
Left Ventricular Systolic Dysfunction in NBCe1-B/C-Knockout Mice
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
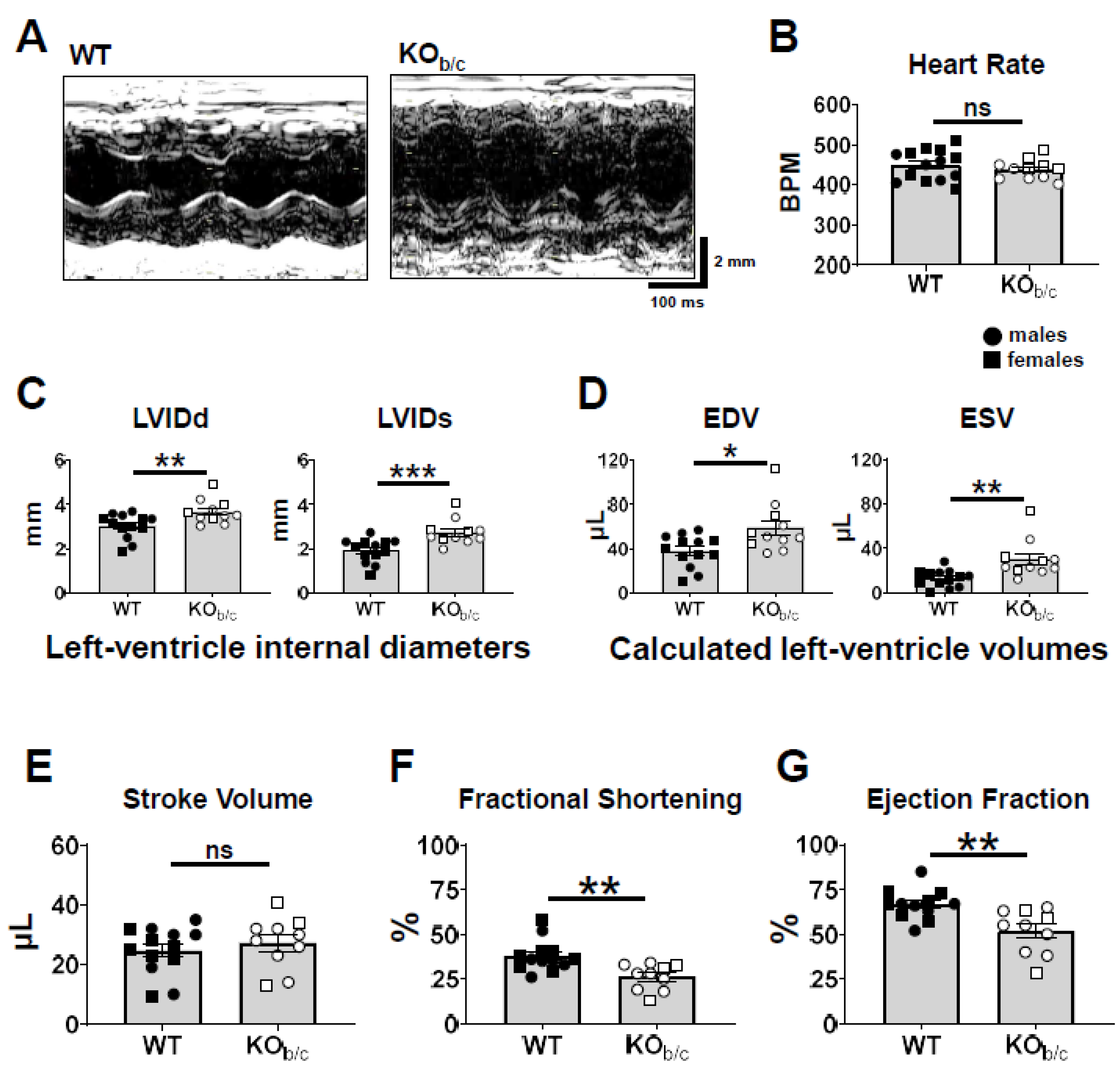
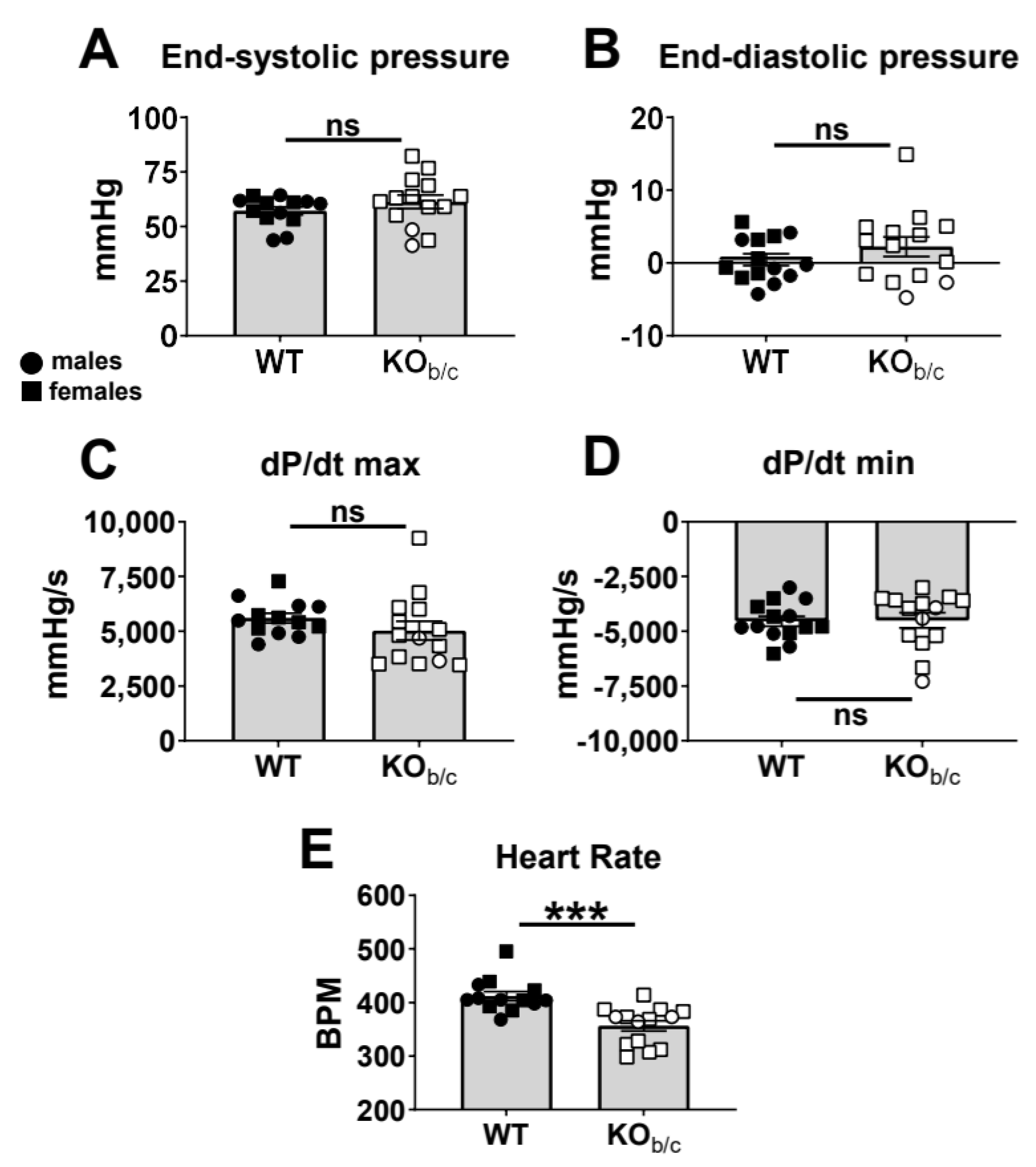
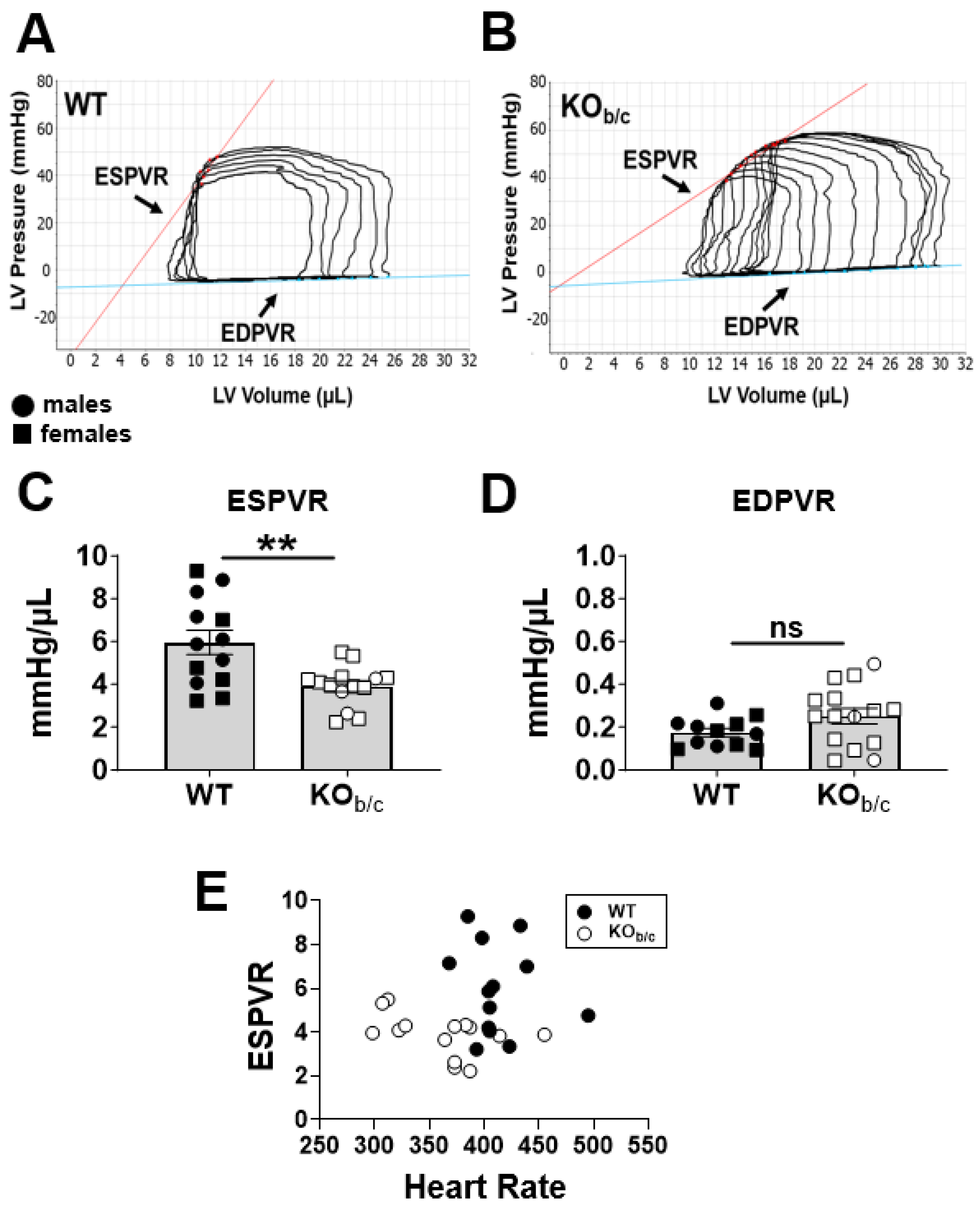
2.1. Reduced Ejection Fraction with Diminished Systolic Function in KOb/c Mice
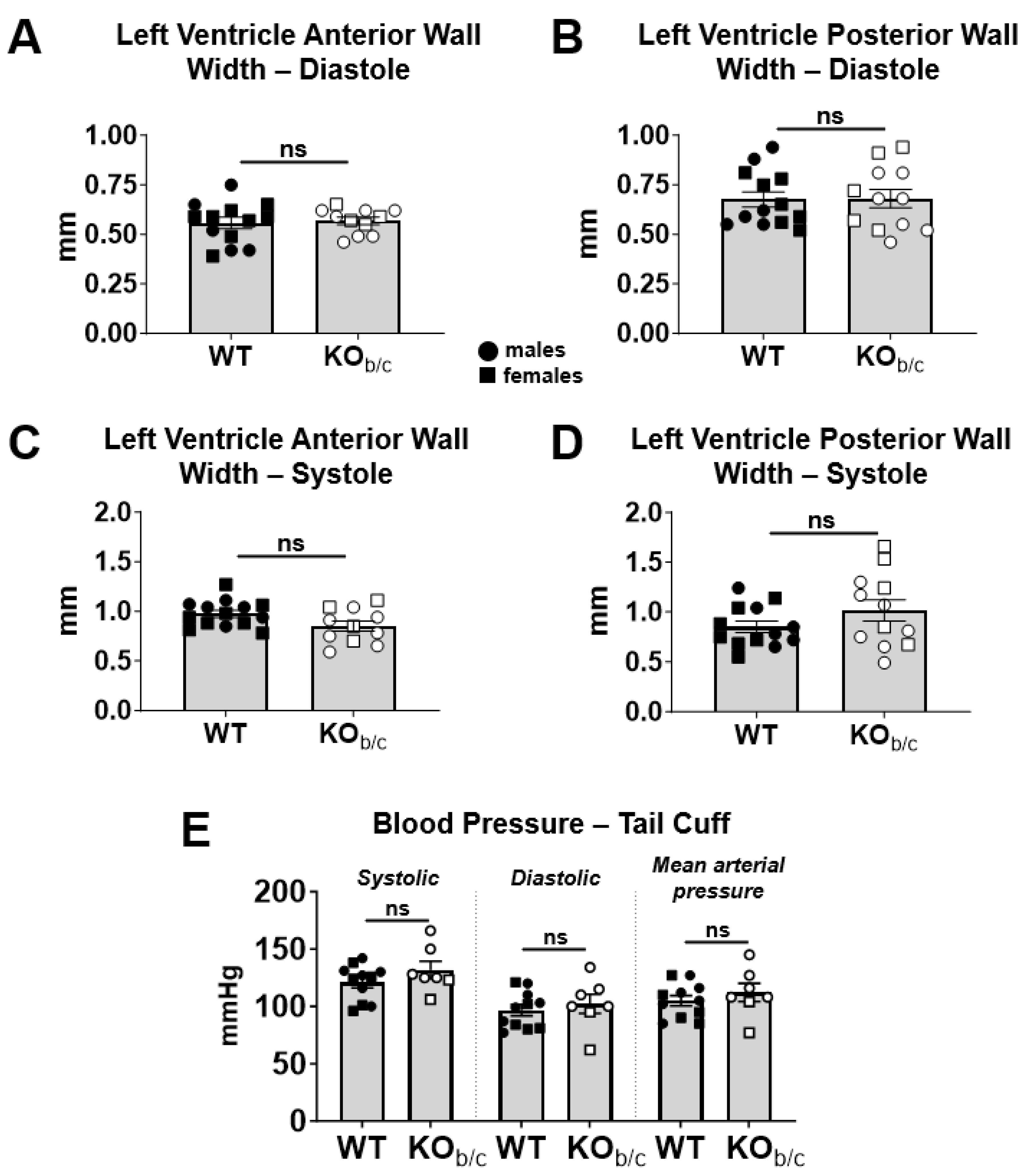
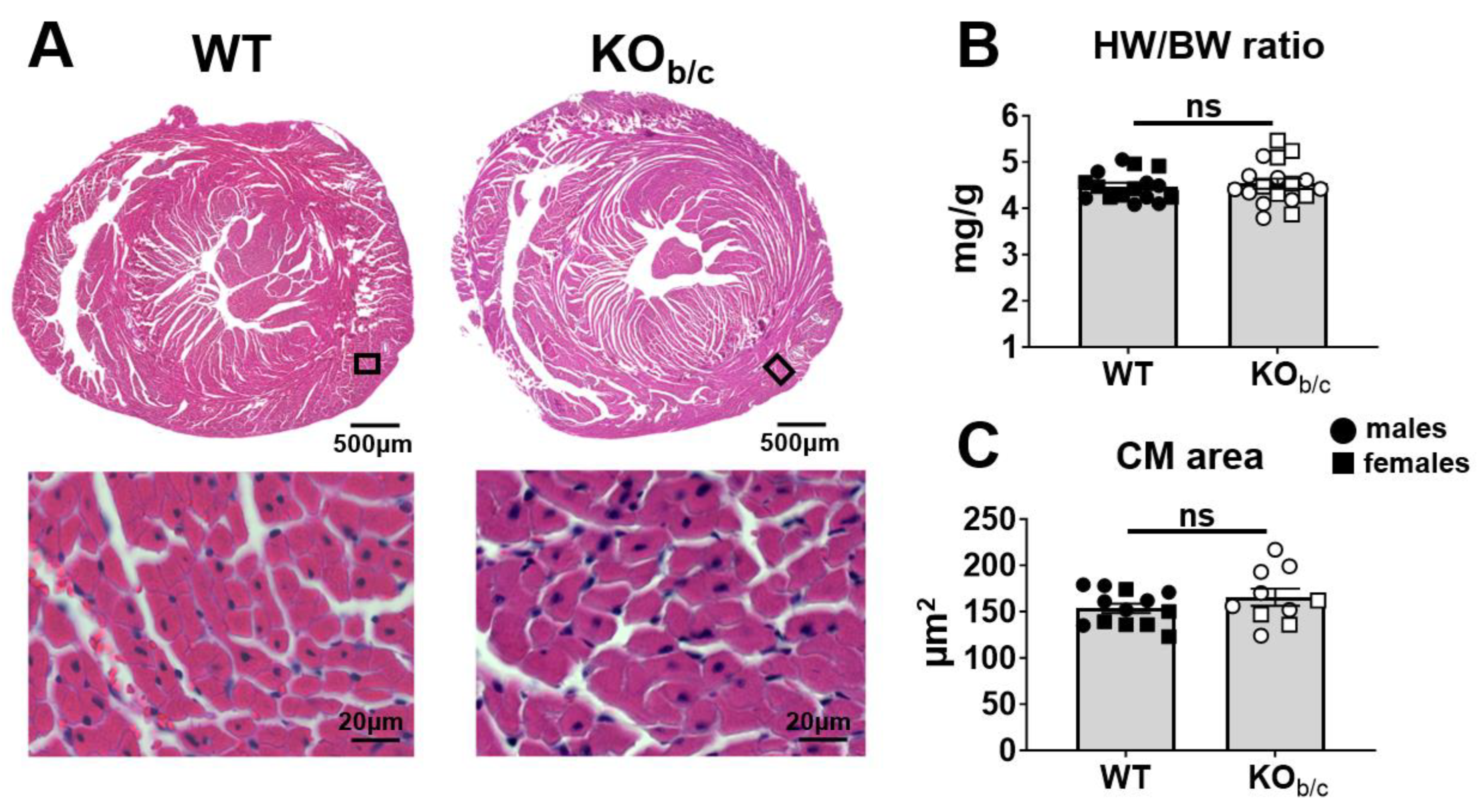
2.2. KOb/c Hearts Are Not Hypertrophic at 4–5 Weeks of Age
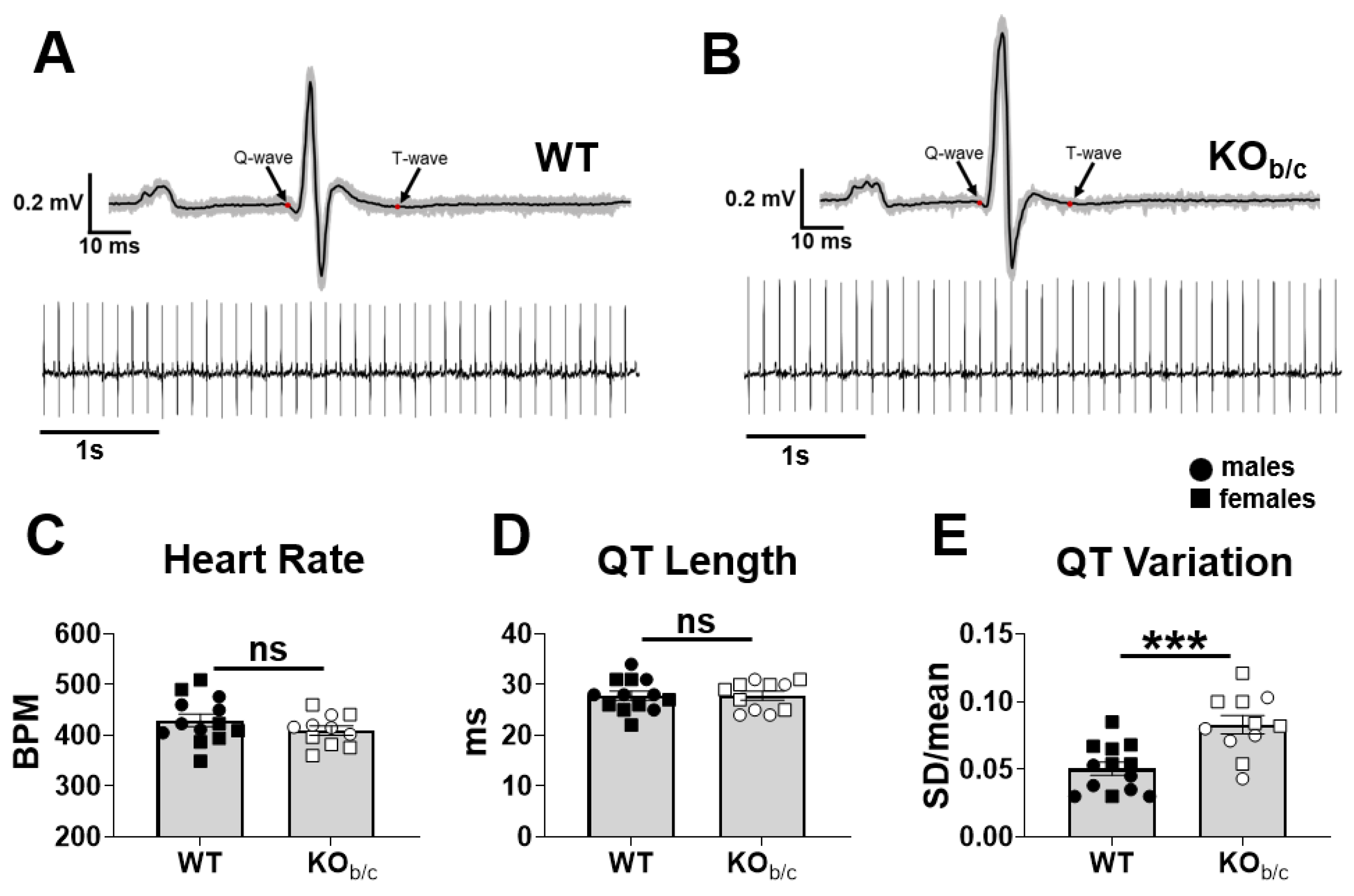
2.3. KOb/c Mice Have No Difference in QT Length but Have Increased QT Length Variation
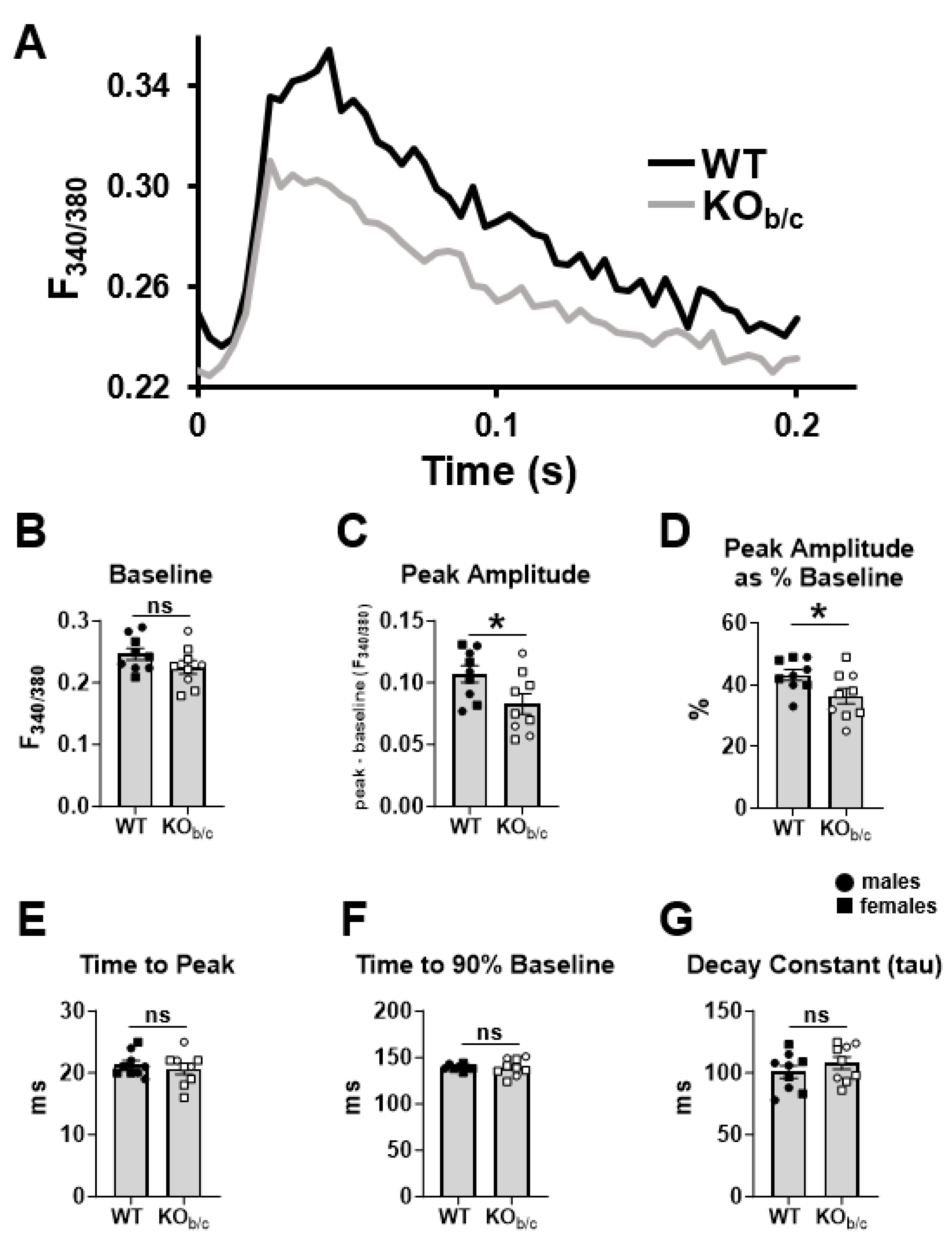
2.4. Cardiomyocytes from KOb/c Mice Have Diminished Ca2+-Transient Amplitude
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Blood Pressure
4.3. Echocardiography
4.4. Electrocardiogram (ECG) Recording and Analysis
4.5. Intraventricular Admittance Catheter-Derived Pressure–Volume (PV) Analysis
4.6. Histological Analysis
4.7. Cardiomyocyte Isolation
4.8. Ca2+ Transient Recordings and Analysis
4.9. Real-Time PCR
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Y.; Sheng, W.; Wu, J.; Zheng, J.; Zhi, X.; Zhang, S.; Gu, C.; Guo, D.; Wang, W. Case Report: Altered Pre-mRNA Splicing Caused by Intronic Variant c.1499 + 1G > A in the SLC4A4 Gene. Front. Pediatr. 2022, 10, 890147. [Google Scholar] [CrossRef]
- Gil-Perotín, S.; Jaijo, T.; Verdú, A.G.; Rubio, P.; Mazón, M.; Gasqué-Rubio, R.; Díaz, S. Epilepsy, Status Epilepticus, and Hemiplegic Migraine Coexisting with a Novel SLC4A4 Mutation. Neurol. Sci. 2021, 42, 3647–3654. [Google Scholar] [CrossRef]
- Yan, L.; Wenhong, W.; Meifang, L.; Wenyu, C. Severe proximal renal tubular acidosis with ocular abnormalities caused by SLC4A4 gene variation: A case report. Chin. J. Pediatr. 2020, 58, 241–242. [Google Scholar] [CrossRef]
- Khan, A.O.; Basamh, O.S. Pediatric Primary Calcific Band Keratopathy with or without Glaucoma from Biallelic SLC4A4 Mutations. Ophthalmic Genet. 2018, 39, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Horita, S.; Simsek, E.; Simsek, T.; Yildirim, N.; Ishiura, H.; Nakamura, M.; Satoh, N.; Suzuki, A.; Tsukada, H.; Mizuno, T.; et al. SLC4A4 Compound Heterozygous Mutations in Exon–Intron Boundary Regions Presenting with Severe Proximal Renal Tubular Acidosis and Extrarenal Symptoms Coexisting with Turner’s Syndrome: A Case Report. BMC Med. Genet. 2018, 19, 103. [Google Scholar] [CrossRef]
- Patel, N.; Khan, A.O.; Al-Saif, M.; Moghrabi, W.N.; AlMaarik, B.M.; Ibrahim, N.; Abdulwahab, F.; Hashem, M.; Alshidi, T.; Alobeid, E.; et al. A Novel Mechanism for Variable Phenotypic Expressivity in Mendelian Diseases Uncovered by an AU-Rich Element (ARE)-Creating Mutation. Genome Biol. 2017, 18, 144. [Google Scholar] [CrossRef]
- Myers, E.J.; Yuan, L.; Felmlee, M.A.; Lin, Y.; Jiang, Y.; Pei, Y.; Wang, O.; Li, M.; Xing, X.; Marshall, A.; et al. A Novel Mutant Na+/HCO3− Cotransporter NBCe1 in a Case of Compound-heterozygous Inheritance of Proximal Renal Tubular Acidosis. J. Physiol. 2016, 594, 6267–6286. [Google Scholar] [CrossRef]
- Kari, J.A.; Desoky, S.M.E.; Singh, A.K.; Gari, M.A.; Kleta, R.; Bockenhauer, D. The Case | Renal Tubular Acidosis and Eye Findings. Kidney Int. 2014, 86, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.-F.; Yang, S.-S.; Seki, G.; Yamada, H.; Horita, S.; Yamazaki, O.; Fujita, T.; Usui, T.; Tsai, J.-D.; Yu, I.-S.; et al. Severe Metabolic Acidosis Causes Early Lethality in NBC1 W516X Knock-in Mice as a Model of Human Isolated Proximal Renal Tubular Acidosis. Kidney Int. 2011, 79, 730–741. [Google Scholar] [CrossRef]
- Suzuki, M.; Paesschen, W.V.; Stalmans, I.; Horita, S.; Yamada, H.; Bergmans, B.A.; Legius, E.; Riant, F.; Jonghe, P.D.; Li, Y.; et al. Defective Membrane Expression of the Na+-HCO3− Cotransporter NBCe1 Is Associated with Familial Migraine. Proc. Natl. Acad. Sci. USA 2010, 107, 15963–15968. [Google Scholar] [CrossRef]
- Suzuki, M.; Vaisbich, M.H.; Yamada, H.; Horita, S.; Li, Y.; Sekine, T.; Moriyama, N.; Igarashi, T.; Endo, Y.; Cardoso, T.P.; et al. Functional Analysis of a Novel Missense NBC1 Mutation and of Other Mutations Causing Proximal Renal Tubular Acidosis. Pflug. Arch. Eur. J. Physiol. 2008, 455, 583–593. [Google Scholar] [CrossRef]
- Toye, A.M.; Parker, M.D.; Daly, C.M.; Lu, J.; Virkki, L.V.; Pelletier, M.F.; Boron, W.F. The Human NBCe1-A Mutant R881C, Associated with Proximal Renal Tubular Acidosis, Retains Function but Is Mistargeted in Polarized Renal Epithelia. Am. J. Physiol.-Cell Physiol. 2006, 291, C788–C801. [Google Scholar] [CrossRef]
- Demirci, F.Y.; Chang, M.; Mah, T.S.; Romero, M.E.F.; Gorin, M.B. Proximal Renal Tubular Acidosis and Ocular Pathology: A Novel Missense Mutation in the Gene (SLC4A4) for Sodium Bicarbonate Cotransporter Protein (NBCe1). Mol. Vis. 2006, 12, 324–330. [Google Scholar]
- Horita, S.; Yamada, H.; Inatomi, J.; Moriyama, N.; Sekine, T.; Igarashi, T.; Endo, Y.; Dasouki, M.; Ekim, M.; Al-Gazali, L.; et al. Functional Analysis of NBC1 Mutants Associated with Proximal Renal Tubular Acidosis and Ocular Abnormalities. JASN 2005, 16, 2270–2278. [Google Scholar] [CrossRef]
- Inatomi, J.; Horita, S.; Braverman, N.; Sekine, T.; Yamada, H.; Suzuki, Y.; Kawahara, K.; Moriyama, N.; Kudo, A.; Kawakami, H.; et al. Mutational and Functional Analysis of SLC4A4 in a Patient with Proximal Renal Tubular Acidosis. Pflug. Arch. Eur. J. Physiol. 2004, 448, 438–444. [Google Scholar] [CrossRef]
- Dinour, D.; Chang, M.-H.; Satoh, J.; Smith, B.L.; Angle, N.; Knecht, A.; Serban, I.; Holtzman, E.J.; Romero, M.F. A Novel Missense Mutation in the Sodium Bicarbonate Cotransporter (NBCe1/SLC4A4) Causes Proximal Tubular Acidosis and Glaucoma through Ion Transport Defects. J. Biol. Chem. 2004, 279, 52238–52246. [Google Scholar] [CrossRef]
- Igarashi, T.; Inatomi, J.; Sekine, T.; Seki, G.; Shimadzu, M.; Tozawa, F.; Takeshima, Y.; Takumi, T.; Takahashi, T.; Yoshikawa, N.; et al. Novel Nonsense Mutation in the Na+/HCO3− Cotransporter Gene (SLC4A4) in a Patient with Permanent Isolated Proximal Renal Tubular Acidosis and Bilateral Glaucoma. JASN 2001, 12, 713–718. [Google Scholar] [CrossRef]
- Deda, G.; Ekim, M.; Gfven, A.; Karagöl, U.; Tümer, N. Hypopotassemic Paralysis: A Rare Presentation of Proximal Renal Tubular Acidosis. J. Child. Neurol. 2001, 16, 770–771. [Google Scholar] [CrossRef]
- Shiohara, M.; Igarashi, T.; Mori, T.; Komiyama, A. Genetic and Long-Term Data on a Patient with Permanent Isolated Proximal Renal Tubular Acidosis. Eur. J. Pediatr. 2000, 159, 892–894. [Google Scholar] [CrossRef]
- Igarashi, T.; Inatomi, J.; Sekine, T.; Cha, S.H.; Kanai, Y.; Kunimi, M.; Tsukamoto, K.; Satoh, H.; Shimadzu, M.; Tozawa, F.; et al. Mutations in SLC4A4 Cause Permanent Isolated Proximal Renal Tubular Acidosis with Ocular Abnormalities. Nat. Genet. 1999, 23, 264–266. [Google Scholar] [CrossRef]
- Parker, M.D.; Boron, W.F. The Divergence, Actions, Roles, and Relatives of Sodium-Coupled Bicarbonate Transporters. Physiol. Rev. 2013, 93, 803–959. [Google Scholar] [CrossRef]
- Lee, H.-W.; Osis, G.; Harris, A.N.; Fang, L.; Romero, M.F.; Handlogten, M.E.; Verlander, J.W.; Weiner, I.D. NBCe1-A Regulates Proximal Tubule Ammonia Metabolism under Basal Conditions and in Response to Metabolic Acidosis. JASN 2018, 29, 1182–1197. [Google Scholar] [CrossRef]
- Brady, C.T.; Marshall, A.; Zhang, C.; Parker, M.D. NBCe1-B/C-Knockout Mice Exhibit an Impaired Respiratory Response and an Enhanced Renal Response to Metabolic Acidosis. Front. Physiol. 2023, 14, 1201034. [Google Scholar] [CrossRef]
- Choi, I.; Romero, M.F.; Khandoudi, N.; Bril, A.; Boron, W.F. Cloning and Characterization of a Human Electrogenic Na+-HCO3− Cotransporter Isoform (hhNBC). Am. J. Physiol.-Cell Physiol. 1999, 276, C576–C584. [Google Scholar] [CrossRef]
- Alsufayan, T.A.; Myers, E.J.; Quade, B.N.; Brady, C.T.; Marshall, A.; Haque, N.; Duffey, M.E.; Parker, M.D. Revisiting the Role of Ser982 Phosphorylation in Stoichiometry Shift of the Electrogenic Na+/qHCO3−; Cotransporter NBCe1. Int. J. Mol. Sci. 2021, 22, 12817. [Google Scholar] [CrossRef]
- Bevensee, M.O.; Schmitt, B.M.; Choi, I.; Romero, M.F.; Boron, W.F. An Electrogenic Na+-HCO3− Cotransporter (NBC) with a Novel COOH-Terminus, Cloned from Rat Brain. Am. J. Physiol.-Cell Physiol. 2000, 278, C1200–C1211. [Google Scholar] [CrossRef]
- Virreira, M.; Jin, L.; Djerbib, S.; De Deken, X.; Miot, F.; Massart, C.; Svoboda, M.; Van Sande, J.; Beauwens, R.; Dumont, J.-E.; et al. Expression, Localization, and Regulation of the Sodium Bicarbonate Cotransporter NBCe1 in the Thyroid. Thyroid 2019, 29, 290–301. [Google Scholar] [CrossRef]
- Gawenis, L.R.; Bradford, E.M.; Prasad, V.; Lorenz, J.N.; Simpson, J.E.; Clarke, L.L.; Woo, A.L.; Grisham, C.; Sanford, L.P.; Doetschman, T.; et al. Colonic Anion Secretory Defects and Metabolic Acidosis in Mice Lacking the NBC1 Cotransporter. J. Biol. Chem. 2007, 282, 9042–9052. [Google Scholar] [CrossRef]
- Salerno, E.E.; Patel, S.P.; Marshall, A.; Marshall, J.; Alsufayan, T.; Mballo, C.S.A.; Quade, B.N.; Parker, M.D. Extrarenal Signs of Proximal Renal Tubular Acidosis Persist in Nonacidemic Nbce1b/c-Null Mice. JASN 2019, 30, 979–989. [Google Scholar] [CrossRef]
- De Giusti, V.C.; Ciancio, M.C.; Orlowski, A.; Aiello, E.A. Modulation of the Cardiac Sodium/Bicarbonate Cotransporter by the Renin Angiotensin Aldosterone System: Pathophysiological Consequences. Front. Physiol. 2014, 4, 411. [Google Scholar] [CrossRef]
- Lee, S.-K.; Occhipinti, R.; Moss, F.J.; Parker, M.D.; Grichtchenko, I.I.; Boron, W.F. Distinguishing among HCO3−, CO3=, and H+ as Substrates of Proteins That Appear To Be “Bicarbonate” Transporters. JASN 2022, 34, 40–54. [Google Scholar] [CrossRef]
- Di Mattia, R.A.; Diaz-Zegarra, L.A.; Blanco, P.G.; Valverde, C.A.; Gonano, L.A.; De Giusti, C.J.; Portiansky, E.L.; Vila-Petroff, M.G.; Aiello, E.A.; Orlowski, A. The specific inhibition of the cardiac electrogenic sodium/bicarbonate cotransporter leads to cardiac hypertrophy. Life Sci. 2023, 312, 121219. [Google Scholar] [CrossRef]
- Orlowski, A.; Ciancio, M.C.; Caldiz, C.I.; Giusti, D.C.V.; Aiello, E.A. Reduced Sarcolemmal Expression and Function of the NBCe1 Isoform of the Na+–HCO3− Cotransporter in Hypertrophied Cardiomyocytes of Spontaneously Hypertensive Rats: Role of the Renin–Angiotensin System. Cardiovasc. Res. 2014, 101, 211–219. [Google Scholar] [CrossRef]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Baumert, M.; Porta, A.; Vos, M.A.; Malik, M.; Couderc, J.-P.; Laguna, P.; Piccirillo, G.; Smith, G.L.; Tereshchenko, L.G.; Volders, P.G.A. QT Interval Variability in Body Surface ECG: Measurement, Physiological Basis, and Clinical Value: Position Statement and Consensus Guidance Endorsed by the European Heart Rhythm Association Jointly with the ESC Working Group on Cardiac Cellular Electrophysiology. EP Eur. 2016, 18, 925–944. [Google Scholar] [CrossRef]
- Johnson, D.M.; Heijman, J.; Bode, E.F.; Greensmith, D.J.; van der Linde, H.; Abi-Gerges, N.; Eisner, D.A.; Trafford, A.W.; Volders, P.G.A. Diastolic Spontaneous Calcium Release From the Sarcoplasmic Reticulum Increases Beat-to-Beat Variability of Repolarization in Canine Ventricular Myocytes After β-Adrenergic Stimulation. Circ. Res. 2013, 112, 246–256. [Google Scholar] [CrossRef]
- Alim, C.C.; Ko, C.Y.; Mira Hernandez, J.; Shen, E.Y.; Baidar, S.; Chen-Izu, Y.; Bers, D.M.; Bossuyt, J. Nitrosylation of Cardiac CaMKII at Cys290 Mediates Mechanical Afterload-Induced Increases in Ca2+ Transient and Ca2+ Sparks. J. Physiol. 2022, 600, 4865–4879. [Google Scholar] [CrossRef]
- van Oort, R.J.; McCauley, M.D.; Dixit, S.S.; Pereira, L.; Yang, Y.; Respress, J.L.; Wang, Q.; De Almeida, A.C.; Skapura, D.G.; Anderson, M.E.; et al. Ryanodine Receptor Phosphorylation by Calcium/Calmodulin-Dependent Protein Kinase II Promotes Life-Threatening Ventricular Arrhythmias in Mice With Heart Failure. Circulation 2010, 122, 2669–2679. [Google Scholar] [CrossRef]
- Uchinoumi, H.; Yang, Y.; Oda, T.; Li, N.; Alsina, K.M.; Puglisi, J.L.; Chen-Izu, Y.; Cornea, R.L.; Wehrens, X.H.T.; Bers, D.M. CaMKII-Dependent Phosphorylation of RyR2 Promotes Targetable Pathological RyR2 Conformational Shift. J. Mol. Cell Cardiol. 2016, 98, 62–72. [Google Scholar] [CrossRef]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/Calmodulin–Dependent Protein Kinase Modulates Cardiac Ryanodine Receptor Phosphorylation and Sarcoplasmic Reticulum Ca2+ Leak in Heart Failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef]
- Díaz, M.E.; Graham, H.K.; O’Neill, S.C.; Trafford, A.W.; Eisner, D.A. The Control of Sarcoplasmic Reticulum Ca Content in Cardiac Muscle. Cell Calcium 2005, 38, 391–396. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Terentyeva, R.; Nishijima, Y.; Sridhar, A.; Hamlin, R.L.; Carnes, C.A.; Györke, S. The Relationship between Arrhythmogenesis and Impaired Contractility in Heart Failure: Role of Altered Ryanodine Receptor Function. Cardiovasc. Res. 2011, 90, 493–502. [Google Scholar] [CrossRef]
- Vairamani, K.; Prasad, V.; Wang, Y.; Huang, W.; Chen, Y.; Medvedovic, M.; Lorenz, J.N.; Shull, G.E. NBCe1 Na+-HCO3− Cotransporter Ablation Causes Reduced Apoptosis Following Cardiac Ischemia-Reperfusion Injury in Vivo. World J. Cardiol. 2018, 10, 97–109. [Google Scholar] [CrossRef]
- Ibañez, A.M.; Espejo, M.S.; Zavala, M.R.; Villa-Abrille, M.C.; Lofeudo, J.M.; Aiello, E.A.; De Giusti, V.C. Regulation of Intracellular pH Is Altered in Cardiac Myocytes of Ovariectomized Rats. JAHA 2019, 8, e011066. [Google Scholar] [CrossRef]
- Yamamoto, T.; Shirayama, T.; Sakatani, T.; Takahashi, T.; Tanaka, H.; Takamatsu, T.; Spitzer, K.W.; Matsubara, H. Enhanced activity of ventricular Na+-HCO3− cotransport in pressure overload hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1254–H1264. [Google Scholar] [CrossRef]
- Teichholz, L.E.; Cohen, M.V.; Sonnenblick, E.H.; Gorlin, R. Study of Left Ventricular Geometry and Function by B-Scan Ultrasonography in Patients with and without Asynergy. N. Engl. J. Med. 1974, 291, 1220–1226. [Google Scholar] [CrossRef]
- Dele-Michael, A.O.; Fujikura, K.; Devereux, R.B.; Islam, F.; Hriljac, I.; Wilson, S.R.; Lin, F.; Weinsaft, J.W. Left Ventricular Stroke Volume Quantification by Contrast Echocardiography-Comparison of Linear and Flow-Based Methods to Cardiac Magnetic Resonance. Echocardiography 2013, 30, 880–888. [Google Scholar] [CrossRef]
- Sedghamiz, H. BioSigKit: A Matlab Toolbox and Interface for Analysis of BioSignals. JOSS 2018, 3, 671. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, J.; King, J.H.; Huang, C.L.-H.; Fraser, J.A. Measurement and Interpretation of Electrocardiographic QT Intervals in Murine Hearts. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1553–H1557. [Google Scholar] [CrossRef]
- Ackers-Johnson, M.; Li, P.Y.; Holmes, A.P.; O’Brien, S.-M.; Pavlovic, D.; Foo, R.S. A Simplified, Langendorff-Free Method for Concomitant Isolation of Viable Cardiac Myocytes and Nonmyocytes From the Adult Mouse Heart. Circ. Res. 2016, 119, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, G.E.; McLellan, M.A.; Weeks, K.L.; Matsumoto, A.; Cohen, C.D.; Krstevski, C.; Gaynor, T.L.; Parslow, A.C.; McMullen, J.R.; Pinto, A.R. A Protocol for Rapid and Parallel Isolation of Myocytes and Non-Myocytes from Multiple Mouse Hearts. STAR Protoc. 2021, 2, 100866. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hu, T.; Chen, L.; Sun, J.; Xie, J.; Li, R.; Xu, B. Selection of reference genes for normalization of quantitative polymerase chain reaction data in mouse models of heart failure. Mol. Med. Rep. 2015, 11, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Diester, C.M.; Banks, M.L.; Neigh, G.N.; Negus, S.S. Experimental design and analysis for consideration of sex as a biological variable. Neuropsychopharmacology 2019, 44, 2159–2162. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brady, C.T.; Marshall, A.; Eagler, L.A.; Pon, T.M.; Duffey, M.E.; Weil, B.R.; Lang, J.K.; Parker, M.D. Left Ventricular Systolic Dysfunction in NBCe1-B/C-Knockout Mice. Int. J. Mol. Sci. 2024, 25, 9610. https://doi.org/10.3390/ijms25179610
Brady CT, Marshall A, Eagler LA, Pon TM, Duffey ME, Weil BR, Lang JK, Parker MD. Left Ventricular Systolic Dysfunction in NBCe1-B/C-Knockout Mice. International Journal of Molecular Sciences. 2024; 25(17):9610. https://doi.org/10.3390/ijms25179610
Chicago/Turabian StyleBrady, Clayton T., Aniko Marshall, Lisa A. Eagler, Thomas M. Pon, Michael E. Duffey, Brian R. Weil, Jennifer K. Lang, and Mark D. Parker. 2024. "Left Ventricular Systolic Dysfunction in NBCe1-B/C-Knockout Mice" International Journal of Molecular Sciences 25, no. 17: 9610. https://doi.org/10.3390/ijms25179610