
ToxDAR: A Workflow Software for Analyzing Toxicologically Relevant Proteomic and Transcriptomic Data, from Data Preparation to Toxicological Mechanism Elucidation

Abstract
:1. Introduction
2. Result
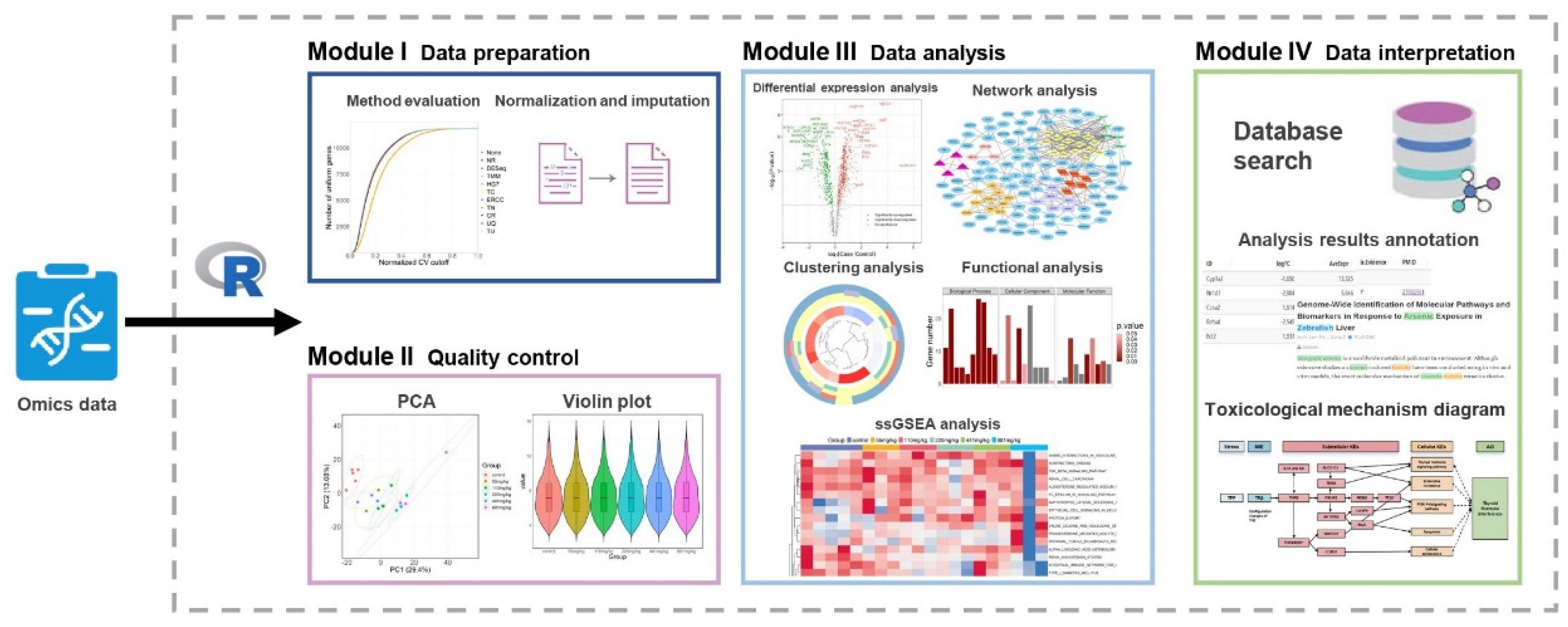
2.1. Software Framework
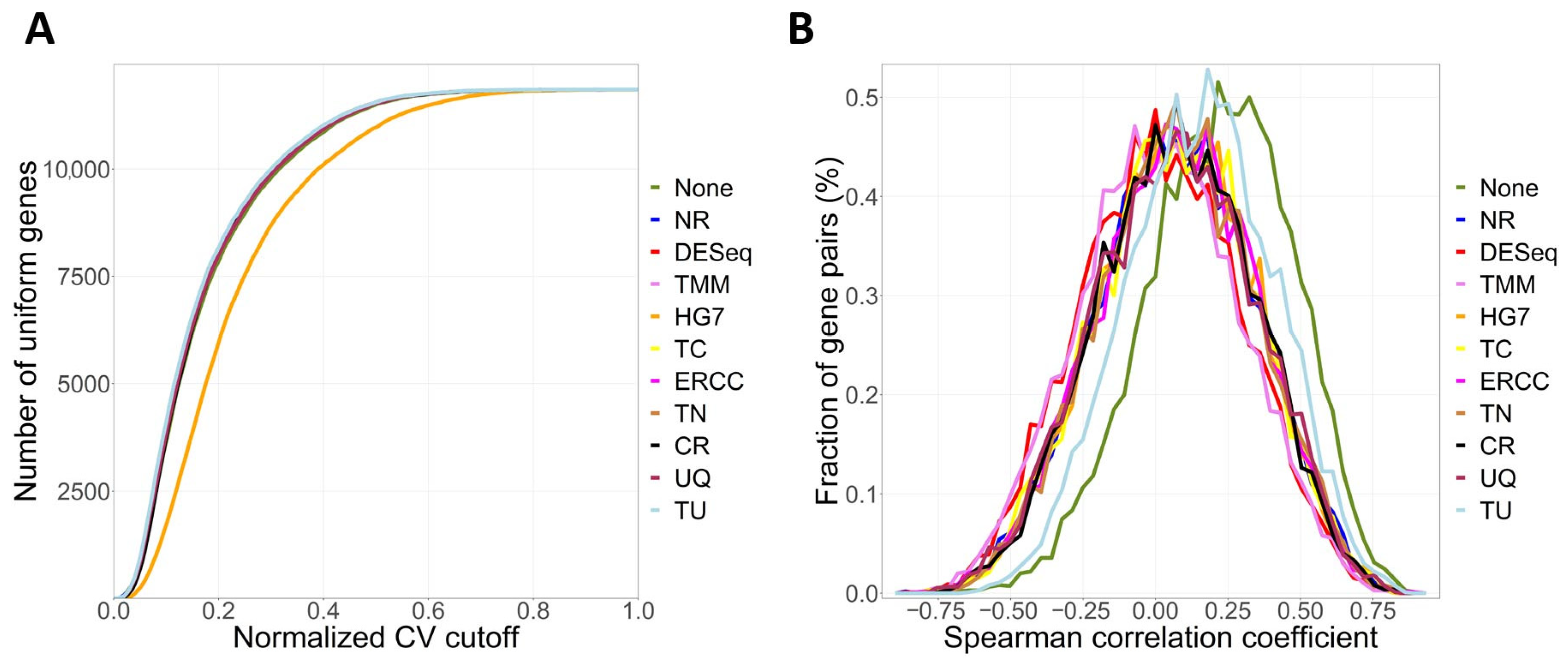
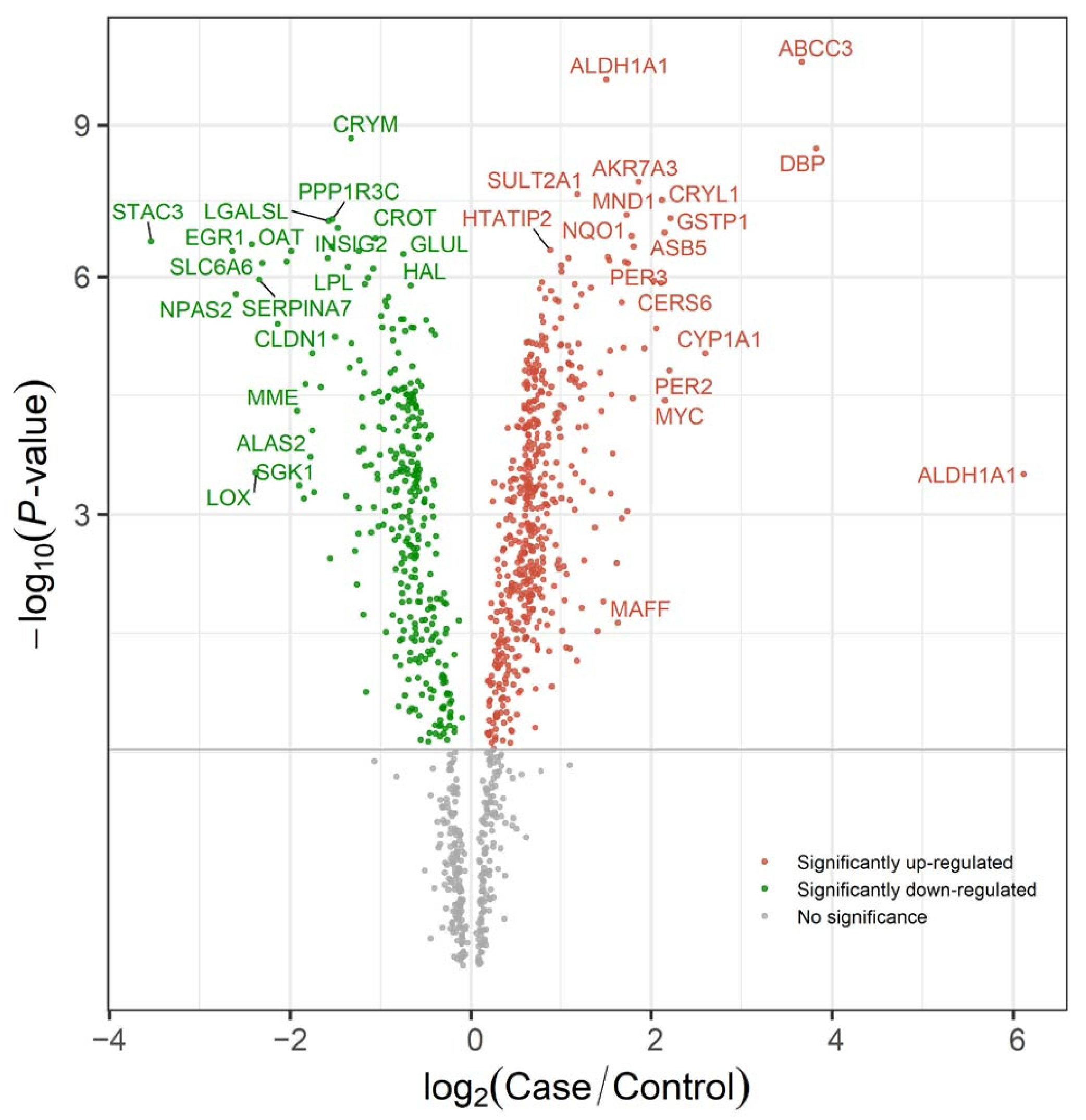
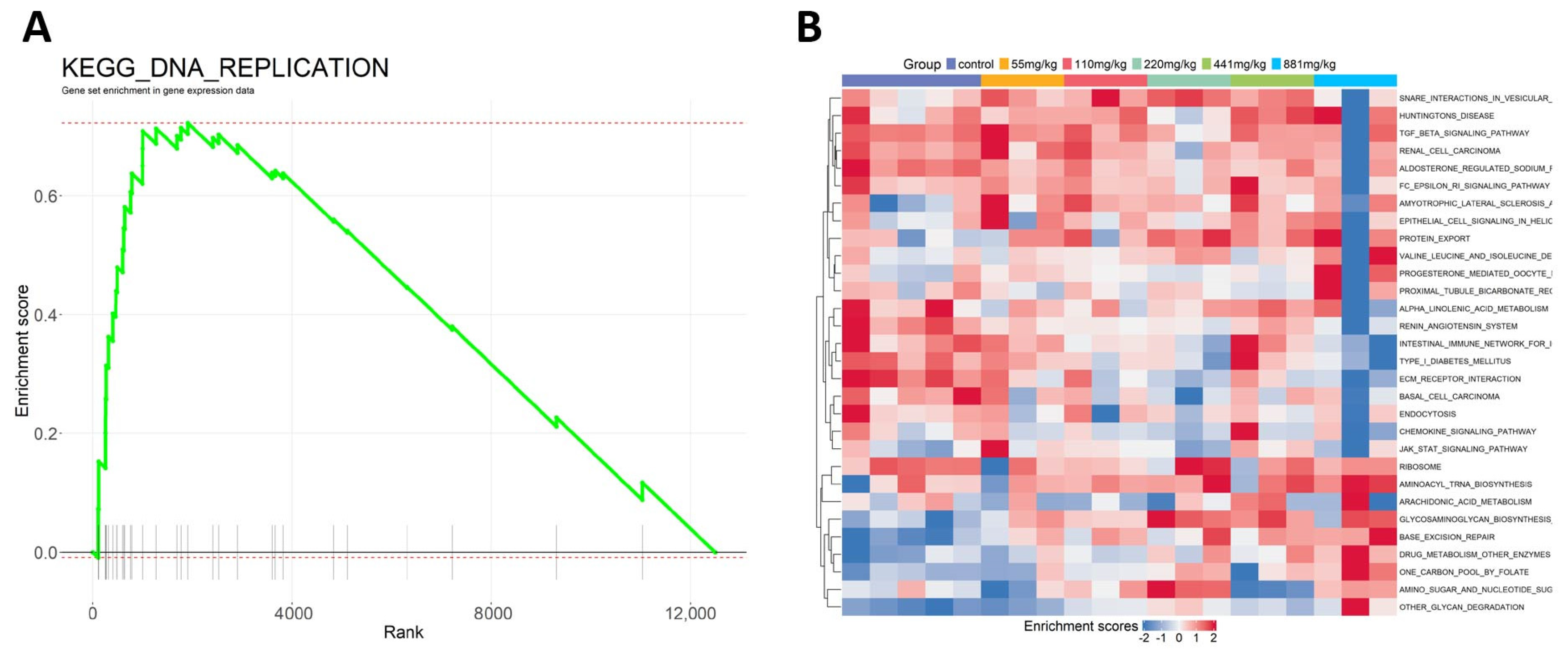
2.2. Software Function
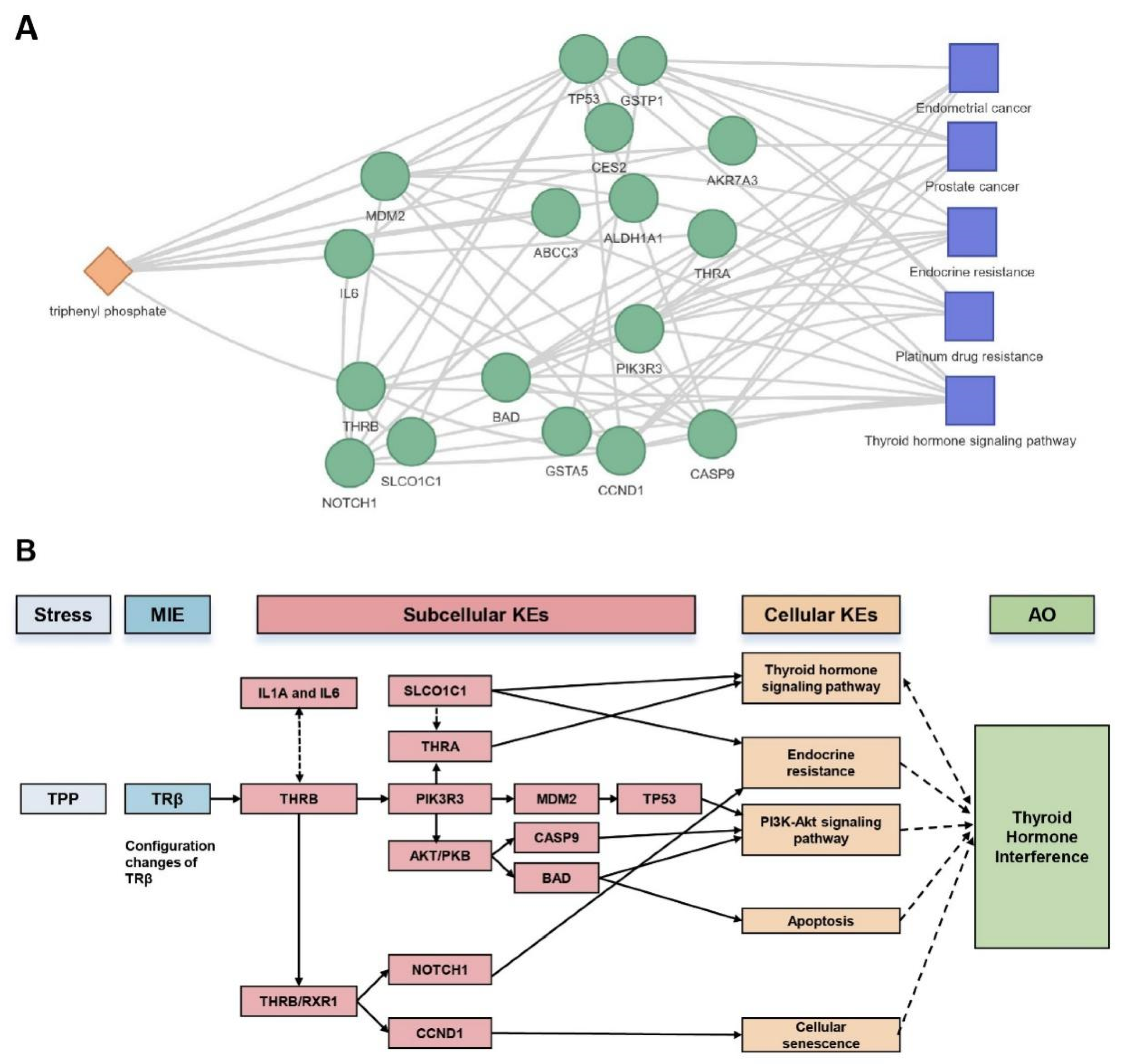
2.3. Research Case: Toxicological Mechanism Analysis of Public Transcriptome Data in L02 Cell Line Post-Triphenyl Phosphate (TPP) Exposure
3. Discussion
4. Materials and Methods
4.1. Primary Functions
4.2. Data Sources
4.3. Toxicological Classification System
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krewski, D.; Andersen, M.E.; Mantus, E.; Zeise, L. Toxicity testing in the 21st century: Implications for human health risk assessment. Risk Anal. 2009, 29, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Yan, K.; Sang, R.; Wang, W.; Liu, X.; Yu, M.; Liu, X.; Qiu, Q.; Zhang, X. Integrated network toxicology, molecular docking, and in vivo experiments to elucidate molecular mechanism of aflatoxin B1 hepatotoxicity. Ecotoxicol. Environ. Saf. 2024, 275, 116278. [Google Scholar] [CrossRef] [PubMed]
- Krewski, D.; Andersen, M.E.; Tyshenko, M.G.; Krishnan, K.; Hartung, T.; Boekelheide, K.; Wambaugh, J.F.; Jones, D.; Whelan, M.; Thomas, R.; et al. Toxicity testing in the 21st century: Progress in the past decade and future perspectives. Arch. Toxicol. 2019, 94, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Pognan, F.; Beilmann, M.; Boonen, H.C.M.; Czich, A.; Dear, G.; Hewitt, P.; Mow, T.; Oinonen, T.; Roth, A.; Steger-Hartmann, T.; et al. The evolving role of investigative toxicology in the pharmaceutical industry. Nat. Rev. Drug Discov. 2023, 22, 317–335. [Google Scholar] [CrossRef]
- Hartung, T. Toxicology for the twenty-first century. Nature 2009, 460, 208–212. [Google Scholar] [CrossRef]
- Tujios, S.; Fontana, R.J. Mechanisms of drug-induced liver injury: From bedside to bench. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 202–211. [Google Scholar] [CrossRef]
- Schork, N.J. Personalized medicine: Time for one-person trials. Nature 2015, 520, 609–611. [Google Scholar] [CrossRef]
- Duan, L.; Guo, L.; Wang, L.; Yin, Q.; Zhang, C.-M.; Zheng, Y.-G.; Liu, E.H. Application of metabolomics in toxicity evaluation of traditional Chinese medicines. Chin. Med. 2018, 13, 60. [Google Scholar] [CrossRef] [PubMed]
- Inadera, H.; Uchida, M.; Shimomura, A. Advances in “omics” technologies for toxicological research. Nippon. Eiseigaku Zasshi 2007, 62, 18–31. [Google Scholar] [CrossRef]
- Goh, H.-H.; Ng, C.L.; Loke, K.-K. Functional Genomics. Adv. Exp. Med. Biol. 2018, 1102, 11–30. [Google Scholar] [CrossRef]
- Hermansen, G.M.M.; Sazinas, P.; Kofod, D.; Millard, A.; Andersen, P.S.; Jelsbak, L. Transcriptomic profiling of interacting nasal staphylococci species reveals global changes in gene and non-coding RNA expression. FEMS Microbiol. Lett. 2018, 365, fny004. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Yang, Q.; Xu, L.; Rao, Z.; Cao, D.; Gao, M.; Liu, S. Protein target identification and toxicological mechanism investigation of silver nanoparticles-induced hepatotoxicity by integrating proteomic and metallomic strategies. Part. Fibre Toxicol. 2019, 16, 46. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.; Serchi, T.; Murk, A.J.; Gutleb, A.C. The Added Value of Proteomics for Toxicological Studies. J. Toxicol. Environ. Health Part B Crit. Rev. 2014, 17, 225–246. [Google Scholar] [CrossRef]
- Aardema, M.J.; MacGregor, J.T. Toxicology and genetic toxicology in the new era of “toxicogenomics”: Impact of “-omics” technologies. Mutat. Res. /Fundam. Mol. Mech. Mutagen. 2002, 499, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Saarimäki, L.A.; Pavel, A.; Del Giudice, G.; Fratello, M.; Cattelani, L.; Federico, A.; Laurino, O.; Marwah, V.S.; Fortino, V.; et al. Nextcast: A software suite to analyse and model toxicogenomics data. Comput. Struct. Biotechnol. J. 2022, 20, 1413–1426. [Google Scholar] [CrossRef]
- Aniba, M.R.; Poch, O.; Thompson, J.D. Issues in bioinformatics benchmarking: The case study of multiple sequence alignment. Nucleic Acids Res. 2010, 38, 7353–7363. [Google Scholar] [CrossRef]
- Richard, A.M.; Judson, R.S.; Houck, K.A.; Grulke, C.M.; Volarath, P.; Thillainadarajah, I.; Yang, C.; Rathman, J.; Martin, M.T.; Wambaugh, J.F.; et al. ToxCast Chemical Landscape: Paving the Road to 21st Century Toxicology. Chem. Res. Toxicol. 2016, 29, 1225–1251. [Google Scholar] [CrossRef]
- Igarashi, Y.; Nakatsu, N.; Yamashita, T.; Ono, A.; Ohno, Y.; Urushidani, T.; Yamada, H. Open TG-GATEs: A large-scale toxicogenomics database. Nucleic Acids Res. 2015, 43, D921–D927. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef]
- Samur, M.K. RTCGAToolbox: A new tool for exporting TCGA Firehose data. PLoS ONE 2014, 9, e106397. [Google Scholar] [CrossRef]
- Gao, J.; Mazor, T.; de Bruijn, I.; Abeshouse, A.; Baiceanu, D.; Erkoc, Z.; Gross, B.; Higgins, D.; Jagannathan, P.K.; Kalletla, K.; et al. Abstract 207: The cBioPortal for Cancer Genomics. Cancer Res. 2021, 81, 207. [Google Scholar] [CrossRef]
- Marukatat, S. Tutorial on PCA and approximate PCA and approximate kernel PCA. Artif. Intell. Rev. 2022, 56, 5445–5477. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [PubMed]
- Waters, M. Systems Toxicology and the Chemical Effects in Biological Systems (CEBS) Knowledge Base. Environ. Health Perspect. 2003, 111, 15–28. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2021. Nucleic Acids Res. 2020, 49, D1138–D1143. [Google Scholar] [CrossRef]
- Olker, J.H.; Elonen, C.M.; Pilli, A.; Anderson, A.; Kinziger, B.; Erickson, S.; Skopinski, M.; Pomplun, A.; LaLone, C.A.; Russom, C.L.; et al. The ECOTOXicology Knowledgebase: A Curated Database of Ecologically Relevant Toxicity Tests to Support Environmental Research and Risk Assessment. Environ. Toxicol. Chem. 2022, 41, 1520–1539. [Google Scholar] [CrossRef]
- Wang, Y.; LêCao, K.-A. Managing batch effects in microbiome data. Brief. Bioinform. 2019, 21, 1954–1970. [Google Scholar] [CrossRef]
- Goh, W.W.B.; Wang, W.; Wong, L. Why Batch Effects Matter in Omics Data, and How to Avoid Them. Trends Biotechnol. 2017, 35, 498–507. [Google Scholar] [CrossRef]
- Li, X.; Brock, G.N.; Rouchka, E.C.; Cooper, N.G.F.; Wu, D.; O’Toole, T.E.; Gill, R.S.; Eteleeb, A.M.; O’Brien, L.; Rai, S.N. A comparison of per sample global scaling and per gene normalization methods for differential expression analysis of RNA-seq data. PLoS ONE 2017, 12, e0176185. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed]
- Bushel, P.R.; Ferguson, S.S.; Ramaiahgari, S.C.; Paules, R.S.; Auerbach, S.S. Comparison of Normalization Methods for Analysis of TempO-Seq Targeted RNA Sequencing Data. Front. Genet. 2020, 11, 594. [Google Scholar] [CrossRef]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.-C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M. TPM, FPKM, or Normalized Counts? A Comparative Study of Quantification Measures for the Analysis of RNA-seq Data from the NCI Patient-Derived Models Repository. J. Transl. Med. 2021, 19, 269. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, W.; Jin, X.; Ji, H.; Wang, H.; Glusman, G.; Robinson, M.; Liu, L.; Ruan, J.; Gao, S. NormExpression: An R Package to Normalize Gene Expression Data Using Evaluated Methods. Front. Genet. 2019, 10, 400. [Google Scholar] [CrossRef]
- null, n. The External RNA Controls Consortium: A progress report. Nat. Methods 2005, 2, 731–734. [Google Scholar] [CrossRef]
- Devonshire, A.S.; Elaswarapu, R.; Foy, C.A. Evaluation of external RNA controls for the standardisation of gene expression biomarker measurements. BMC Genom. 2010, 11, 662. [Google Scholar] [CrossRef] [PubMed]
- Kouadjo, K.E.; Nishida, Y.; Cadrin-Girard, J.F.; Yoshioka, M.; St-Amand, J. Housekeeping and tissue-specific genes in mouse tissues. BMC Genom. 2007, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, D.; Deng, W.-M. Standardization of Single-Cell RNA-Sequencing Analysis Workflow to Study Drosophila Ovary. Methods Mol. Biol. 2023, 2677, 151–171. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- Salkovic, E.; Bensmail, H. A Novel Bayesian Outlier Score Based on the Negative Binomial Distribution for Detecting Aberrantly Expressed Genes in RNA-Seq Gene Expression Count Data. IEEE Access 2021, 9, 75789–75800. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010. [Google Scholar] [CrossRef]
- Bleazard, T.; Lamb, J.A.; Griffiths-Jones, S. Bias in microRNA functional enrichment analysis. Bioinformatics 2015, 31, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Valls-Margarit, J.; Piñero, J.; Füzi, B.; Cerisier, N.; Taboureau, O.; Furlong, L.I. Assessing network-based methods in the context of system toxicology. Front. Pharmacol. 2023, 14, 1225697. [Google Scholar] [CrossRef]
- Wang, X.; Li, F.; Liu, J.; Ji, C.; Wu, H. Transcriptomic, proteomic and metabolomic profiling unravel the mechanisms of hepatotoxicity pathway induced by triphenyl phosphate (TPP). Ecotoxicol. Environ. Saf. 2020, 205, 111126. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, X.; Wang, P.; Zhang, Y.; He, S.; Li, Y.; Li, S.; Liang, K.; Liao, S.; Gao, Y.; et al. Low concentration triphenyl phosphate fuels proliferation and migration of hepatocellular carcinoma cells. Environ. Toxicol. 2022, 37, 2445–2459. [Google Scholar] [CrossRef]
- Hu, C.; Yang, S.; Zhang, T.; Ge, Y.; Chen, Z.; Zhang, J.; Pu, Y.; Liang, G. Organoids and organoids-on-a-chip as the new testing strategies for environmental toxicology-applications & advantages. Environ. Int. 2024, 184, 108415. [Google Scholar] [CrossRef]
- Brooks, A.; Liang, X.; Zhang, Y.; Zhao, C.-X.; Roberts, M.S.; Wang, H.; Zhang, L.; Crawford, D.H.G. Liver organoid as a 3D in vitro model for drug validation and toxicity assessment. Pharmacol. Res. 2021, 169, 105608. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Ning, W.; Acharya, A.; Li, S.; Schmalz, G.; Huang, S. Identification of Key Pyroptosis-Related Genes and Distinct Pyroptosis-Related Clusters in Periodontitis. Front. Immunol. 2022, 13, 862049. [Google Scholar] [CrossRef]
- Valero-Mora, P.M. ggplot2:Elegant Graphics for Data Analysis. J. Stat. Softw. 2010, 35, 1–3. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Davis, A.P.; Wiegers, T.C.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): Update 2023. Nucleic Acids Res. 2023, 51, D1257–D1262. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Evelo, C.T.; Willighagen, E.L. Providing Adverse Outcome Pathways from the AOP-Wiki in a Semantic Web Format to Increase Usability and Accessibility of the Content. Appl. Vitr. Toxicol. 2022, 8, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2016, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Schriml, L.M.; Arze, C.; Nadendla, S.; Chang, Y.-W.W.; Mazaitis, M.; Felix, V.; Feng, G.; Kibbe, W.A. Disease Ontology: A backbone for disease semantic integration. Nucleic Acids Res. 2012, 40, D940–D946. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2020, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Peters, F.T. Principles of Forensic Toxicology, 5th ed.; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, M.H.; Zhao, X.; Feng, J.H. Ammonia exposure induced intestinal inflammation injury mediated by intestinal microbiota in broiler chickens via TLR4/TNF-α signaling pathway. Ecotoxicol. Environ. Saf. 2021, 226, 112832. [Google Scholar] [CrossRef]
- Borroni, E.; Pesatori, A.C.; Bollati, V.; Buoli, M.; Carugno, M. Air pollution exposure and depression: A comprehensive updated systematic review and meta-analysis. Environ. Pollut. 2022, 292 Pt A, 118332. [Google Scholar] [CrossRef]
- Tang, S.L.; Xie, J.J.; Wu, W.D.; Yi, B.; Liu, L.; Zhang, H.F. High ammonia exposure regulates lipid metabolism in the pig skeletal muscle via mTOR pathway. Sci. Total Environ. 2020, 740, 139917. [Google Scholar] [CrossRef]
- Liu, M.J.; Guo, H.Y.; Zhu, K.C.; Liu, B.S.; Liu, B.; Guo, L.; Zhang, N.; Yang, J.W.; Jiang, S.G.; Zhang, D.-C. Effects of acute ammonia exposure and recovery on the antioxidant response and expression of genes in the Nrf2-Keap1 signaling pathway in the juvenile golden pompano (Trachinotus ovatus). Aquat. Toxicol. 2021, 240, 105969. [Google Scholar] [CrossRef]
- Liang, L.Y.; Huang, Z.B.; Li, N.; Wang, D.M.; Ding, L.; Shi, H.T.; Hong, M.L. Effects of ammonia exposure on antioxidant function, immune response and NF-κB pathway in Chinese Strip-necked Turtle (Mauremys sinensis). Aquat. Toxicol. 2020, 229, 105621. [Google Scholar] [CrossRef] [PubMed]
- Hindfelt, B.; Plum, F.; Duffy, T.E. Effect of acute ammonia intoxication on cerebral metabolism in rats with portacaval shunts. J. Clin. Investig. 1977, 59, 386–396. [Google Scholar] [CrossRef]
- Kosenko, E.; Montoliu, C.; Giordano, G.; Kaminsky, Y.; Venediktova, N.; Buryanov, Y.; Felipo, V. Acute ammonia intoxication induces an NMDA receptor-mediated increase in poly(ADP-ribose) polymerase level and NAD metabolism in nuclei of rat brain cells. J. Neurochem. 2004, 89, 1101–1110. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Z.; Zhu, M.; Qiu, F.; Zhou, Y.B.; Shu, P.; Liu, N.; Wei, C.X.; Xiang, S.L. TNFAIP1 Mediates Formaldehyde-Induced Neurotoxicity by Inhibiting the Akt/CREB Pathway in N2a Cells. Neurotox Res. 2020, 38, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.X.; Tian, P.; Zhai, M.M.; Lei, X.D.; Yang, Z.H.; Liu, Y.; Liu, M.T.; Huang, H.; Zhang, X.R.; Yang, X.; et al. Formaldehyde regulates vascular tensions through nitric oxide-cGMP signaling pathway and ion channels. Chemosphere 2018, 193, 60–73. [Google Scholar] [CrossRef]
- Park, J.; Kang, G.H.; Kim, Y.; Lee, J.Y.; Song, J.A.; Hwang, J.H. Formaldehyde exposure induces differentiation of regulatory T cells via the NFAT-mediated T cell receptor signalling pathway in Yucatan minipigs. Sci. Rep. 2022, 12, 8149. [Google Scholar] [CrossRef]
- Medda, N.; De, S.K.; Maiti, S. Different mechanisms of arsenic related signaling in cellular proliferation, apoptosis and neo-plastic transformation. Ecotoxicol. Environ. Saf. 2021, 208, 111752. [Google Scholar] [CrossRef]
- Ding, X.X.; Ding, E.M.; Yin, H.Y.; Mei, P.; Chen, H.; Han, L.; Zhang, H.D.; Wang, J.F.; Wang, H.; Zhu, B.L. Serum hsa-circ-0025244 as a biomarker in Chinese occupational mercury-exposed population and mediate apoptosis through JNK/p38 MAPK signaling pathway. J. Trace Elem. Med. Biol. 2022, 74, 127057. [Google Scholar] [CrossRef]
- Li, N.; Wen, L.D.; Wang, F.Y.; Li, T.G.; Zheng, H.D.; Wamg, T.L.; Qiao, M.W.; Huang, X.Q.; Song, L.J.; Erkigul, B. Alleviating effects of pea peptide on oxidative stress injury induced by lead in PC12 cells via Keap1/Nrf2/TXNIP signaling pathway. Ecotoxicol. Environ. Saf. 2021, 207, 111231. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Lin, C.-Y.; Farmand, F.; Sindhu, R.K. Superoxide dismutase, catalase, glutathione peroxidase and NADPH oxidase in lead-induced hypertension. Kidney Int. 2003, 63, 186–194. [Google Scholar] [CrossRef]
- Osorio-Rico, L.; Santamaria, A.; Galván-Arzate, S. Thallium Toxicity: General Issues, Neurological Symptoms, and Neurotoxic Mechanisms. Adv. Neurobiol. 2017, 18, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Kaviyarasi, R.; Rituraj, C.; Haritha, M.; Rajeshwari, K.; Ademola, C.F.; Harishkumar, M.; Balachandar, V.; Alex, G.; Abilash, V.G. Molecular mechanism of heavy metals (Lead, Chromium, Arsenic, Mercury, Nickel and Cadmium)—Induced hepatotoxicity—A review. Chemosphere 2021, 271, 129735. [Google Scholar] [CrossRef]
- Bian, X.K.; Guo, J.L.; Xu, S.X.; Han, Y.W.; Lee, S.C.; Zhao, J.Z. Hexavalent chromium induces centrosome amplification through ROS-ATF6-PLK4 pathway in colon cancer cells. Cell Biol. Int. 2022, 46, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Liu, H.; Prabhakaran, K.; Zhang, X.; Borowitz, J.L.; Isom, G.E. HIF-1α activation by a redox-sensitive pathway mediates cyanide-induced BNIP3 upregulation and mitochondrial-dependent cell death. Free. Radic. Biol. Med. 2007, 43, 117–127. [Google Scholar] [CrossRef]
- Camacho-Pérez, M.R.; Covantes-Rosales, C.E.; Toledo-Ibarra, G.A.; Mercado-Salgado, U.; Ponce-Regalado, M.D.; Díaz-Resendiz, K.J.G.; Girón-Pérez, M.I. Organophosphorus Pesticides as Modulating Substances of Inflammation through the Cholinergic Pathway. Int. J. Mol. Sci. 2022, 23, 4523. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Ma, K.C.; Choi, A.M.K. Carbon monoxide in lung cell physiology and disease. Am. J. Physiol. Cell Physiol. 2018, 314, C211–C227. [Google Scholar] [CrossRef]
- Zhang, T.T.; Ma, P.; Yin, X.Y.; Yang, D.Y.; Li, D.P.; Tang, R. Acute Nitrite Exposure Induces Dysfunction and Oxidative Damage in Grass Carp Isolated Hemocytes. J. Aquat. Anim. Health 2022, 34, 58–68. [Google Scholar] [CrossRef]
- Zheng, S.; Jin, X.; Chen, M.; Shi, Q.; Zhang, H.; Xu, S. Hydrogen sulfide exposure induces jejunum injury via CYP450s/ROS pathway in broilers. Chemosphere 2019, 214, 25–34. [Google Scholar] [CrossRef]
- Chi, Q.; Wang, D.; Hu, X.; Li, S.; Li, S. Hydrogen Sulfide Gas Exposure Induces Necroptosis and Promotes Inflammation through the MAPK/NF-κB Pathway in Broiler Spleen. Oxidative Med. Cell. Longev. 2019, 2019, 8061823. [Google Scholar] [CrossRef]
- Jamshidifard, S.; Koushkbaghi, S.; Hosseini, S.; Rezaei, S.; Karamipour, A.; Jafari, R.A.; Irani, M. Incorporation of UiO-66-NH2 MOF into the PAN/chitosan nanofibers for adsorption and membrane filtration of Pb(II), Cd(II) and Cr(VI) ions from aqueous solutions. J. Hazard. Mater. 2019, 368, 10–20. [Google Scholar] [CrossRef]
- Chi, Q.; Chi, X.; Hu, X.; Wang, S.; Zhang, H.; Li, S. The effects of atmospheric hydrogen sulfide on peripheral blood lymphocytes of chickens: Perspectives on inflammation, oxidative stress and energy metabolism. Environ. Res. 2018, 167, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Birková, A.; Hubková, B.; Čižmárová, B.; Bolerázska, B. Current View on the Mechanisms of Alcohol-Mediated Toxicity. Int. J. Mol. Sci. 2021, 22, 9686. [Google Scholar] [CrossRef] [PubMed]
- Mellerick, D.M.; Liu, H. Methanol exposure interferes with morphological cell movements in the Drosophila embryo and causes increased apoptosis in the CNS. J. Neurobiol. 2004, 60, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.; Guo, T.; Shah, P.; Moorthy, B.; Ghose, R. Chlorpromazine-induced hepatotoxicity during inflammation is mediated by TIRAP-dependent signaling pathway in mice. Toxicol. Appl. Pharmacol. 2013, 266, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Soon, Y.S.; Kyoung, S.L.; Yang-Kyu, C.; Hyunjung, J.L.; Hong, G.L.; Yoongho, L.; Young, H.L. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis 2013, 34, 2080–2089. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, H.; Zou, M.; Yuan, Q.; Huang, Z.; Pan, X.; Zhang, W. Nicotine in Inflammatory Diseases: Anti-Inflammatory and Pro-Inflammatory Effects. Front. Immunol. 2022, 13, 826889. [Google Scholar] [CrossRef]
- Chapman, A.G.; Nordström, C.H.; Siesjö, B.K. Influence of phenobarbital anesthesia on carbohydrate and amino acid metabolism in rat brain. Anesthesiology 1978, 48, 175–182. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| External Software Package | Version | Functionality |
|---|---|---|
| NormExpression | V0.1.0 | getNormMatrix; gatherCVs |
| ggord | V1.1.7 | ggord.pca |
| limma | V3.54.2 | model.matrix; lmFit; eBayes |
| clusterProfiler | V4.6.2 | enricher |
| org.Hs.eg.db | V3.16.0 | org.Hs.eg.db |
| gprofiler2 | V0.2.1 | gconvert |
| fgsea | V1.24.0 | fgsea; plotEnrichment |
| igraph | V1.3.5 | graph_from_edgelist; clusters;layout |
| msigdbr | V7.5.1 | msigdbr |
| ComplexHeatmap | V2.14.0 | rowAnnotation; Heatmap |
| dplyr | V1.0.10 | mutate;select; group_by |
| ggplot2 | V3.4.0 | ggplot; ggtitle; theme; geom_point; geom_hline |
| Source | Version | URL |
|---|---|---|
| CTD | v2021-10 | http://ctdbase.org/, accessed on 13 October 2021 |
| AOP-Wiki | v2022-12 | https://aopwiki.org/, accessed on 10 December 2022. |
| KEGG | v0.7.2 | https://www.kegg.jp/, accessed on 5 October 2020. |
| DrugBank | v2020-12-15 | https://go.drugbank.com/, accessed on 15 December 2020. |
| DisGeNet | v7.0 | https://www.disgenet.org/, accessed on 15 October 2022. |
| Disease Ontology | v2021-10-11 | https://disease-ontology.org/, accessed on 11 October 2022. |
| Human Phenotype Ontology | v2021-10-10 | https://hpo.jax.org/app/, accessed on 10 October 2022. |
| PhosphoSitePlus | v6.6 | https://www.phosphosite.org/ |
| UbiBrowser | v2.0 | http://ubibrowser.ncpsb.org.cn, accessed on 18 October 2022. |
| ENCODE | v120 | https://www.encodeproject.org/, accessed on 8 October 2022. |
| STRINGdb | v11.0 | https://string-db.org/, accessed on 18 December 2020. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, P.; Zhang, Z.; Yu, Q.; Wang, Z.; Diao, L.; Li, D. ToxDAR: A Workflow Software for Analyzing Toxicologically Relevant Proteomic and Transcriptomic Data, from Data Preparation to Toxicological Mechanism Elucidation. Int. J. Mol. Sci. 2024, 25, 9544. https://doi.org/10.3390/ijms25179544
Jiang P, Zhang Z, Yu Q, Wang Z, Diao L, Li D. ToxDAR: A Workflow Software for Analyzing Toxicologically Relevant Proteomic and Transcriptomic Data, from Data Preparation to Toxicological Mechanism Elucidation. International Journal of Molecular Sciences. 2024; 25(17):9544. https://doi.org/10.3390/ijms25179544
Chicago/Turabian StyleJiang, Peng, Zuzhen Zhang, Qing Yu, Ze Wang, Lihong Diao, and Dong Li. 2024. "ToxDAR: A Workflow Software for Analyzing Toxicologically Relevant Proteomic and Transcriptomic Data, from Data Preparation to Toxicological Mechanism Elucidation" International Journal of Molecular Sciences 25, no. 17: 9544. https://doi.org/10.3390/ijms25179544