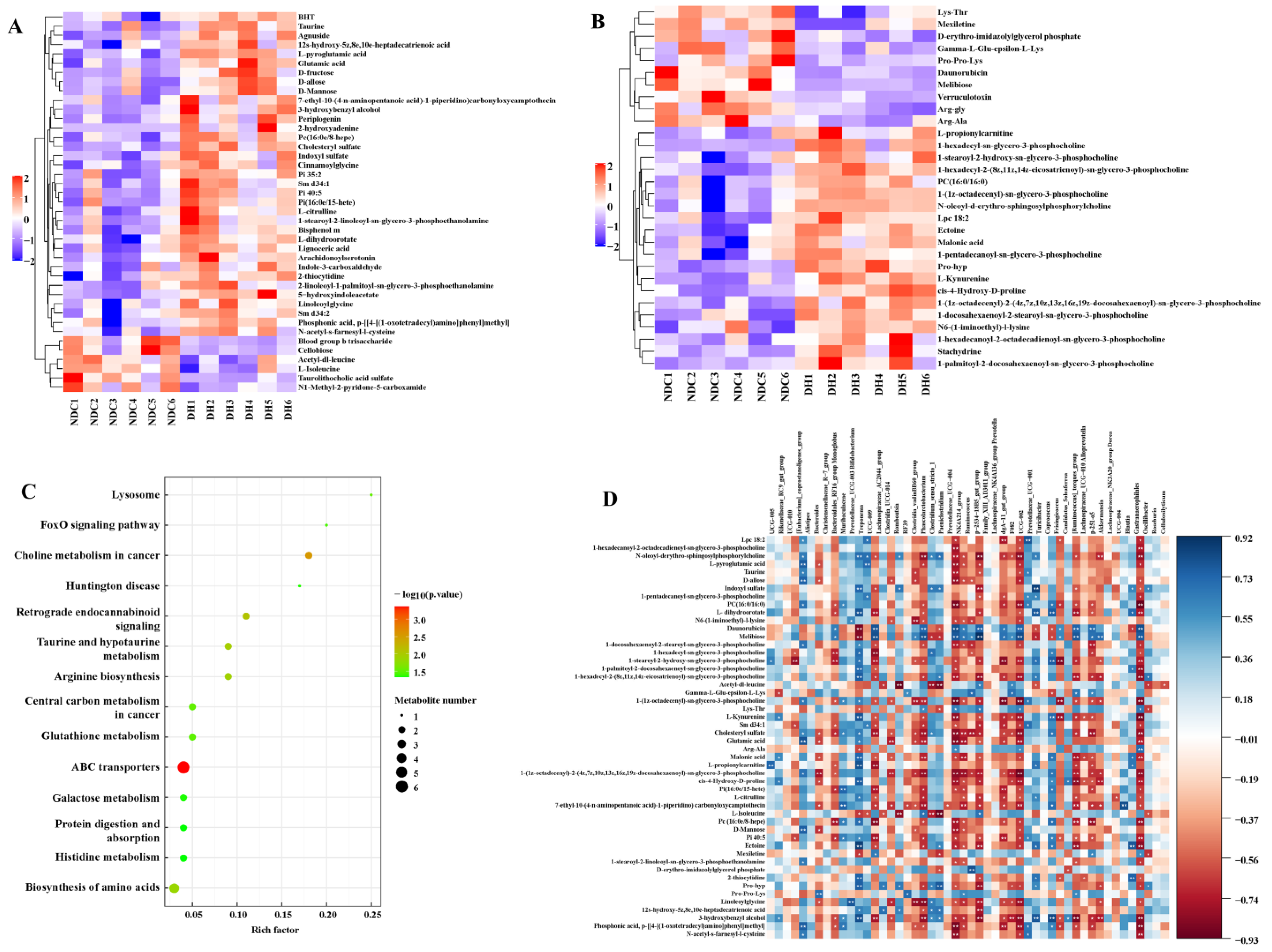
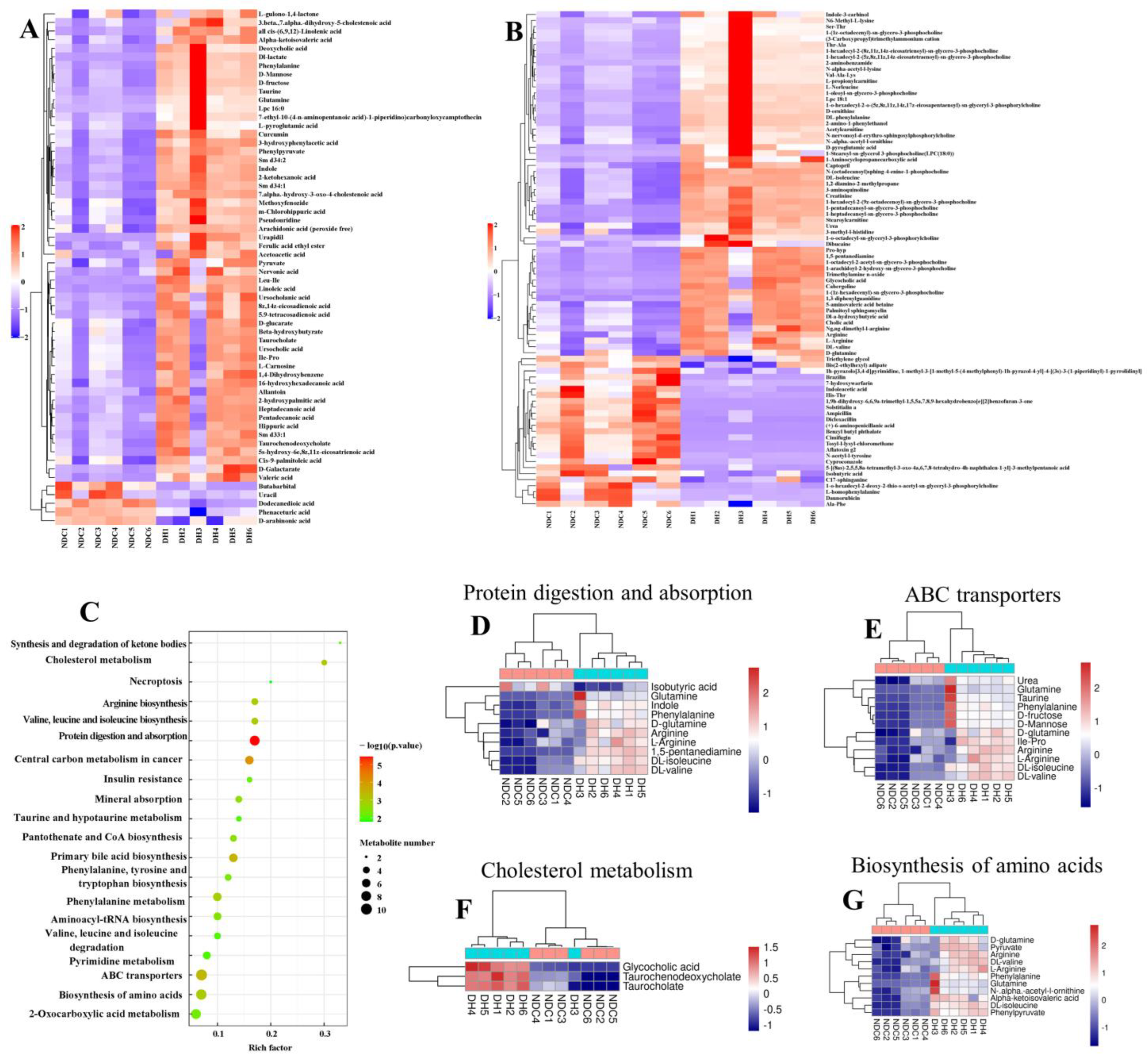
2.5. Metabolomic Analysis of FF in the DH and NDC Groups
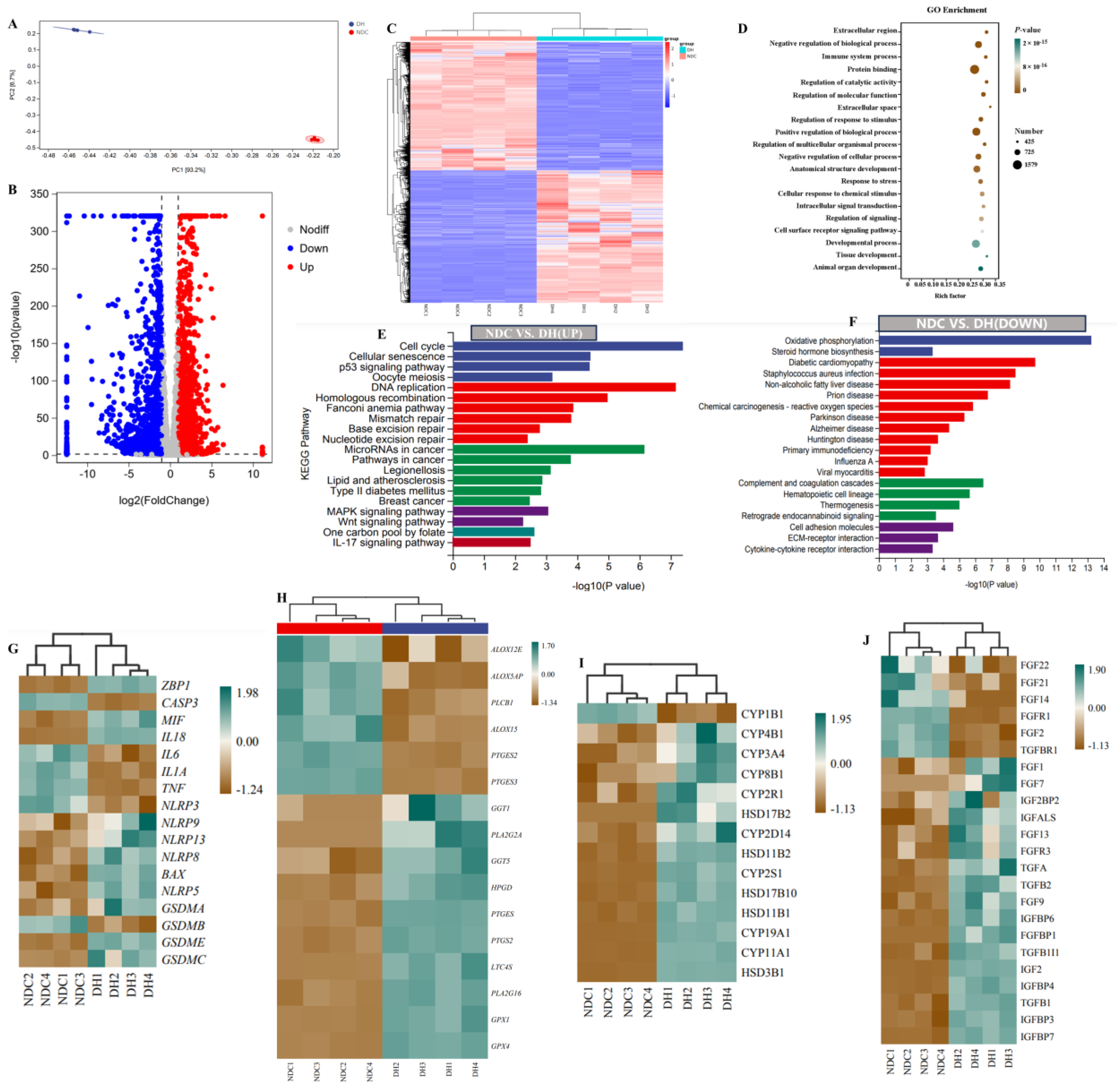
2.6. Transcriptome Profiles Reveal Functional Gene Expression in GCs
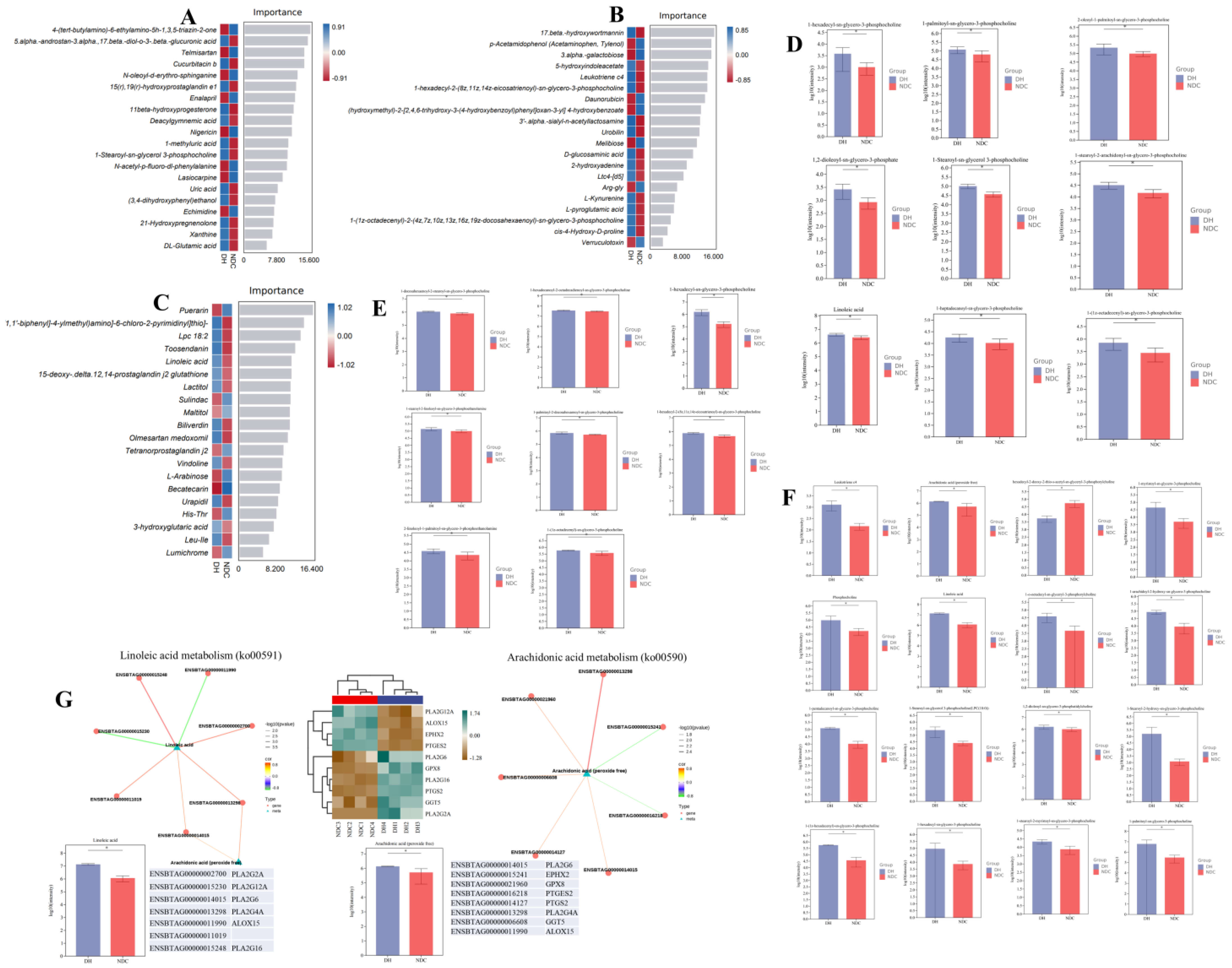
2.7. Comprehensive Analysis of Different Metabolites in Gut Microbiota, Serum, and FF, and Differentially Expressed Genes
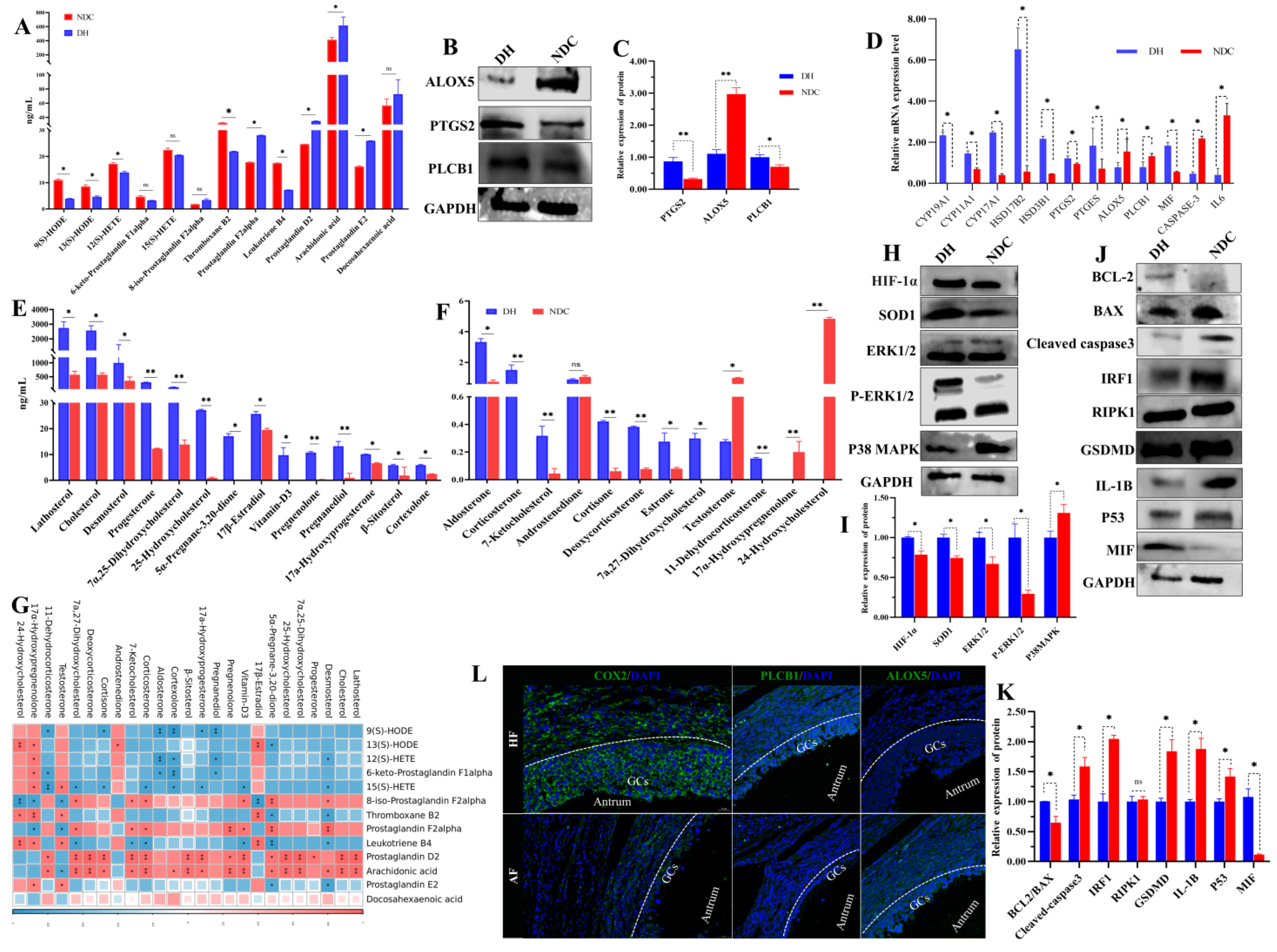
2.8. PLCB1/PTGS2 Axis Promotes Follicular Development via COX/PGD2 Pathway Dependent on Arachidonic Acid Metabolism
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Animals
5.2. Samples Collection and Study Design
5.3. Superovulation Synchronization and In Vitro Fertilization (IVF)
5.4. Fecal 16S rRNA Sequencing and Processing
5.5. Untargeted Metabolomic Analysis for Gut Microbiota, Serum, and FF
5.6. Transcriptomic Analysis of Follicular GCs
5.7. Quantitative Real-Time PCR Analysis
5.8. Western Blotting
5.9. Immunofluorescence Staining
5.10. Blood Physiological Indices and Hormone Analysis
5.11. Targeted Metabolite Analysis
5.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ferré, L.B.; Kjelland, M.E.; Strøbech, L.B.; Hyttel, P.; Mermillod, P.; Ross, P.J. Review: Recent advances in bovine in vitro embryo production: Reproductive biotechnology history and methods. Animal 2020, 14, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.J. Implications of assisted reproductive technologies for pregnancy outcomes in mammals. Annu. Rev. Anim. Biosci. 2020, 8, 395–413. [Google Scholar] [CrossRef]
- Krisher, R.L. In vivo and in vitro environmental effects on mammalian oocyte quality. Annu. Rev. Anim. Biosci. 2013, 1, 393–417. [Google Scholar] [CrossRef] [PubMed]
- Lonergan, P.; Fair, T. Maturation of oocytes in vitro. Annu. Rev. Anim. Biosci. 2016, 4, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Demetrio, D.G.B.; Benedetti, E.; Demetrio, C.G.B.; Fonseca, J.; Oliveira, M.; Magalhaes, A.; Santos, R.M.D. How can we improve embryo production and pregnancy outcomes of Holstein embryos produced in vitro? (12 years of practical results at a California dairy farm). Anim. Reprod. 2020, 17, e20200053. [Google Scholar] [CrossRef] [PubMed]
- Lollato, J.P.M.; Souza, A.C.C.; Silva, R.C.P.; Marques, M.O.; Crozara, A.S.; Gonçalves, R.L.; Cunha, M.; Seneda, M.; Morotti, F. In vivo, embryo production in bovine donors with low and high antral follicle counts was superovulated with low and high FSH doses. Livest. Sci. 2022, 262, 104985. [Google Scholar] [CrossRef]
- Leroy, J.L.; Rizos, D.; Sturmey, R.; Bossaert, P.; Gutierrez-Adan, A.; Van Hoeck, V.; Valckx, S.; Bols, P.E.J. Intrafollicular conditions as a major link between maternal metabolism and oocyte quality: A focus on dairy cow fertility. Reprod. Fertil. Dev. 2011, 24, 1–12. [Google Scholar] [CrossRef]
- Ribeiro, E.S.; Gomes, G.; Greco, L.F.; Cerri, R.; Vieira-Neto, A.; Monteiro, P.J., Jr.; Lima, F.S.; Bisinotto, R.S.; Thatcher, W.W.; Santos, J.E.P. Carryover effect of postpartum inflammatory diseases on developmental biology and fertility in lactating dairy cows. J. Dairy Sci. 2016, 99, 2201–2220. [Google Scholar] [CrossRef]
- Alward, K.J.; Cockrum, R.R.; Ealy, A.D. Associations of antral follicle count with fertility in cattle: A review. JDS Commun. 2023, 4, 132–137. [Google Scholar] [CrossRef]
- Sangsritavong, S.; Combs, D.K.; Sartori, R.; Armentano, L.E.; Wiltbank, M.C. High feed intake increases liver blood flow and metabolism of progesterone and estradiol-17beta in dairy cattle. J. Dairy Sci. 2002, 85, 2831–2842. [Google Scholar] [CrossRef] [PubMed]
- Wiltbank, M.; Lopez, H.; Sartori, R.; Sangsritavong, S.; Gumen, A. Changes in reproductive physiology of lactating dairy cows due to elevated steroid metabolism. Theriogenology 2006, 65, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Gabor, G.; Kastelic, J.P.; Abonyi-Toth, Z.; Gabor, P.; Endrodi, T.; Balogh, O.G. Pregnancy loss in dairy cattle: Relationship of ultrasound, blood pregnancy-specific protein b, progesterone, and production variables. Reprod. Domest. Anim. 2016, 51, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Lopez, H.; Caraviello, D.Z.; Satter, L.D.; Fricke, P.M.; Wiltbank, M.C. Relationship between level of milk production and multiple ovulations in lactating dairy cows. J. Dairy Sci. 2005, 88, 2783–2793. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Meldrum, D.R.; Katz-Jaffe, M.G.; Krisher, R.L.; Schoolcraft, W.B. Oocyte environment: Follicular fluid and cumulus cells are critical for oocyte health. Fertil. Steril. 2015, 103, 303–316. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, W.; Li, N.; Dai, S.; Wu, H.; Wu, Z.; Zeng, S. Integration analysis of metabolome and transcriptome reveals the effect of exogenous supplementation with mixtures of vitamins ADE, zinc, and selenium on follicular growth and granulosa cells molecular metabolism in donkeys (Equus asinus). Front. Vet. Sci. 2022, 9, 993426. [Google Scholar] [CrossRef] [PubMed]
- Bender, K.; Walsh, S.; Evans, A.C.; Fair, T.; Brennan, L. Metabolite concentrations in follicular fluid may explain differences in fertility between heifers and lactating cows. Reproduction 2010, 139, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Seli, E.; Babayev, E.; Collins, S.C.; Nemeth, G.; Horvath, T.L. Minireview: Metabolism of female reproduction: Regulatory mechanisms and clinical implications. Mol. Endocrinol. 2014, 28, 790–804. [Google Scholar] [CrossRef]
- Qi, X.; Yun, C.; Pang, Y.; Qiao, J. The impact of the gut microbiota on the reproductive and metabolic endocrine system. Gut Microbes 2021, 13, 1894070. [Google Scholar] [CrossRef]
- Chadchan, S.B.; Singh, V.; Kommagani, R. Female reproductive dysfunctions and the gut microbiota. J. Mol. Endocrinol. 2022, 69, R81–R94. [Google Scholar] [CrossRef]
- Mccallum, G.; Tropini, C. The gut microbiota and its biogeography. Nat. Rev. Microbiol. 2024, 22, 105–118. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, P.; Geng, Q.; Fan, H.; Gong, Y.; Hu, Y.; Shan, L.; Sun, Y.; Shen, W.; Zhou, Y. Disrupted spermatogenesis in a metabolic syndrome model: The role of vitamin A metabolism in the gut–testis axis. Gut 2021, 71, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.P. Regulation of signal transduction pathways by estrogen and progesterone. Annu. Rev. Physiol. 2005, 67, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; Von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, L.; Yi, K.; Zhang, B.; Li, C.; Zhou, X. The effects of microbiota on reproductive health: A review. Crit. Rev. Food Sci. 2022. ahead of print. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen–gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; Ma, X.; Du, L.; Jia, Z.; Cui, X.; Yu, L.; Yang, J.; Xiao, L.; Zhang, B.; et al. Translocation of vaginal microbiota is involved in impairment and protection of uterine health. Nat. Commun. 2021, 12, 4191. [Google Scholar] [CrossRef]
- Franasiak, J.M.; Scott, R.J. Introduction: Microbiome in human reproduction. Fertil. Steril. 2015, 104, 1341–1343. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; Li, C.; Jing, J.; Li, Z.; Sun, S.; Xue, T.; Zhang, K.; Xue, M.; Cao, C.; et al. Effects of gut microbiota on omega-3-mediated ovary and metabolic benefits in polycystic ovary syndrome mice. J. Ovarian Res. 2023, 16, 138. [Google Scholar] [CrossRef]
- Menon, R.; Watson, S.E.; Thomas, L.N.; Allred, C.D.; Dabney, A.; Azcarate-Peril, M.A.; Sturino, J.M. Diet complexity and estrogen receptor beta status affect the composition of the murine intestinal microbiota. Appl. Environ. Microbiol. 2013, 79, 5763–5773. [Google Scholar] [CrossRef]
- Egbase, P.E.; Al-Sharhan, M.; Al-Othman, S.; Al-Mutawa, M.; Udo, E.E.; Grudzinskas, J.G. Incidence of microbial growth from the tip of the embryo transfer catheter after embryo transfer in relation to clinical pregnancy rate following in-vitro fertilization and embryo transfer. Hum. Reprod. 1996, 11, 1687–1689. [Google Scholar] [CrossRef]
- Luo, Z.; Yong, K.; Luo, Q.; Du, Z.; Ma, L.; Huang, Y.; Zhou, T.; Yao, X.; Shen, L.; Yu, S.; et al. Altered fecal microbiome and correlations of the metabolome with plasma metabolites in dairy cows with left displaced abomasum. Microbiol. Spectr. 2022, 10, e197222. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Gao, H.; Fan, Y.; Chen, S.; Li, Y.; Liu, R.; Li, T.; Yin, C. Gut microbiota disorder induces liver dysfunction in polycystic ovary syndrome rats’ model by regulating metabolite rosmarinic acid. Life Sci. 2023, 330, 121912. [Google Scholar] [CrossRef] [PubMed]
- Weigel, K.A. Prospects for improving reproductive performance through genetic selection. Anim. Reprod. Sci. 2006, 96, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Hyttel, P.; Greve, T.; Callesen, H. Ultrastructure of oocyte maturation and fertilization in superovulated cattle. Prog. Clin. Biol. Res. 1989, 296, 287–297. [Google Scholar] [PubMed]
- Sirard, M.A.; Richard, F.; Blondin, P.; Robert, C. Contribution of the oocyte to embryo quality. Theriogenology 2006, 65, 126–136. [Google Scholar] [CrossRef]
- Guo, Y.; Qi, Y.; Yang, X.; Zhao, L.; Wen, S.; Liu, Y.; Tang, L. Association between polycystic ovary syndrome and gut microbiota. PLoS ONE 2016, 11, e153196. [Google Scholar] [CrossRef]
- Sommer, F.; Backhed, F. The gut microbiota-masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Kriebs, A. IL-22 links gut microbiota to PCOS. Nat. Rev. Endocrinol. 2019, 15, 565. [Google Scholar] [CrossRef] [PubMed]
- Consortium, H.M.P. Structure, function, and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Manor, O.; Dai, C.L.; Kornilov, S.A.; Smith, B.; Price, N.D.; Lovejoy, J.C.; Gibbons, S.M.; Magis, A.T. Health and disease markers correlate with gut microbiome composition across thousands of people. Nat. Commun. 2020, 11, 5206. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zheng, Z.; Yu, L.; Wu, S.; Sun, L.; Wu, S.; Xu, Q.; Cai, S.; Qin, N.; Bao, W. Examination of the temporal and spatial dynamics of the gut microbiome in newborn piglets reveals distinct microbial communities in six intestinal segments. Sci. Rep. 2019, 9, 3453. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M.C. what is the healthy gut microbiota composition? a changing ecosystem across age, environment, diet, and diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Zierer, J.; Jackson, M.A.; Kastenmuller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef] [PubMed]
- de la Barca, J.M.C.; Boueilh, T.; Simard, G.; Boucret, L.; Ferre-L’Hotellier, V.; Tessier, L.; Gadras, C.; E Bouet, P.; Descamps, P.; Procaccio, V.; et al. Targeted metabolomics reveals reduced levels of polyunsaturated choline plasmalogens and a smaller dimethylarginine/arginine ratio in the follicular fluid of patients with a diminished ovarian reserve. Hum. Reprod. 2017, 32, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Pacella-Ince, L.; Zander-Fox, D.L.; Lane, M. Mitochondrial SIRT5 is present in follicular cells and is altered by reduced ovarian reserve and advanced maternal age. Reprod. Fertil. Dev. 2014, 26, 1072–1083. [Google Scholar] [CrossRef]
- Revelli, A.; Delle, P.L.; Casano, S.; Molinari, E.; Massobrio, M.; Rinaudo, P. Follicular fluid content and oocyte quality: From single biochemical markers to metabolomics. Reprod. Biol. Endocrinol. 2009, 7, 40. [Google Scholar] [CrossRef]
- Burns, D.S.; Jimenez-Krassel, F.; Ireland, J.; Knight, P.G.; Ireland, J.J. Numbers of antral follicles during follicular waves in cattle: Evidence for high variation among animals, very high repeatability in individuals, and an inverse association with serum follicle-stimulating hormone concentrations. Biol. Reprod. 2005, 73, 54–62. [Google Scholar] [CrossRef]
- Sakaguchi, K.; Yanagawa, Y.; Yoshioka, K.; Suda, T.; Katagiri, S.; Nagano, M. Relationships between the antral follicle count, steroidogenesis, and secretion of follicle-stimulating hormone and anti-Mullerian hormone during follicular growth in cattle. Reprod. Biol. Endocrinol. 2019, 17, 88. [Google Scholar] [CrossRef]
- Demetz, E.; Schroll, A.; Auer, K.; Heim, C.; Patsch, J.R.; Eller, P.; Theurl, M.; Theurl, I.; Theurl, M.; Seifert, M.; et al. The arachidonic acid metabolome serves as a conserved regulator of cholesterol metabolism. Cell Metab. 2014, 20, 787–798. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, M.; Wang, X. Evolutionary conservation analysis of human arachidonic acid metabolism pathway genes. Life Med. 2023, 2, 9. [Google Scholar] [CrossRef]
- Oh, M.; Jang, S.Y.; Lee, J.; Kim, J.W.; Jung, Y.; Kim, J.; Seo, J.; Han, T.-S.; Jang, E.; Son, H.Y.; et al. The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism. Nat. Commun. 2023, 14, 5728. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B.; Lee, C.S.; Jang, J.H.; Ghim, J.; Kim, Y.J.; You, S.; Hwang, D.; Suh, P.-G.; Ryu, S.H. Phospholipase signalling networks in cancer. Nat. Rev. Cancer 2012, 12, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Edwards, E.M.; Holmes, B.B.; Falck, J.R.; Campbell, W.B. Role of phospholipase C and diacylglyceride lipase pathway in arachidonic acid release and acetylcholine-induced vascular relaxation in rabbit aorta. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H37–H45. [Google Scholar] [CrossRef]
- Martín-Vázquez, E.; Cobo-Vuilleumier, N.; López-Noriega, L.; Lorenzo, P.I.; Gauthier, B.R. The PTGS2/COX2-PGE2 signaling cascade in inflammation: Pro or anti? A case study with type 1 diabetes mellitus. Int. J. Biol. Sci. 2023, 19, 4157–4165. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, T.; Song, J.; Deng, J.; Sun, Z. Study on follicular fluid metabolomics components at different ages based on lipid metabolism. Reprod. Biol. Endocrin. 2020, 18, 42. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Li, S.; Zhang, Y.; Feng, R.; Huang, R.; Chen, M.; Qian, Y. Metabolic signatures in human follicular fluid identify lysophosphatidylcholine as a predictor of follicular development. Commun. Biol. 2022, 5, 763. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Sun, L.; Cheng, J.; Lu, Y.; Wei, Y.; Qin, G.; Liang, J.; Lan, G. Non-targeted metabolomics reveals metabolic characteristics of porcine atretic follicles. Front. Vet. Sci. 2021, 8, 679947. [Google Scholar] [CrossRef] [PubMed]
- Vrablik, T.L.; Watts, J.L. Polyunsaturated fatty acid derived signaling in reproduction and development: Insights from Caenorhabditis elegans and Drosophila melanogaster. Mol. Reprod. Dev. 2013, 80, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhu, S.; Shu, W.; Guo, Y.; Guan, Y.; Zeng, J.; Wang, H.; Han, L.; Zhang, J.; Liu, X.; et al. Characterization of metabolic patterns in mouse oocytes during meiotic maturation. Mol. Cell 2020, 80, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Homa, S.T.; Brown, C.A. Changes in linoleic acid during follicular development and inhibition of spontaneous breakdown of germinal vesicles in cumulus-free bovine oocytes. J. Reprod. Fertil. 1992, 94, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Marei, W.F.; Wathes, D.C.; Fouladi-Nashta, A.A. Impact of linoleic acid on bovine oocyte maturation and embryo development. Reproduction 2010, 139, 979–988. [Google Scholar] [CrossRef]
- Mckenzie, L.J.; Pangas, S.A.; Carson, S.A.; Kovanci, E.; Cisneros, P.; Buster, J.E.; Amato, P.; Matzuk, M.M. Human cumulus granulosa cell gene expression: A predictor of fertilization and embryo selection in women undergoing IVF. Hum. Reprod. 2004, 19, 2869–2874. [Google Scholar] [CrossRef] [PubMed]
- Nuttinck, F.; Gall, L.; Ruffini, S.; Laffont, L.; Clement, L.; Reinaud, P.; Adenot, P.; Grimard, B.; Charpigny, G.; Guienne, B.M.-L. PTGS2-related PGE2 affects oocyte MAPK phosphorylation and meiosis progression in cattle: Late effects on early embryonic development. Biol. Reprod. 2011, 84, 1248–1257. [Google Scholar] [CrossRef]
- Marei, W.F.; Abayasekara, D.R.; Wathes, D.C.; Fouladi-Nashta, A.A. Role of PTGS2-generated PGE2 during gonadotrophin-induced bovine oocyte maturation and cumulus cell expansion. Reprod. Biomed. Online 2014, 28, 388–400. [Google Scholar] [CrossRef]
- Boruszewska, D.; Kowalczyk-Zieba, I.; Suwik, K.; Staszkiewicz-Chodor, J.; Jaworska, J.; Lukaszuk, K.; Woclawek-Potocka, I. Prostaglandin E(2) affects in vitro maturation of bovine oocytes. Reprod. Biol. Endocrinol. 2020, 18, 40. [Google Scholar] [CrossRef]
- Rojas, J.; Chavez-Castillo, M.; Olivar, L.C.; Calvo, M.; Mejias, J.; Rojas, M.; Morillo, J.; Bermúdez, V. Physiologic course of female reproductive function: A molecular look into the prologue of life. J. Pregnancy 2015, 2015, 715735. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Kornman, K.S.; Loesche, W.J. Effects of estradiol and progesterone on Bacteroides melaninogenicus and Bacteroides gingivalis. Infect. Immun. 1982, 35, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Nuriel-Ohayon, M.; Neuman, H.; Ziv, O.; Belogolovski, A.; Barsheshet, Y.; Bloch, N.; Uzan, A.; Lahav, R.; Lahav, A.; Frishman, S. Progesterone increases bifidobacterium relative abundance during late pregnancy. Cell Rep. 2019, 27, 730–736. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Wang, S.; Wu, X.; Zhao, R.; Chang, S.; Ma, C.; Song, S.; Zeng, S. Multi-Omics Reveals the Role of Arachidonic Acid Metabolism in the Gut–Follicle Axis for the Antral Follicular Development of Holstein Cows. Int. J. Mol. Sci. 2024, 25, 9521. https://doi.org/10.3390/ijms25179521
Guo Y, Wang S, Wu X, Zhao R, Chang S, Ma C, Song S, Zeng S. Multi-Omics Reveals the Role of Arachidonic Acid Metabolism in the Gut–Follicle Axis for the Antral Follicular Development of Holstein Cows. International Journal of Molecular Sciences. 2024; 25(17):9521. https://doi.org/10.3390/ijms25179521
Chicago/Turabian StyleGuo, Yajun, Shiwei Wang, Xuan Wu, Rong Zhao, Siyu Chang, Chen Ma, Shuang Song, and Shenming Zeng. 2024. "Multi-Omics Reveals the Role of Arachidonic Acid Metabolism in the Gut–Follicle Axis for the Antral Follicular Development of Holstein Cows" International Journal of Molecular Sciences 25, no. 17: 9521. https://doi.org/10.3390/ijms25179521