
Body Composition and Alzheimer’s Disease: A Holistic Review
 ,
,  ,
,  , ,
, , 

Abstract
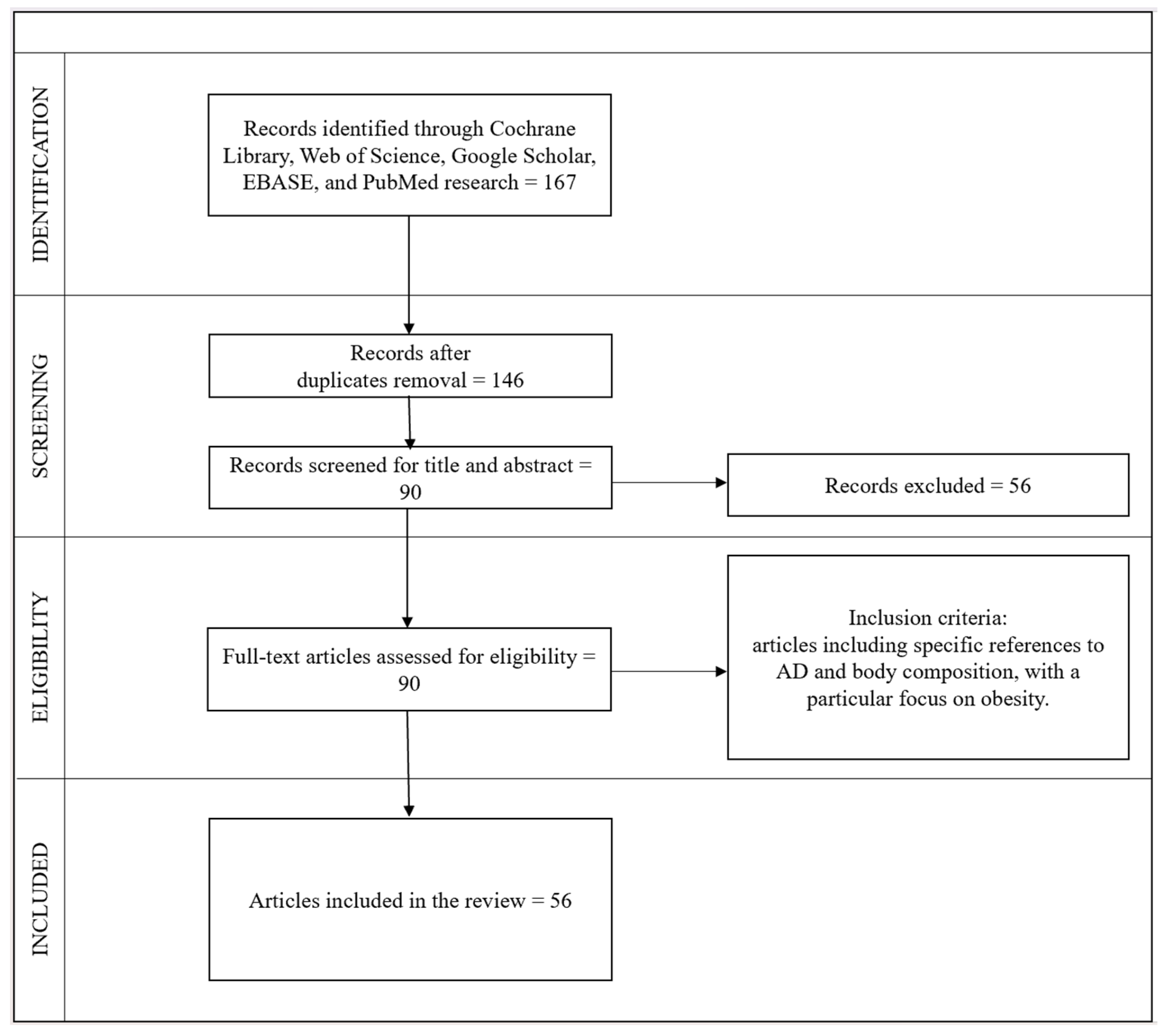
:1. Introduction
2. Results
3. Discussion
3.1. Body Composition and Main Findings in Alzheimer’s Disease
3.2. Lean Mass and Main Findings in Alzheimer’s Disease
3.3. Bone Mass and Main Findings in Alzheimer’s Disease
3.4. Fat Mass and Main Findings in Alzheimer’s Disease from an Endocrinological Perspective
3.5. Fat Mass and Main Findings in Alzheimer’s Disease from an Immunological Perspective
3.6. Fat Mass and Main Findings in Alzheimer’s Disease from a Molecular and Genetic Perspective
3.7. Limitations
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brayne, C.; Miller, B. Dementia and Aging Populations-A Global Priority for Contextualized Research and Health Policy. PLoS Med. 2017, 14, e1002275. [Google Scholar] [CrossRef]
- Scheltens, P.; Strooper, B.D.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s Disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Therriault, J.; Pascoal, T.A.; Benedet, A.L.; Tissot, C.; Savard, M.; Chamoun, M.; Lussier, F.; Kang, M.S.; Berzgin, G.; Wang, T.; et al. Frequency of Biologically Defined Alzheimer Disease in Relation to Age, Sex, APOE Ε4, and Cognitive Impairment. Neurology 2021, 96, e975–e985. [Google Scholar] [CrossRef] [PubMed]
- Maisam, M.; Khan, M.T.; Lodhi, M.S.; Mou, K.; Liu, Z.; Wei, D. Alzheimer’s Disease; Mechanism, Mutations, and Applications of Nano-Medicine. Front. Biosci. 2023, 28, 258. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, M.; Li, W.; Liu, X.; Zhu, M.; Qin, H. Biomarkers Associated with the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2023, 17, 1279046. [Google Scholar] [CrossRef]
- Hazan, J.; Liu, K.Y.; Fox, N.C.; Howard, R. Online Clinical Tools to Support the Use of New Plasma Biomarker Diagnostic Technology in the Assessment of Alzheimer’s Disease: A Narrative Review. Brain Commun. 2023, 5, fcad322. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Singh S, A.; Rushendran, R.; Vellapandian, C.; Prajapati, B. Alzheimer’s Disease: The Role of Extrinsic Factors in Its Development, an Investigation of the Environmental Enigma. Front. Neurol. 2023, 14, 1303111. [Google Scholar] [CrossRef]
- Cao, C.; Fu, G.; Xu, R.; Li, N. Coupling of Alzheimer’s Disease Genetic Risk Factors with Viral Susceptibility and Inflammation. Aging Dis. 2023, 15, 2028–2050. [Google Scholar] [CrossRef]
- Lozupone, M.; Dibello, V.; Sardone, R.; Castellana, F.; Zupo, R.; Lampignano, L.; Bortone, I.; Daniele, A.; Bellomo, A.; Solfrizzi, V.; et al. The Impact of Apolipoprotein E (APOE) Epigenetics on Aging and Sporadic Alzheimer’s Disease. Biology 2023, 12, 1529. [Google Scholar] [CrossRef]
- Huang, W.; Huang, J.; Huang, N.; Luo, Y. The Role of TREM2 in Alzheimer’s Disease: From the Perspective of Tau. Front. Cell Dev. Biol. 2023, 11, 1280257. [Google Scholar] [CrossRef]
- Wen, L.; Bi, D.; Shen, Y. Complement-Mediated Synapse Loss in Alzheimer’s Disease: Mechanisms and Involvement of Risk Factors. Trends Neurosci. 2023, 47, 135–149. [Google Scholar] [CrossRef]
- Gui, W.; Qiu, C.; Shao, Q.; Li, J. Associations of Vascular Risk Factors, APOE and TOMM40 Polymorphisms with Cognitive Function in Dementia-Free Chinese Older Adults: A Community-Based Study. Front. Psychiatry 2021, 12, 617773. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.; Armstrong, N.; Bis, J.C.; Bressler, J.; Chouraki, V.; Giddaluru, S.; Hofer, E.; Ibrahim-Verbaas, C.A.; Kirin, M.; Lahti, J.; et al. Genetic Contributions to Variation in General Cognitive Function: A Meta-Analysis of Genome-Wide Association Studies in the CHARGE Consortium (N=53949). Mol. Psychiatry 2015, 20, 183–192. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, J.; Xu, B.; Ma, X.; Dai, Q.; Li, T.; Xue, F.; Chen, B. The TOMM40 Gene Rs2075650 Polymorphism Contributes to Alzheimer’s Disease in Caucasian, and Asian Populations. Neurosci. Lett. 2016, 628, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Nianogo, R.A.; Rosenwohl-Mack, A.; Yaffe, K.; Carrasco, A.; Hoffmann, C.M.; Barnes, D.E. Risk Factors Associated with Alzheimer Disease and Related Dementias by Sex and Race and Ethnicity in the US. JAMA Neurol. 2022, 79, 584–591. [Google Scholar] [CrossRef]
- Abdelmaksoud, N.M.; Sallam, A.-A.M.; Abulsoud, A.I.; El-Dakroury, W.A.; Abdel Mageed, S.S.; AL-Noshokaty, T.M.; Elrebehy, M.A.; Elshaer, S.S.; Mahmoud, N.A.; Fathi, D.; et al. Unraveling the Role of miRNAs in the Diagnosis, Progression, and Therapeutic Intervention of Alzheimer’s Disease. Pathol. Res. Pract. 2024, 253, 155007. [Google Scholar] [CrossRef] [PubMed]
- Planche, V.; Bouteloup, V.; Pellegrin, I.; Mangin, J.-F.; Dubois, B.; Ousset, P.-J.; Pasquier, F.; Blanc, F.; Paquet, C.; Hanon, O.; et al. Validity and Performance of Blood Biomarkers for Alzheimer Disease to Predict Dementia Risk in a Large Clinic-Based Cohort. Neurology 2023, 100, e473–e484. [Google Scholar] [CrossRef] [PubMed]
- Sperling, R.A.; Donohue, M.C.; Raman, R.; Rafii, M.S.; Johnson, K.; Masters, C.L.; van Dyck, C.H.; Iwatsubo, T.; Marshall, G.A.; Yaari, R.; et al. Trial of Solanezumab in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2023, 389, 1096–1107. [Google Scholar] [CrossRef]
- Yi, L.X.; Tan, E.K.; Zhou, Z.D. Passive Immunotherapy for Alzheimer’s Disease: Challenges & Future Directions. J. Transl. Med. 2024, 22, 430. [Google Scholar] [CrossRef]
- Kosyreva, A.; Sentyabreva, A.; Tsvetkov, I.; Makarova, O. Alzheimer’s Disease and Inflammaging. Brain Sci. 2022, 12, 1237. [Google Scholar] [CrossRef]
- Zhuang, Q.-S.; Meng, L.; Wang, Z.; Shen, L.; Ji, H.-F. Associations Between Obesity and Alzheimer’s Disease: Multiple Bioinformatic Analyses. J. Alzheimer’s Dis. 2021, 80, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Borda, M.G.; Castellanos-Perilla, N.; Tovar-Rios, D.A.; Ferreira, D.; Duque, G.; Aarsland, D. Tongue Muscle Mass Is Associated with Total Grey Matter and Hippocampal Volumes in Dementia with Lewy Bodies. Arch. Gerontol. Geriatr. 2022, 100, 104647. [Google Scholar] [CrossRef] [PubMed]
- Więckowska-Gacek, A.; Mietelska-Porowska, A.; Wydrych, M.; Wojda, U. Western Diet as a Trigger of Alzheimer’s Disease: From Metabolic Syndrome and Systemic Inflammation to Neuroinflammation and Neurodegeneration. Ageing Res. Rev. 2021, 70, 101397. [Google Scholar] [CrossRef] [PubMed]
- Birkic, V.; Celeste, T.; Cochrane, L. Which Review Is That? A Guide to Review Types; The University of Melbourne: Melbourne, Australia, 2020. [Google Scholar]
- Wu, M.-Y.; Zou, W.-J.; Lee, D.; Mei, L.; Xiong, W.-C. APP in the Neuromuscular Junction for the Development of Sarcopenia and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 7809. [Google Scholar] [CrossRef] [PubMed]
- Correa-de-Araujo, R.; Addison, O.; Miljkovic, I.; Goodpaster, B.H.; Bergman, B.C.; Clark, R.V.; Elena, J.W.; Esser, K.A.; Ferrucci, L.; Harris-Love, M.O.; et al. Myosteatosis in the Context of Skeletal Muscle Function Deficit: An Interdisciplinary Workshop at the National Institute on Aging. Front. Physiol. 2020, 11, 963. [Google Scholar] [CrossRef]
- Ingenbleek, Y. Implications of Protein Malnutrition and Inflammatory Disorders in the Pathophysiology of Alzheimer’s Disease. Asia Pac. J. Clin. Nutr. 2020, 29, 450–461. [Google Scholar] [CrossRef]
- Suryadevara, V.; Klüppel, M.; Monte, F.D.; Willis, M.S. The Unraveling. Am. J. Pathol. 2020, 190, 1609–1621. [Google Scholar] [CrossRef]
- Halon-Golabek, M.; Borkowska, A.; Herman-Antosiewicz, A.; Antosiewicz, J. Iron Metabolism of the Skeletal Muscle and Neurodegeneration. Front. Neurosci. 2019, 13, 165. [Google Scholar] [CrossRef]
- Han, X.; Ashraf, M.; Tipparaju, S.M.; Xuan, W. Muscle-Brain Crosstalk in Cognitive Impairment. Front. Aging Neurosci. 2023, 15, 1221653. [Google Scholar] [CrossRef]
- Brisendine, M.H.; Drake, J.C. Early-Stage Alzheimer’s Disease: Are Skeletal Muscle and Exercise the Key? J. Appl. Physiol. 2023, 134, 515–520. [Google Scholar] [CrossRef]
- García-Llorente, A.M.; Casimiro-Andújar, A.J.; Linhares, D.G.; De Souza Vale, R.G.; Marcos-Pardo, P.J. Multidomain Interventions for Sarcopenia and Cognitive Flexibility in Older Adults for Promoting Healthy Aging: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Aging Clin. Exp. Res. 2024, 36, 47. [Google Scholar] [CrossRef] [PubMed]
- Jodeiri Farshbaf, M.; Alviña, K. Multiple Roles in Neuroprotection for the Exercise Derived Myokine Irisin. Front. Aging Neurosci. 2021, 13, 649929. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, K.; Wang, T. Protective Effect of Irisin against Alzheimer’s Disease. Front. Psychiatry 2022, 13, 967683. [Google Scholar] [CrossRef]
- Cao, X.; Thyfault, J.P. Exercise Drives Metabolic Integration between Muscle, Adipose and Liver Metabolism and Protects against Aging-Related Diseases. Exp. Gerontol. 2023, 176, 112178. [Google Scholar] [CrossRef]
- Karnik, S.J.; Margetts, T.J.; Wang, H.S.; Movila, A.; Oblak, A.L.; Fehrenbacher, J.C.; Kacena, M.A.; Plotkin, L.I. Mind the Gap: Unraveling the Intricate Dance Between Alzheimer’s Disease and Related Dementias and Bone Health. Curr. Osteoporos. Rep. 2024, 22, 165–176. [Google Scholar] [CrossRef]
- Frame, G.; Bretland, K.A.; Dengler-Crish, C.M. Mechanistic Complexities of Bone Loss in Alzheimer’s Disease: A Review. Connect. Tissue Res. 2020, 61, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-N.; Zhang, Q.; Li, M. Alzheimer’s Disease and Its Associated Risk of Bone Fractures: A Narrative Review. Front. Endocrinol. 2023, 14, 1190762. [Google Scholar] [CrossRef]
- Ruggiero, C.; Baroni, M.; Xenos, D.; Parretti, L.; Macchione, I.G.; Bubba, V.; Laudisio, A.; Pedone, C.; Ferracci, M.; Magierski, R.; et al. Dementia, Osteoporosis and Fragility Fractures: Intricate Epidemiological Relationships, Plausible Biological Connections, and Twisted Clinical Practices. Ageing Res. Rev. 2024, 93, 102130. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, H.; Qiu, F.; He, J.; Chen, J. Cognitive Impairment and Risks of Osteoporosis: A Systematic Review and Meta-Analysis. Arch. Gerontol. Geriatr. 2023, 106, 104879. [Google Scholar] [CrossRef]
- Sindzingre, L.; Bouaziz-Amar, E.; Mouton-Liger, F.; Cognat, E.; Dumurgier, J.; Vrillon, A.; Paquet, C.; Lilamand, M. The Role of Adiponectin in Alzheimer’s Disease: A Translational Review. J. Nutr. Health Aging 2024, 28, 100166. [Google Scholar] [CrossRef]
- Casado, M.E.; Collado-Pérez, R.; Frago, L.M.; Barrios, V. Recent Advances in the Knowledge of the Mechanisms of Leptin Physiology and Actions in Neurological and Metabolic Pathologies. Int. J. Mol. Sci. 2023, 24, 1422. [Google Scholar] [CrossRef] [PubMed]
- Andrade, L.J.D.O.; Oliveira, L.M.D.; Bittencourt, A.M.V.; Lourenço, L.G.D.C.; Oliveira, G.C.M.D. Brain Insulin Resistance and Alzheimer’s Disease: A Systematic Review. Dement. Neuropsychol. 2024, 18, e20230032. [Google Scholar] [CrossRef] [PubMed]
- Huber, K.; Szerenos, E.; Lewandowski, D.; Toczylowski, K.; Sulik, A. The Role of Adipokines in the Pathologies of the Central Nervous System. Int. J. Mol. Sci. 2023, 24, 14684. [Google Scholar] [CrossRef]
- Neto, A.; Fernandes, A.; Barateiro, A. The Complex Relationship between Obesity and Neurodegenerative Diseases: An Updated Review. Front. Cell. Neurosci. 2023, 17, 1294420. [Google Scholar] [CrossRef]
- Abdalla, M.M.I. Insulin Resistance as the Molecular Link between Diabetes and Alzheimer’s Disease. World J. Diabetes 2024, 15, 1430–1447. [Google Scholar] [CrossRef]
- Arjunan, A.; Sah, D.K.; Woo, M.; Song, J. Identification of the Molecular Mechanism of Insulin-like Growth Factor-1 (IGF-1): A Promising Therapeutic Target for Neurodegenerative Diseases Associated with Metabolic Syndrome. Cell. Biosci. 2023, 13, 16. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, N.; Khan, N.; Gupta, A.; Neupane, P.; Mehta, A.; Shah, S.A.; Dey, R.C. Exploring the Effects of Adiponectin and Leptin in Correlating Obesity with Cognitive Decline: A Systematic Review. Ann. Med. Surg. 2023, 85, 2906–2915. [Google Scholar] [CrossRef] [PubMed]
- Cimini, F.A.; Perluigi, M.; Barchetta, I.; Cavallo, M.G.; Barone, E. Role of Biliverdin Reductase A in the Regulation of Insulin Signaling in Metabolic and Neurodegenerative Diseases: An Update. Int. J. Mol. Sci. 2022, 23, 5574. [Google Scholar] [CrossRef]
- Flores-Cordero, J.A.; Pérez-Pérez, A.; Jiménez-Cortegana, C.; Alba, G.; Flores-Barragán, A.; Sánchez-Margalet, V. Obesity as a Risk Factor for Dementia and Alzheimer’s Disease: The Role of Leptin. Int. J. Mol. Sci. 2022, 23, 5202. [Google Scholar] [CrossRef]
- Chung, K.W. Advances in Understanding of the Role of Lipid Metabolism in Aging. Cells 2021, 10, 880. [Google Scholar] [CrossRef]
- Khoramipour, K.; Chamari, K.; Hekmatikar, A.A.; Ziyaiyan, A.; Taherkhani, S.; Elguindy, N.M.; Bragazzi, N.L. Adiponectin: Structure, Physiological Functions, Role in Diseases, and Effects of Nutrition. Nutrients 2021, 13, 1180. [Google Scholar] [CrossRef] [PubMed]
- Komleva, Y.; Chernykh, A.; Lopatina, O.; Gorina, Y.; Lokteva, I.; Salmina, A.; Gollasch, M. Inflamm-Aging and Brain Insulin Resistance: New Insights and Role of Life-Style Strategies on Cognitive and Social Determinants in Aging and Neurodegeneration. Front. Neurosci. 2021, 14, 618395. [Google Scholar] [CrossRef]
- Polito, R.; Di Meo, I.; Barbieri, M.; Daniele, A.; Paolisso, G.; Rizzo, M.R. Adiponectin Role in Neurodegenerative Diseases: Focus on Nutrition Review. Int. J. Mol. Sci. 2020, 21, 9255. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Barua, S.; Jeong, Y.J.; Lee, J.E. Adiponectin: The Potential Regulator and Therapeutic Target of Obesity and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6419. [Google Scholar] [CrossRef] [PubMed]
- Forny-Germano, L.; De Felice, F.G.; do Nascimento Vieira, M.N. The Role of Leptin and Adiponectin in Obesity-Associated Cognitive Decline and Alzheimer’s Disease. Front. Neurosci. 2018, 12, 1027. [Google Scholar] [CrossRef]
- Uddin, M.S.; Rahman, M.M.; Sufian, M.A.; Jeandet, P.; Ashraf, G.M.; Bin-Jumah, M.N.; Mousa, S.A.; Abdel-Daim, M.M.; Akhtar, M.F.; Saleem, A.; et al. Exploring the New Horizon of AdipoQ in Obesity-Related Alzheimer’s Dementia. Front. Physiol. 2020, 11, 567678. [Google Scholar] [CrossRef]
- Huang, X.; Wang, Y.-J.; Xiang, Y. Bidirectional Communication between Brain and Visceral White Adipose Tissue: Its Potential Impact on Alzheimer’s Disease. eBioMedicine 2022, 84, 104263. [Google Scholar] [CrossRef]
- Farruggia, M.C.; Small, D.M. Effects of Adiposity and Metabolic Dysfunction on Cognition: A Review. Physiol. Behav. 2019, 208, 112578. [Google Scholar] [CrossRef]
- Kueck, P.J.; Morris, J.K.; Stanford, J.A. Current Perspectives: Obesity and Neurodegeneration—Links and Risks. Degener. Neurol. Neuromuscul. Dis. 2023, 13, 111–129. [Google Scholar] [CrossRef]
- Woo, A.; Botta, A.; Shi, S.S.W.; Paus, T.; Pausova, Z. Obesity-Related Neuroinflammation: Magnetic Resonance and Microscopy Imaging of the Brain. Int. J. Mol. Sci. 2022, 23, 8790. [Google Scholar] [CrossRef]
- Litwiniuk, A.; Bik, W.; Kalisz, M.; Baranowska-Bik, A. Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 5603. [Google Scholar] [CrossRef] [PubMed]
- Pichiah, P.B.T.; Sankarganesh, D.; Arunachalam, S.; Achiraman, S. Adipose-Derived Molecules-Untouched Horizons in Alzheimer’s Disease Biology. Front. Aging Neurosci. 2020, 12, 17. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Zivkovic, A.M. The Potential Utility of Prebiotics to Modulate Alzheimer’s Disease: A Review of the Evidence. Microorganisms 2021, 9, 2310. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhang, P.; Xu, F.; Zheng, Y.; Zhao, H. Advances in the Study of IL-17 in Neurological Diseases and Mental Disorders. Front. Neurol. 2023, 14, 1284304. [Google Scholar] [CrossRef]
- Natale, G.; Zhang, Y.; Hanes, D.W.; Clouston, S.A. Obesity in Late-Life as a Protective Factor Against Dementia and Dementia-Related Mortality. Am. J. Alzheimers Dis. Other Demen. 2023, 38, 15333175221111658. [Google Scholar] [CrossRef]
- Flores-Dorantes, M.T.; Díaz-López, Y.E.; Gutiérrez-Aguilar, R. Environment and Gene Association with Obesity and Their Impact on Neurodegenerative and Neurodevelopmental Diseases. Front. Neurosci. 2020, 14, 863. [Google Scholar] [CrossRef]
- Cianci, R.; Franza, L.; Massaro, M.G.; Borriello, R.; Tota, A.; Pallozzi, M.; De Vito, F.; Gambassi, G. The Crosstalk between Gut Microbiota, Intestinal Immunological Niche and Visceral Adipose Tissue as a New Model for the Pathogenesis of Metabolic and Inflammatory Diseases: The Paradigm of Type 2 Diabetes Mellitus. Curr. Med. Chem. 2022, 29, 3189–3201. [Google Scholar] [CrossRef]
- Kuneš, J.; Hojná, S.; Mráziková, L.; Montezano, A.; Touyz, R.; Maletínská, L. Obesity, Cardiovascular and Neurodegenerative Diseases: Potential Common Mechanisms. Physiol. Res. 2023, 72, S73–S90. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alsayegh, A.A.; Hakami, Z.H.; Khamjan, N.A.; Saad, H.M.; Batiha, G.E.-S.; De Waard, M. A Potential Link Between Visceral Obesity and Risk of Alzheimer’s Disease. Neurochem. Res. 2023, 48, 745–766. [Google Scholar] [CrossRef]
- Ingenbleek, Y. Plasma Transthyretin as A Biomarker of Sarcopenia in Elderly Subjects. Nutrients 2019, 11, 895. [Google Scholar] [CrossRef]
- Ioannou, A.; Fontana, M.; Gillmore, J.D. Patisiran for the Treatment of Transthyretin-Mediated Amyloidosis with Cardiomyopathy. Heart Int. 2023, 17, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Ingenbleek, Y. Revisiting PINI Scoring in Light of Recent Biological Advances. Nutrients 2023, 15, 1846. [Google Scholar] [CrossRef]
- Obradovic, M.; Sudar-Milovanovic, E.; Soskic, S.; Essack, M.; Arya, S.; Stewart, A.J.; Gojobori, T.; Isenovic, E.R. Leptin and Obesity: Role and Clinical Implication. Front. Endocrinol. 2021, 12, 585887. [Google Scholar] [CrossRef]
- Sarapultsev, A.; Gusev, E.; Komelkova, M.; Utepova, I.; Luo, S.; Hu, D. JAK-STAT Signaling in Inflammation and Stress-Related Diseases: Implications for Therapeutic Interventions. Mol. Biomed. 2023, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Amontree, M.; Deasy, S.; Turner, R.S.; Conant, K. Matrix Disequilibrium in Alzheimer’s Disease and Conditions That Increase Alzheimer’s Disease Risk. Front. Neurosci. 2023, 17, 1188065. [Google Scholar] [CrossRef]
- Reyna, N.C.; Clark, B.J.; Hamilton, D.A.; Pentkowski, N.S. Anxiety and Alzheimer’s Disease Pathogenesis: Focus on 5-HT and CRF Systems in 3xTg-AD and TgF344-AD Animal Models. Front. Aging Neurosci. 2023, 15, 1251075. [Google Scholar] [CrossRef]
- Gómez-Apo, E.; Mondragón-Maya, A.; Ferrari-Díaz, M.; Silva-Pereyra, J. Structural Brain Changes Associated with Overweight and Obesity. J. Obes. 2021, 2021, 6613385. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, L.O.; Gaspar, J.M. Obesity-Induced Brain Neuroinflammatory and Mitochondrial Changes. Metabolites 2023, 13, 86. [Google Scholar] [CrossRef]
- Herrmann, M.J.; Tesar, A.; Beier, J.; Berg, M.; Warrings, B. Grey Matter Alterations in Obesity: A Meta-analysis of Whole-brain Studies. Obes. Rev. 2019, 20, 464–471. [Google Scholar] [CrossRef]
- Boccara, E.; Golan, S.; Beeri, M.S. The Association between Regional Adiposity, Cognitive Function, and Dementia-Related Brain Changes: A Systematic Review. Front. Med. 2023, 10, 1160426. [Google Scholar] [CrossRef]
- Larsson, S.C.; Burgess, S. Causal Role of High Body Mass Index in Multiple Chronic Diseases: A Systematic Review and Meta-Analysis of Mendelian Randomization Studies. BMC Med. 2021, 19, 320. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhong, T.; Zhang, M.; Xu, Y.; Zhang, M.; Chen, L. Alzheimer’s Disease: Causal Effect between Obesity and APOE Gene Polymorphisms. Int. J. Mol. Sci. 2023, 24, 13531. [Google Scholar] [CrossRef]
- Liu, C.-C.; Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Linton, M.F.; Gish, R.; Hubl, S.T.; Bütler, E.; Esquivel, C.; Bry, W.I.; Boyles, J.K.; Wardell, M.R.; Young, S.G. Phenotypes of Apolipoprotein B and Apolipoprotein E after Liver Transplantation. J. Clin. Invest. 1991, 88, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.M.; Holmes, M.V.; Davey Smith, G. Reading Mendelian Randomisation Studies: A Guide, Glossary, and Checklist for Clinicians. BMJ 2018, 362, k601. [Google Scholar] [CrossRef] [PubMed]
- Charisis, S.; Lin, H.; Ray, R.; Joehanes, R.; Beiser, A.S.; Levy, D.; Seshadri, S.; Sargurupremraj, M.; Satizabal, C.L. Obesity Impacts the Expression of Alzheimer’s Disease-Related Genes: The Framingham Heart Study. Alzheimers Dement. 2023, 19, 3496–3505. [Google Scholar] [CrossRef]
- Von Bank, H.; Kirsh, C.; Simcox, J. Aging Adipose: Depot Location Dictates Age-Associated Expansion and Dysfunction. Ageing Res. Rev. 2021, 67, 101259. [Google Scholar] [CrossRef]
- Cavaliere, G.; Trinchese, G.; Penna, E.; Cimmino, F.; Pirozzi, C.; Lama, A.; Annunziata, C.; Catapano, A.; Mattace Raso, G.; Meli, R.; et al. High-Fat Diet Induces Neuroinflammation and Mitochondrial Impairment in Mice Cerebral Cortex and Synaptic Fraction. Front. Cell. Neurosci. 2019, 13, 509. [Google Scholar] [CrossRef]
- Caldarelli, M.; Rio, P.; Marrone, A.; Ocarino, F.; Chiantore, M.; Candelli, M.; Gasbarrini, A.; Gambassi, G.; Cianci, R. Gut–Brain Axis: Focus on Sex Differences in Neuroinflammation. Int. J. Mol. Sci. 2024, 25, 5377. [Google Scholar] [CrossRef]
- Solanki, R.; Karande, A.; Ranganathan, P. Emerging Role of Gut Microbiota Dysbiosis in Neuroinflammation and Neurodegeneration. Front. Neurol. 2023, 14, 1149618. [Google Scholar] [CrossRef]
- Grey, A.; Bolland, M.J.; Avenell, A.; Klein, A.A.; Gunsalus, C.K. Check for Publication Integrity before Misconduct. Nature 2020, 577, 167–169. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Authors | Title | Type of Article | Date | Key Findings |
|---|---|---|---|---|
| Wu, M.Y. et al. [25] | APP in the neuromuscular junction for the development of sarcopenia and Alzheimer’s disease. | Review | 2023 | APP, a key protein in AD development, is expressed in various tissues, including skeletal muscle, where its metabolism is crucial for NMJ synapse development and maintenance. The association of APP/βA pathology in muscles with myopathy in neurodegenerative disorders, such as AD and ALS, suggests a connection between muscular APP and brain pathology, shedding light on muscle–brain crosstalk and potential contributions to age-related degeneration. |
| Correa-de-Araujo, R. et al. [26] | Myosteatosis in the context of skeletal muscle function deficit: an interdisciplinary workshop at the National Institute on Aging. | Review | 2020 | The role of myosteatosis in muscle aging and metabolic diseases is highlighted, and determinants, consequences, and methods of evaluation are explored. Innovative topics, such as the impact of circadian rhythms on skeletal muscle and the relationship with myosteatosis, are addressed. The muscle–bone interaction perspective highlights preventive approaches, such as physical activity, myostatin treatment, and caloric restriction. The impact of myosteatosis on cancer survivors opens up new perspectives for identifying its role through interdisciplinary collaborations. |
| Ingenbleek, Y. et al. [27] | Implications of protein malnutrition and inflammatory disorders in the pathophysiology of Alzheimer’s disease. | Review | 2020 | Inflammatory states and malnutrition result in a reduced LBM and, consequently, in reduced nitrogen stores. Nitrogen and sulfur depletion from LBM stores could contribute to brain deterioration. |
| Suryadevara, V. et al. [28] | The unraveling: cardiac and musculoskeletal defects and their role in common Alzheimer disease morbidity and mortality. | Review | 2020 | Skeletal muscle dysfunction could precede the onset of AD-related symptoms and serve as a predictor of cognitive decline. Sarcopenia and dynapenia correlate with the severity of AD. Unsteady gait is considered a prodromal sign of cognitive decline. |
| Halon-Golabek, M. et al. [29] | Iron metabolism of the skeletal muscle and neurodegeneration. | Review | 2019 | Skeletal muscle has endocrine functions essential for a long and healthy life. Iron accumulation in muscle increases oxidative stress and is neurotoxic. Exercise stimulates the release of myokines, aids skeletal muscle wellness, reduces skeletal muscle iron stores, and consequently lowers the risk of AD. |
| Han, X. et al. [30] | Muscle–brain crosstalk in cognitive impairment. | Review | 2023 | The authors suggest a strong association between sarcopenia, cognitive impairment, and AD. They identify factors that maintain muscle trophism and that are neuroprotective, including IGF-1, BDNF, irisin, and SPARC. Aerobic and resistance exercise maintains muscle mass and would appear to have positive effects in maintaining cognitive health. |
| Brisendine, M.H. et al. [31] | Early-stage Alzheimer’s disease: are skeletal muscle and exercise the key? | Review | 2023 | The presence of a close relationship between skeletal muscles and cognitive function is emphasized. Resistance exercise appears to preserve cognitive function and reduce AD-related morbidity. |
| García-Llorente, A.M. et al. [32] | Mutidomain interventions for sarcopenia and cognitive flexibility in older adults for promoting healthy aging: a systematic review and meta-analysis of randomized controlled trials. | Review and meta-analysis | 2024 | Cognitive and physical training conducted for at least 8 weeks is shown to be effective in improving muscle strength and cognitive flexibility in the elderly. Therefore, multidomain training can contribute to the prevention and treatment of age-related diseases, including AD. |
| Jodeiri Farshbaf, M. et al. [33] | Multiple roles in neuroprotection for the exercise-derived myokine irisin. | Review | 2021 | FNDC5/Irisin expression has a neuroprotective role against the development of AD, as it reduces the formation of βA deposits, neuronal apoptosis, and neuroinflammation. Its expression is increased during exercise. Irisin administration results in a reduction in depressive and anxiety symptoms and memory impairment. |
| Chen, K. et al. [34] | Protective effect of irisin against Alzheimer’s disease. | Review | 2022 | Irisin has a protective role against AD through several mechanisms. It promotes learning and memory. Mechanisms that reduce the risk of AD include a reduction in the levels of proinflammatory cytokines (IL-6 and IL-1β) and a reduction in insulin resistance. |
| Cao, X. et al. [35] | Exercise drives metabolic integration between muscle, adipose and liver metabolism and protects against aging-related diseases. | Review | 2023 | Exercise reduces the risk of AD through several mechanisms that are not fully elucidated. Irisin would appear to have a protective and therapeutic role in AD; however, further research is needed to clarify its use in clinical practice. |
| Authors | Title | Type of Article | Date | Key Findings |
|---|---|---|---|---|
| Karnik, S.J. et al. [36] | Mind the gap: unraveling the intricate dance between Alzheimer’s disease and related dementias and bone health. | Review | 2024 | AD and bone pathology have a bidirectional cause–effect relationship. The neuroinflammation present in AD seems to cause dysregulation of the hypothalamic–pituitary–adrenal axis with a consequent increase in cortisol, which subsequently leads to a loss of bone density. Conversely, chronic inflammation, resulting from osteoporosis and fractures, contributes to the pathogenesis of AD. AD patients have an increased risk of fractures due to the increased risk of falls and the reduced BMD. On the other hand, although it seems that fractures might promote the progression of AD, there is currently insufficient evidence to support this |
| Frame, G. et al. [37] | Mechanistic complexities of bone loss in Alzheimer’s disease: a review. | Review | 2020 | Bone loss in AD occurs early and is not associated with aging, sex, mobility, or genetics. Despite preliminary research, possible disruptive mechanisms of skeletal homeostasis have emerged, including the effects of amyloid-beta on bone cells and the damage caused by neurofibrillary tau in neural centers that regulate skeletal remodeling, along with deficits in systemic Wnt/β-catenin signaling. Irisin and FNDC5 have implicated roles in AD-related bone loss and neurodegeneration. The current research is considered insufficient, requiring critical attention for possible new diagnostic and therapeutic opportunities. |
| Zhou, B.N. et al. [38] | Alzheimer’s disease and its associated risk of bone fractures: a narrative review. | Review | 2023 | AD patients have a significant risk of bone fractures, which are attributable to multiple factors, such as the direct effects of amyloid pathology on bone cells, an abnormal connection between brain and bone, deficits in Wnt/β-catenin signaling, reduced activity, high risk of falls, frailty, and chronic immune activity. Exercise, fall prevention, and an enriched diet are found to be beneficial in reducing the fracture risk. However, the efficacy of anti-osteoporotic agents needs to be further evaluated in AD patients, as clinical trials are limited. |
| Ruggiero, C. et al. [39] | Dementia, osteoporosis and fragility fractures: intricate epidemiological relationships, plausible biological connections, and twisted clinical practices. | Review | 2023 | AD patients have twice the risk of fragility fractures compared with healthy people. Individuals aged >65 years have a 60% risk of dementia following hip fractures, a 47% risk following vertebral fractures, and a 35% risk following lower limb fractures. Several molecular mechanisms would appear to be common to osteoporosis and cognitive impairment. The pathophysiological pathways underlying the complex interaction between bone and brain involve neuropeptides, osteokines, and hormones. Some of these molecules, including NPY, vitamin D, OCN, leptin and glutamate, are being investigated. However, understanding their interactions in the context of changes in body composition and dietary habits remains a topic of study. The gut microbiota is being studied to create innovative interventions to improve the well-being of older individuals at risk of these interconnected health challenges. |
| Zhao, Y. et al. [40] | Cognitive impairment and risks of osteoporosis: a systematic review and meta-analysis. | Systematic review/ meta-analysis | 2022 | AD patients have a worse bone status than healthy people. Patients with osteoporosis have an increased risk of cognitive impairment (OR = 2.01, 95% CI: 1.63-2.48) and the same is true for patients with reduced BMD. Osteoporosis treatment could prevent or delay the onset of cognitive impairment. This article analyzes possible interactions between osteoporosis and cognitive impairment. Specifically, AD is associated with βA deposition, and it is highlighted that βA can increase RANKL activation, and thus osteoclast activation, by increasing bone resorption. In addition, low levels of osteocalcin, a marker of bone formation, reflect both the severity of osteoporosis and the severity of cognitive impairment. |
| Authors | Title | Type of Article | Date | Key Findings |
|---|---|---|---|---|
| Sindzingre, L. et al. [41] | The role of adiponectin in Alzheimer’s disease: a translational review. | Review | 2024 | While preclinical studies have shown that adiponectin has a neuroprotective role in preventing AD, human studies have shown conflicting results. Therefore, the non-unique results in humans support the need for further research. |
| Casado, M.E. et al. [42] | Recent advances in the knowledge of the mechanisms of leptin physiology and actions in neurological and metabolic pathologies. | Review | 2023 | Excess weight correlates with low-grade inflammation due to the production of adipokines. These include leptin, the secretion of which is directly proportional to the amount of adipose tissue. Leptin is neuroprotective. Therefore, central leptin resistance, which is established in obese individuals, correlates with some neurological diseases, including AD. |
| Andrade, L.J. et al. [43] | Brain insulin resistance and Alzheimer’s disease: a systematic review. | Review | 2023 | Brain insulin resistance is present in several neurodegenerative diseases. Some studies show that impaired brain glucose utilization correlates with reduced neuronal plasticity and cognitive impairment; however, some research studies do not support a relationship between brain insulin resistance and AD. Therefore, further studies are needed to determine whether or not there is a causal relationship between insulin resistance and AD. |
| Huber, K. et al. [44] | The role of adipokines in the pathologies of the central nervous system. | Review | 2023 | The role of thirteen adipokines in the pathologies of the central nervous system are analyzed. In particular, cystatin C, adiponectin, and leptin have a neuroprotective role so they counteract AD development. Chitinase 3-like protein 1 promotes proinflammatory responses and is elevated in the early stages of AD. |
| Neto, A. et al. [45] | The complex relationship between obesity and neurodegenerative diseases: an updated review. | Review | 2023 | Obesity is associated with low-grade inflammation and is a risk factor for AD development and progression. This review analyzes the mechanisms involved in the interaction between obesity and AD, such as leptin resistance, low levels of adiponectin, insulin resistance, oxidative stress, βA disturbances, and brain atrophy. |
| Abdalla, M.M.I. [46] | Insulin resistance as the molecular link between diabetes and Alzheimer’s disease. | Review | 2024 | Increased AD risk has been linked to diabetes mellitus, with insulin resistance playing a key role in AD development. Dysfunctional insulin signaling in the brain may contribute to hallmark AD features like βA plaques and tau protein tangles, suggesting that targeting insulin resistance could offer new treatment possibilities for AD. |
| Arjunan, A. et al. [47] | Identification of the molecular mechanism of insulin-like growth factor-1 (IGF-1): a promising therapeutic target for neurodegenerative diseases associated with metabolic syndrome. | Review | 2023 | Recent evidence links metabolic syndrome to neurodegenerative diseases like AD, with factors such as hyperglycemia and abdominal obesity contributing to disease progression. IGF-1 deficiency, associated with metabolic syndrome-related pathologies, may play a key role in AD, highlighting IGF-1 as a potential therapeutic target for treating metabolic-syndrome-related neurodegeneration. |
| Tasnim, N. et al. [48] | Exploring the effects of adiponectin and leptin in correlating obesity with cognitive decline: a systematic review. | Systematic review/meta-analysis | 2023 |
Obese patients frequently suffer from cognitive decline, dementia, and AD. This review analyzes the neurological effects of adiponectin and leptin in obese patients. Adiponectin and leptin are the two major adipokines that have a preventive role against obesity and dementia, and both are involved in the causal relationship between obesity and cognitive decline.
|
| Cimini, F.A. et al. [49] | Role of biliverdin reductase A in the regulation of insulin signaling in metabolic and neurodegenerative diseases: an update. | Review | 2022 | Insulin resistance is a feature of obesity, metabolic syndrome, and type 2 diabetes, and it is linked with cognitive impairment. Among the mechanisms involved in the development of insulin resistance is that of BVR-A, which acts as a regulator of insulin signaling. |
| Flores-Cordero, J.A. et al. [50] | Obesity as a risk factor for dementia and Alzheimer’s disease: the role of leptin. | Review | 2022 | In obesity, chronic low-grade inflammation is responsible for leptin resistance, which, in turn, correlates with the development of AD. |
| Chung, K.W. et al. [51] | Advances in understanding of the role of lipid metabolism in aging. | Review | 2021 | During aging, the amount of adipose tissue increases, and the formation of ectopic fat occurs. This process is responsible for lipotoxicity, reduced energy availability, and altered cellular signaling mechanisms. These factors contribute to the development of AD. |
| Khoramipour, K. et al. [52] | Adiponectin: structure, physiological functions, role in diseases, and effects of nutrition. | Review | 2021 | Adiponectin regulates energy homeostasis, promotes hippocampal neurogenesis and neuronal plasticity, and has positive effects on cognitive function. Its secretion increases after a healthy diet and is reduced in obese subjects. Reduction of adiponectin promotes the progression of AD and causes cognitive impairment. |
| Komleva, Y. et al. [53] | Inflamm-aging and brain insulin resistance: new insights and role of life-style strategies on cognitive and social determinants in aging and neurodegeneration. | Review | 2021 | AD is associated with aging and is characterized by inflammation; otherwise known as inflammaging. This review analyzes the role of nutrients, obesity, low-grade chronic inflammation linked with obesity in brain aging, neurodegeneration, cognitive decline, and AD. Obesity causes inflammation and insulin resistance, which, in turn, promotes the development of AD. |
| Polito, R. et al. [54] | Adiponectin role in neurodegenerative diseases: focus on nutrition. | Review | 2020 | Adiponectin has a role in the prevention of neurodegenerative diseases such as AD. A reduction of adiponectin correlates with neurodegenerative disease severity, and this is observed in obese patients, suggesting a correlation between AD and obesity. |
| Kim, J.Y. et al. [55] | Adiponectin: the potential regulator and therapeutic target of obesity and Alzheimer’s disease. | Review | 2020 | There is an association between obesity, type 2 diabetes, and AD. Adiponectin increases insulin sensitivity, reduces inflammation, and is neuroprotective. Adiponectin levels are inversely proportional to the amount of central adipose tissue. Reduced levels of adiponectin are found in mild cognitive impairment and AD. |
| Forny-Germano, L. et al. [56] | The Role of leptin and adiponectin in obesity-associated cognitive decline and Alzheimer’s disease. | Review | 2019 | Obesity has been linked to an increased risk of AD due to dysregulated adipokines like leptin and adiponectin, which are critical in brain function. Dysfunctions in these adipokines may contribute to AD-related neuropathological events, including amyloid buildup, tau hyperphosphorylation, and neuroinflammation, suggesting that restoring their proper signaling could be a potential therapeutic strategy for AD. |
| Uddin, M.S. et al. [57] | Exploring the new horizon of adipoQ in obesity-related Alzheimer’s dementia. | Review | 2021 | Obesity is a risk factor for the development and progression of AD. Adiponectin has neuroprotective activity, reduces βA deposit formation, neuroinflammation, and insulin resistance; and increases neuronal plasticity. In obese patients, adiponectin is reduced, and this correlates with an increased risk of developing AD. |
| Huang, X. et al. [58] | Bidirectional communication between brain and visceral white adipose tissue: its potential impact on Alzheimer’s disease. | Review | 2022 | This review highlights that visceral white adipose tissue contributes to the development and progression of AD and analyzes the mechanisms involved. |
| Farruggia, M.C. et al. [59] | Effects of adiposity and metabolic dysfunction on cognition: a review. | Review | 2019 | Adiposity and metabolic dysfunction, such as insulin resistance and type 2 diabetes, are independent risk factors for dementia and AD in animal models, whereas in humans, it remains uncertain whether these risk factors are independent. |
| Authors | Title | Type of Article | Date | Key Findings |
|---|---|---|---|---|
| Kueck, P.J. et al. [60] | Current perspectives: obesity and neurodegeneration—links and risks. | Review | 2023 | Obesity-induced metabolic changes, including insulin resistance and increased oxidative stress, disrupt energy expenditure and mitochondrial function, contributing to cognitive decline and an increased risk of AD. The resulting oxidative stress damages lipids, proteins, and DNA, leading to endothelial dysfunction and reduced white matter integrity, exacerbating neurodegeneration in AD. |
| Woo, A. et al. [61] | Obesity-related neuroinflammation: magnetic resonance and microscopy imaging of the brain. | Mini-review | 2022 | Higher adiposity, especially visceral fat, is associated with structural brain changes such as reduced cortical thickness and increased white matter hyperintensities, contributing to neuroinflammation and potentially impairing cognitive functions related to AD. Microscopy imaging reveals that obesity-related cellular changes, like reduced dendritic spine density and increased activation of microglia and astrocytes, disrupt the blood–brain barrier and promote neuroinflammation, further contributing to AD. |
| Litwiniuk, A. et al. [62] | Inflammasome NLRP3 potentially links obesity-associated low-grade systemic inflammation and insulin resistance with Alzheimer’s disease. | Review | 2021 | AD involves a progressive decline in memory and cognitive functions that is linked to βA accumulation, tau phosphorylation, mitochondrial damage, synaptic loss, and neuroinflammation. The activation of inflammasomes like NLRP3 exacerbates neuroinflammation and contributes to AD pathology, suggesting potential therapeutic avenues targeting these pathways to mitigate cognitive decline. |
| Pichiah, P.B.T. et al. [63] | Derived molecules—untouched horizons in Alzheimer’s disease biology. | Review | 2020 | Adipose-tissue-derived cytokines and proteins, including IL-1β, IL-4, IL-10, IL-18, TNFα, MIF, CRP, chemerin, RANTES, PAI-1, CFH, adiponectin, CETP, LPL, RBP4, and resistin, contribute to AD progression by influencing neuroinflammation, βA accumulation, and potentially affecting autophagy pathways. Their roles underscore the complex interplay between obesity-related factors and AD pathophysiology, suggesting potential therapeutic targets for managing the disease. |
| Kang, J.W. et al. [64] | The potential utility of prebiotics to modulate Alzheimer’s disease: a review of the evidence. | Review | 2021 | Evidence suggests that promoting beneficial microbes, particularly SCFA-producing ones like Bifidobacteria, through prebiotic interventions has the potential to alleviate AD-associated symptoms. Different types of prebiotics, especially fructans, have shown efficacy in modulating gut microbiome composition and metabolite production. |
| Lu, Y. et al. [65] | Advances in the study of IL-17 in neurological diseases and mental disorders. | Review | 2023 | IL-17 is increasingly recognized as a crucial mediator of immune regulation through neuroinflammation and the microbiota–gut–brain axis. The complex interplay of factors influencing the human neurological system and gut microbiota, along with ongoing debates about the translatability of animal models to humans, warrants further investigation into the role of IL-17 in disease pathogenesis and the potential for targeted therapeutic interventions. |
| Natale, G. et al. [66] | Obesity in late-life as a protective factor against dementia and dementia-related mortality. | Review | 2023 | A protective effect of later-life obesity against cognitive decline, dementia, and dementia-related mortality is suggested, challenging the notion that obesity is a direct risk factor for these conditions. The findings emphasize the need for further exploration of the complex relationships between obesity, age, and cardiometabolic risks, especially considering the increasing prevalence of obesity in younger populations and its potential implications for future dementia incidence. |
| Flores-Dorantes, M.T. et al. [67] | Environment and gene association with obesity and their impact on neurodegenerative and neurodevelopmental diseases. | Review | 2020 | The article explores gene–environment interactions in obesity, focusing on genetic variants like FTO, MC4R, LEP, LEPR, POMC, CART, NPY, PCSK1, SIM1, BDNF, and TrKB, which influence obesity susceptibility through pathways affecting energy balance and neurodegenerative/neurodevelopmental diseases. It underscores the shared genetic mechanisms linking obesity with AD, Parkinson’s disease, and schizophrenia, suggesting potential therapeutic targets across these complex disorders. |
| Cianci, R. et al. [68] | The crosstalk between gut microbiota, intestinal immunological niche and visceral adipose tissue as a new model for the pathogenesis of metabolic and inflammatory diseases: the paradigm of type 2 diabetes mellitus. | Review | 2022 | The GM plays a crucial role in a wide range of molecular interactions, influencing endocrine functions, immune responses, and metabolism. The bidirectional interaction between GM and VAT is pivotal, impacting adipokines, hormones, and immune reactions and contributing to conditions such as diabetes and other metabolic disorders, suggesting potential therapeutic avenues for these conditions. |
| Kuneš, J. et al. [69] | Obesity, cardiovascular and neurodegenerative diseases: potential common mechanisms. | Review | 2023 | Obesity, CVD, and neurodegenerative disorders often co-occur and share mechanisms like inflammation and oxidative stress. Emphasizing lifestyle modifications, such as physical activity and a healthy diet, is crucial for prevention, considering the absence of effective drugs for obesity and neurodegenerative diseases, unlike available options for CVD, which may also influence cognitive decline and vascular dementia. |
| Al-Kuraishy, H.M. et al. [70] | A potential link between visceral obesity and risk of Alzheimer’s disease. | Review | 2022 | AD, featuring brain plaques and tangles, may have a connection to visceral obesity with inflammation and adipocytokine irregularities. Visceral obesity-induced oxidative stress and inflammation could contribute to AD development, emphasizing the importance of early recognition and management for potential prevention. |
| Ingenbleek, Y. et al. [71] | Plasma transthyretin as a biomarker of sarcopenia in elderly subjects. | Review | 2019 | Plasma TTR serves not only as a carrier protein for thyroid hormones and retinol but also exhibits neuroprotective properties when secreted by the brain choroid plexus. While TTR has been associated with amyloidogenic processes leading to morbidities in various tissues, recent data contest previous objections and establish that TTR is an active contributor in inflammatory responses, releasing free fractions of thyroxine and retinol in stressful disorders and strengthening the effects initiated by cytokines. |
| Ioannou, A. et al. [72] | Patisiran for the treatment of transthyretin-mediated amyloidosis with cardiomyopathy. | Review | 2023 | Reducing circulating TTR concentrations with patisiran has shown promising results, leading to improvements in various cardiac and neurological measures, as in ATTR-CM. The potential expansion of patisiran’s license to include ATTR-CM could provide an additional disease-modifying treatment for this aggressive cardiomyopathy, although the long-term effects of patisiran-induced TTR depletion still need to be determined. |
| Ingenbleek, Y. et al. [73] | Revisiting PINI scoring in light of recent biological advances. | Review | 2023 | The PINI scoring system involves a ratio between inflammatory biomarkers in the numerator and liver biomarkers assessing the protein nutritional status (TTR and albumin) in the denominator. TTR, which uniquely correlates with lean body mass from birth to old age, plays a crucial role in quantifying protein depletion during inflammatory disorders, making it a versatile indicator in various clinical conditions, including in neonates, critically ill patients, and those with conditions like AD. |
| Authors | Title | Type of Article | Date | Key Findings |
|---|---|---|---|---|
| Obradovic, M. et al. [74] | Leptin and obesity: role and clinical implication. | Review | 2021 | The discovery of leptin has shed light on obesity control, and alterations in its expression and receptor contribute to leptin resistance, a crucial factor in obesity-related complications. Despite its role as a principal appetite suppressor, utilizing leptin-based therapeutics for obesity treatment requires further exploration, emphasizing the need to identify new mechanisms of leptin regulation to design drugs that can reverse leptin resistance and offer alternatives in obesity treatment. |
| Sarapultsev, A. et al. [75] | JAK-STAT signaling in inflammation and stress-related diseases: implications for therapeutic interventions. | Review | 2023 | The multifaceted role of the JAK-STAT signaling pathway in systemic homeostasis, including its involvement in inflammation, meta-inflammation, and aging, prompts an exploration of its relevance to stress-associated disorders. Isoform-specific inhibitors like filgotinib and upadacitinib, along with combination therapies and precision drug delivery systems, offer a new era of targeted pharmacology that enriches therapeutic approaches and deepen our understanding of the pathway’s involvement in the complex interplay between stress and inflammation. |
| Amontree, M. et al. [76] | Matrix disequilibrium in Alzheimer’s disease and conditions that increase Alzheimer’s disease risk. | Review | 2023 | Common risk factors for late-onset AD, including age, APOE genotype, untreated major depressive disorder, and obesity, are associated with chronic inflammation and increased ECM deposition. The review suggests that excess ECM deposition may restrict neuroplasticity, impair cognitive reserves, and alter the excitatory/inhibitory balance, highlighting the importance of studying ECM interventions in AD and related risk factor models. |
| Reyna, N.C. et al. [77] | Anxiety and Alzheimer’s disease pathogenesis: focus on 5-HT and CRF systems in 3xTg-AD and TgF344-AD animal models. | Review | 2023 | Despite behavioral inconsistencies, both mouse models consistently exhibit robust AD pathology, with the TgF344-AD rat model showing more reliable behavioral findings and expressing neuroinflammation and neuronal loss, aspects lacking in mouse AD models. Targeting the 5-HT and CRF systems emerges as a promising strategy, with the 5-HT system offering a feasible preventative measure and CRF1 antagonists presenting a potential avenue to refine interventions and improve clinical outcomes in AD patients. |
| Gómez-Apo, E. et al. [78] | Structural brain changes associated with overweight and obesity. | Review | 2021 | Excessive visceral fat induces chronic inflammation affecting various tissues, including the brain, leading to structural abnormalities and reduced gray matter volume. Obesity is linked to neurodegenerative diseases, influencing synaptic plasticity, cognitive deficits, and behavioral patterns, while physiological changes such as chronic inflammation, insulin resistance, and microangiopathy contribute to neuronal loss and lower cortical thickness, explaining the poor cognitive performance in individuals with overweight and obesity. |
| Schmitt, L.O. et al. [79] | Obesity-induced brain neuroinflammatory and mitochondrial changes. | Review | 2023 | A high-fat and high-caloric diet intake, along with obesity and T2DM, induces brain dysfunction that is characterized by neuroinflammation and mitochondrial dysfunction. These mechanisms are proposed to contribute to cognitive deficits and dementia associated with metabolic disorders, and interventions targeting brain mitochondrial dysfunction and reducing oxidative stress levels in the hippocampus may have beneficial effects on cognitive processes. |
| Herrmann, M.J. et al. [80] | Grey matter alterations in obesity: a meta-analysis of whole-brain studies. | Meta-analysis | 2019 | Notable structural alterations in the GMV within the insula and cerebellum among individuals with obesity are identified that are associated with psychopathological symptoms. However, additional investigations are necessary to ascertain whether these GMV changes play a causal role, result from obesity-related factors, or simply co-occur with the condition. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, G.; Gualtieri, P.; Cianci, R.; Caldarelli, M.; Palma, R.; De Santis, G.L.; Porfilio, C.; Nicoletti, F.; Bigioni, G.; Di Renzo, L. Body Composition and Alzheimer’s Disease: A Holistic Review. Int. J. Mol. Sci. 2024, 25, 9573. https://doi.org/10.3390/ijms25179573
Frank G, Gualtieri P, Cianci R, Caldarelli M, Palma R, De Santis GL, Porfilio C, Nicoletti F, Bigioni G, Di Renzo L. Body Composition and Alzheimer’s Disease: A Holistic Review. International Journal of Molecular Sciences. 2024; 25(17):9573. https://doi.org/10.3390/ijms25179573
Chicago/Turabian StyleFrank, Giulia, Paola Gualtieri, Rossella Cianci, Mario Caldarelli, Roselisa Palma, Gemma Lou De Santis, Chiara Porfilio, Francesco Nicoletti, Giulia Bigioni, and Laura Di Renzo. 2024. "Body Composition and Alzheimer’s Disease: A Holistic Review" International Journal of Molecular Sciences 25, no. 17: 9573. https://doi.org/10.3390/ijms25179573