

Ccn2 Deletion Reduces Cardiac Dysfunction, Oxidative Markers, and Fibrosis Induced by Doxorubicin Administration in Mice
 , , , , , , , , ,
, , , , , , , , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
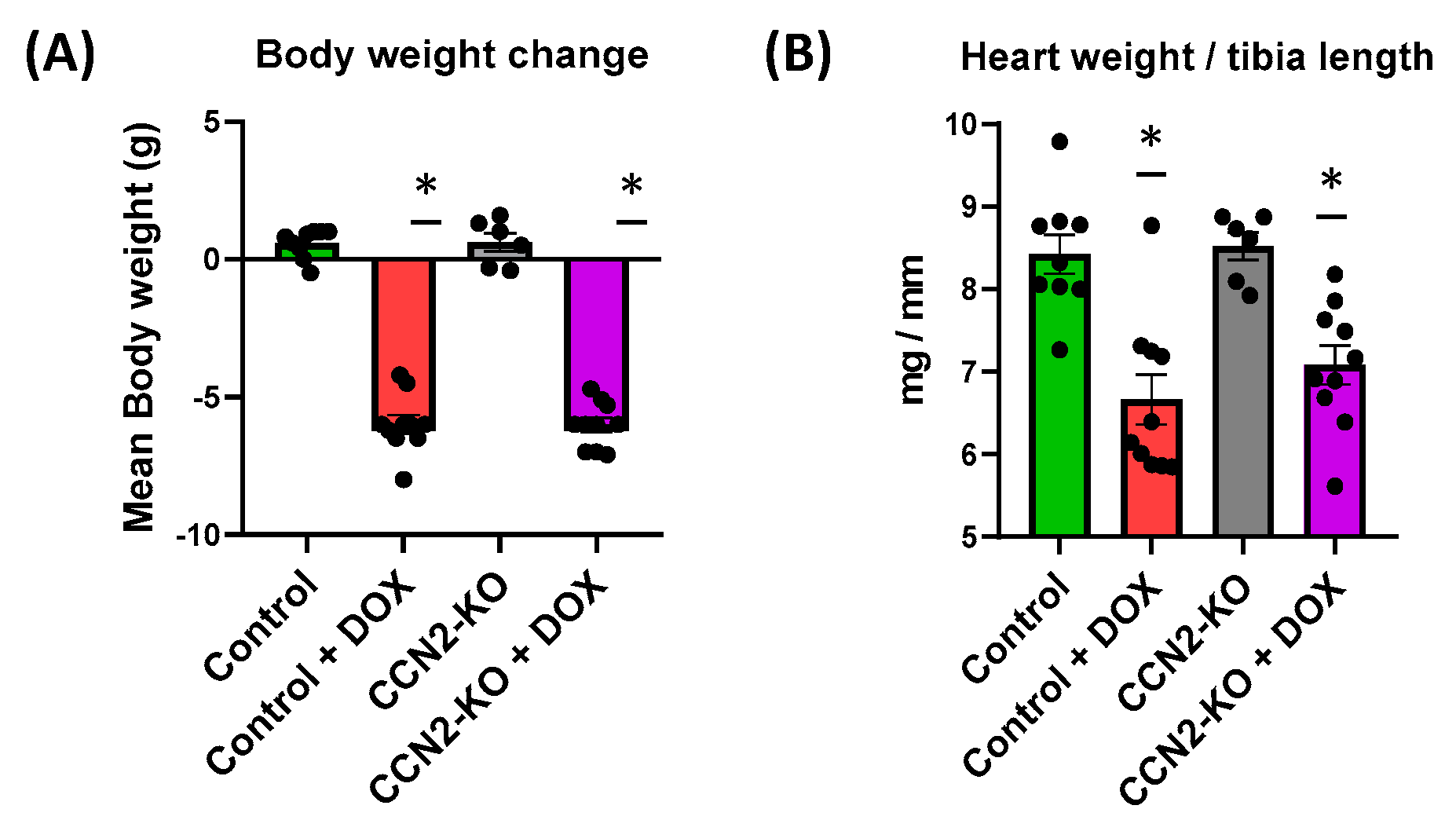
2.1. Absence of CCN2 Does Not Alter DOX-Induced Body Weight and Cardiac Mass Loss in Mice
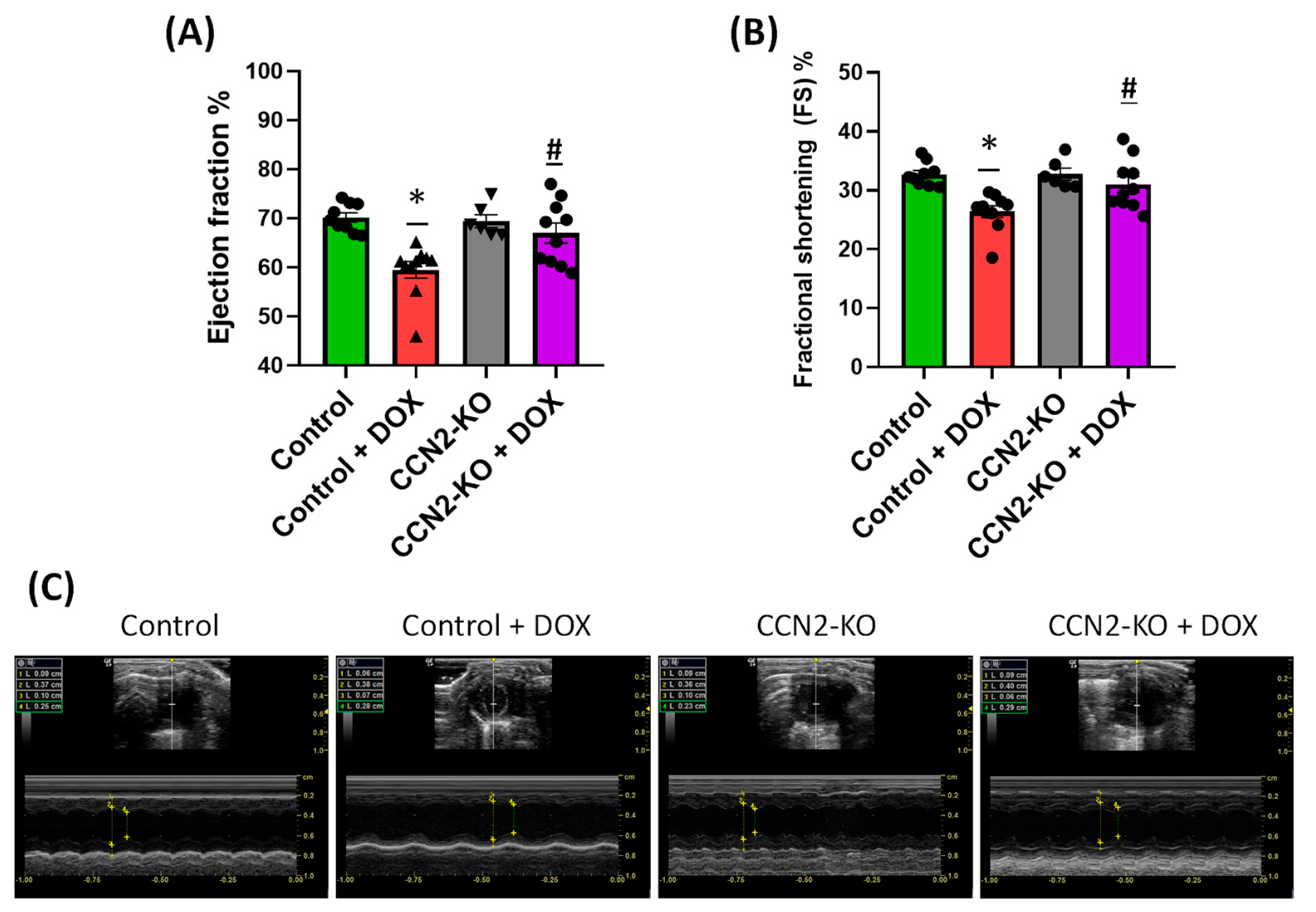
2.2. Deletion of Ccn2 in Mice Reduces DOX-Induced Cardiac Dysfunction
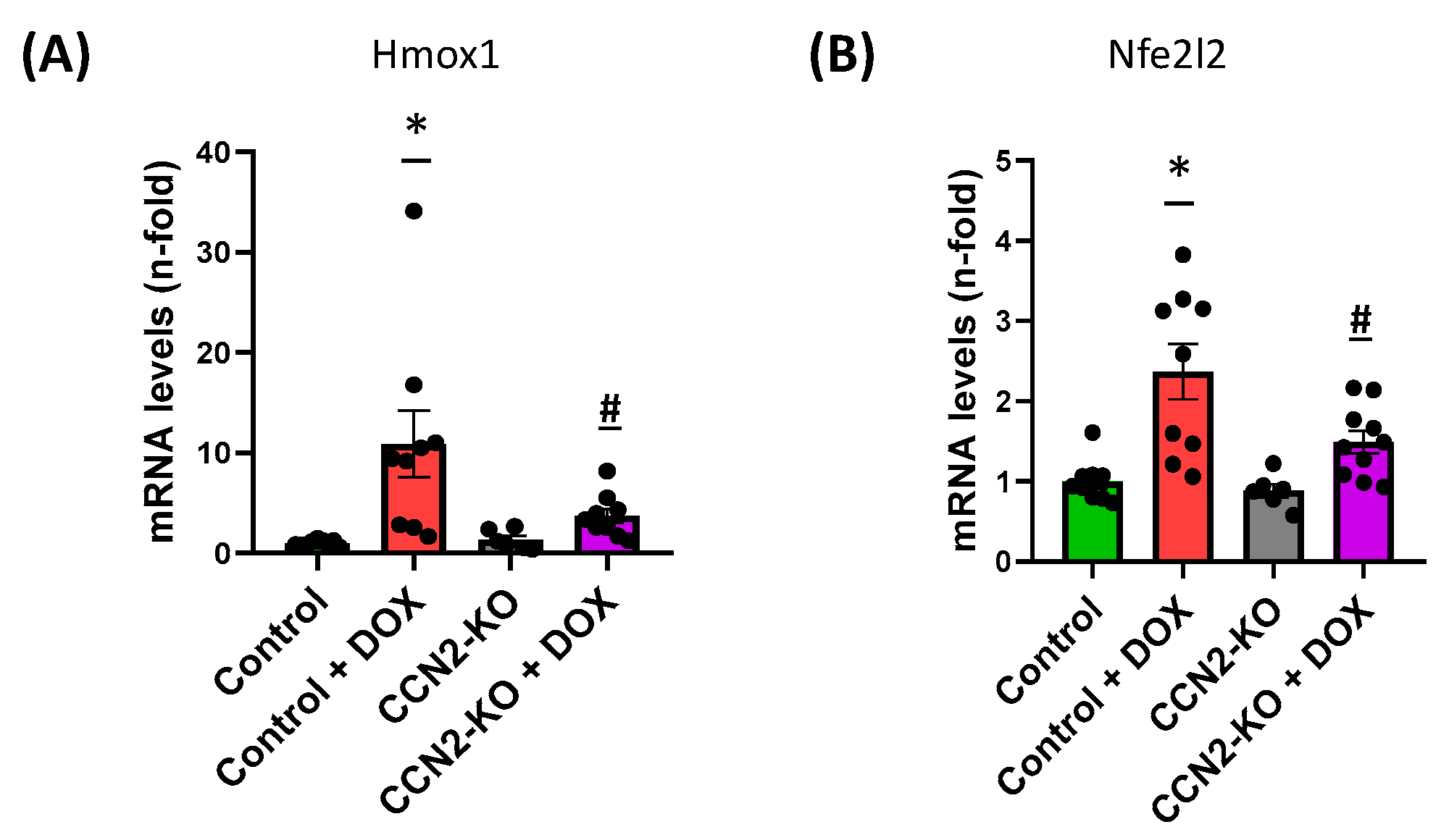
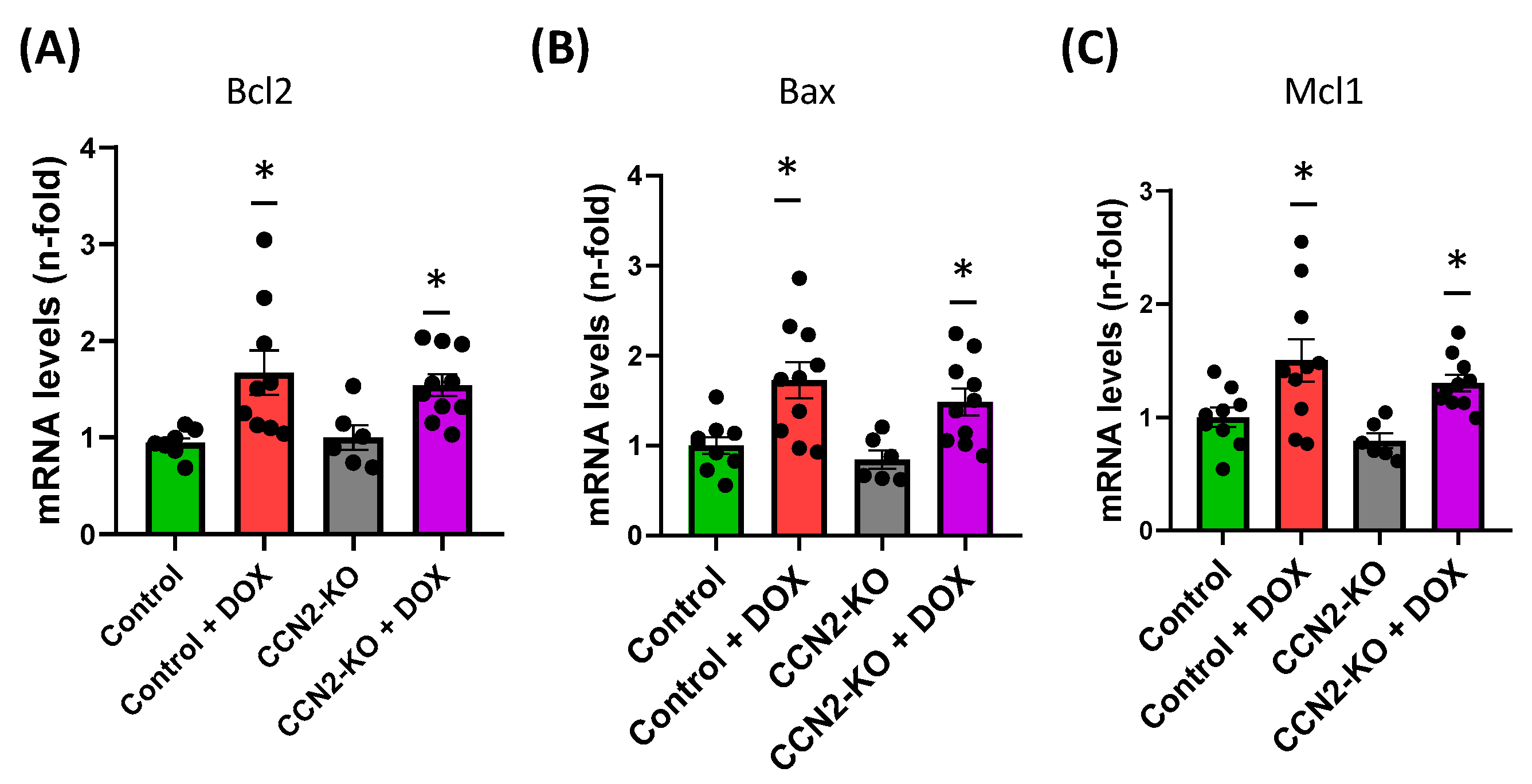
2.3. CCN2 Deletion Modulates the Effects of DOX on Gene Expression Markers of Oxidative Stress and Cardiac Damage in Mouse Heart
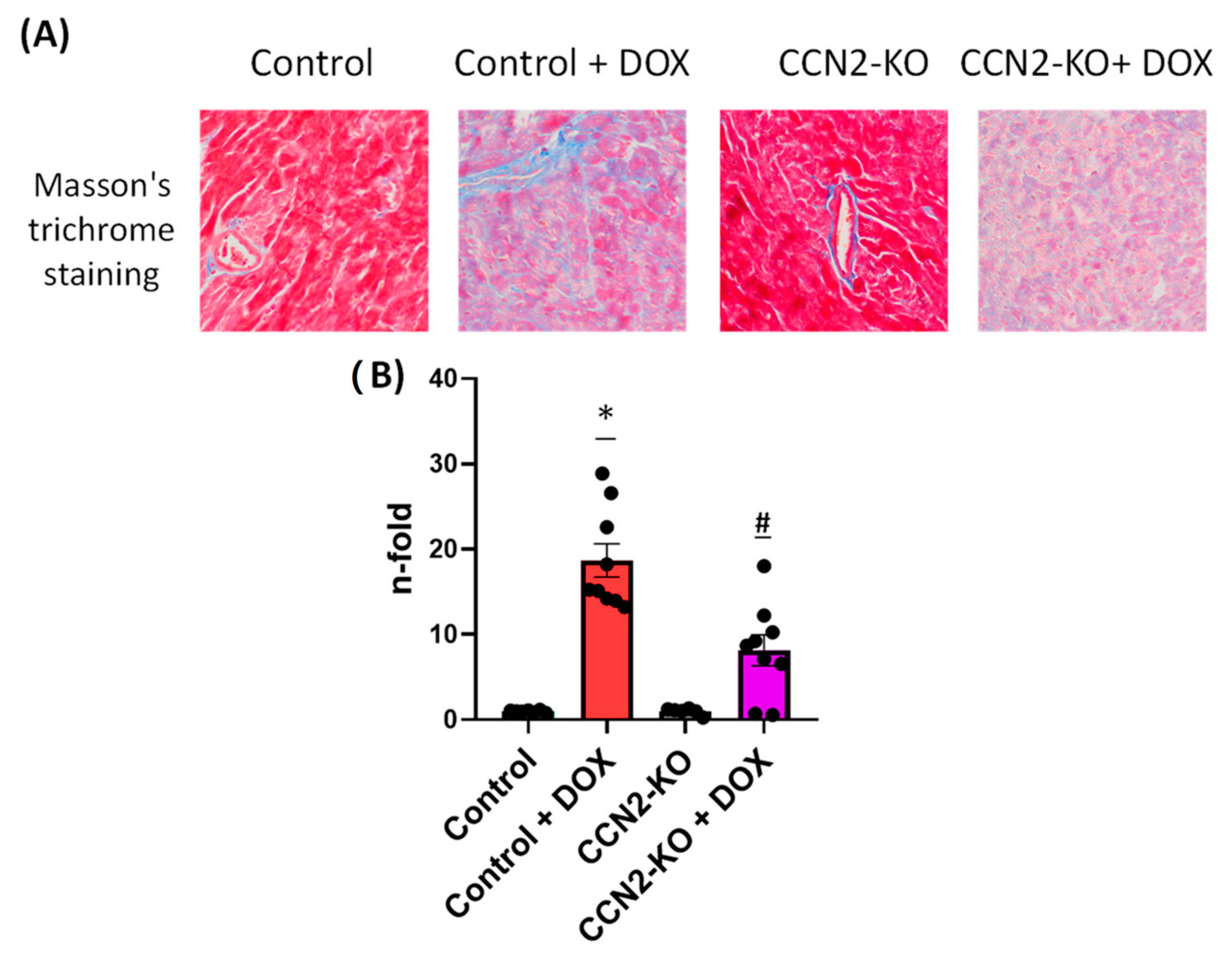
2.4. Absence of CCN2 Reduces DOX-Induced Cardiac Fibrosis
3. Discussion
4. Materials and Methods
4.1. Study Approval
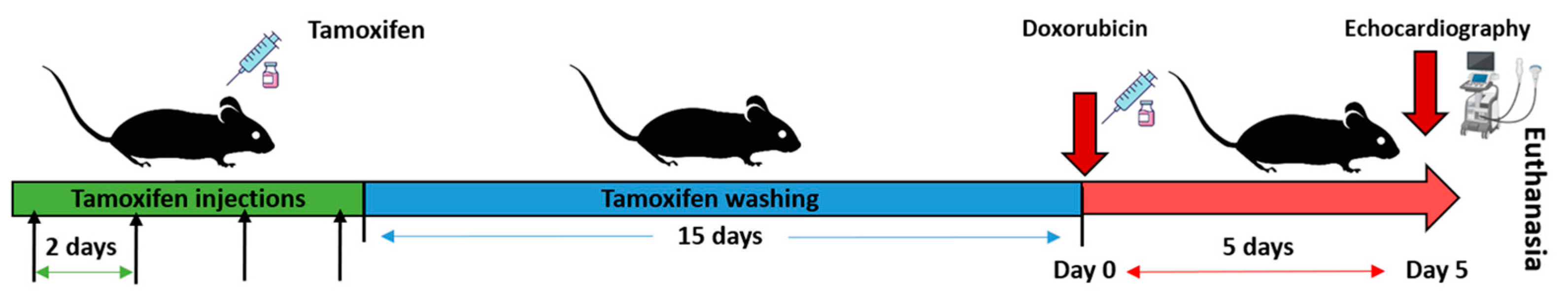
4.2. Generation of Tamoxifen-Inducible CCN2 Full-KO Mice and DOX Administration
4.3. Evaluation of Cardiac Function by Echocardiography
4.4. Gene Expression Studies by RT-qPCR
4.5. Detection of NRF2 Activation by Immunohistochemistry
4.6. Evaluation of Cardiac Fibrosis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; Heuvel, L.v.D.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Zaykov, V.; Chaqour, B. The CCN2/CTGF interactome: An approach to understanding the versatility of CCN2/CTGF molecular activities. J. Cell Commun. Signal. 2021, 15, 567–580. [Google Scholar] [CrossRef]
- Perbal, B. The concept of the CCN protein family revisited: A centralized coordination network. J. Cell Commun. Signal. 2018, 12, 3–12. [Google Scholar] [CrossRef]
- Hashimoto, G.; Inoki, I.; Fujii, Y.; Aoki, T.; Ikeda, E.; Okada, Y. Matrix Metalloproteinases Cleave Connective Tissue Growth Factor and Reactivate Angiogenic Activity of Vascular Endothelial Growth Factor 165. J. Biol. Chem. 2002, 277, 36288–36295. [Google Scholar] [CrossRef]
- Rodrigues-Díez, R.R.; Tejera-Muñoz, A.; Esteban, V.; Steffensen, L.B.; Rodrigues-Díez, R.; Orejudo, M.; Rayego-Mateos, S.; Falke, L.L.; Cannata-Ortiz, P.; Ortiz, A.; et al. CCN2 (Cellular Communication Network Factor 2) Deletion Alters Vascular Integrity and Function Predisposing to Aneurysm Formation. Hypertension 2022, 79, e42–e55. [Google Scholar] [CrossRef]
- Xu, Z.; Jin, Y.; Gao, Z.; Zeng, Y.; Du, J.; Yan, H.; Chen, X.; Ping, L.; Lin, N.; Yang, B.; et al. Autophagic degradation of CCN2 (cellular communication network factor 2) causes cardiotoxicity of sunitinib. Autophagy 2022, 18, 1152–1173. [Google Scholar] [CrossRef]
- Ponticos, M. Connective tissue growth factor (CCN2) in blood vessels. Vasc. Pharmacol. 2013, 58, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Dev. Camb. Engl. 2003, 130, 2779–2791. [Google Scholar] [CrossRef]
- Rupérez, M.; Lorenzo, O.; Blanco-Colio, L.M.; Esteban, V.; Egido, J.; Ruiz-Ortega, M. Connective Tissue Growth Factor Is a Mediator of Angiotensin II–Induced Fibrosis. Circulation 2003, 108, 1499–1505. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ. Res. 2010, 106, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Campillo, S.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marquez-Exposito, L.; Goldschmeding, R.; Rodríguez-Puyol, D.; Calleros, L.; Ruiz-Ortega, M. Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis. Clin. Sci. 2021, 135, 1999–2029. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Conjunction junction, what’s the function? CCN proteins as targets in fibrosis and cancers. Am. J. Physiol. Cell Physiol. 2020, 318, C1046–C1054. [Google Scholar] [CrossRef] [PubMed]
- Birkeness, L.B.; Banerjee, S.; Quadir, M.; Banerjee, S.K. The role of CCNs in controlling cellular communication in the tumor microenvironment. J. Cell Commun. Signal. 2023, 17, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Gene: CCN2 (ENSG00000118523). Available online: http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000118523;r=6:131948176-131951372;t=ENST00000367976 (accessed on 1 April 2024).
- Wang, Y.; Liu, X.; Xu, Q.; Xu, W.; Zhou, X.; Lin, Z. CCN2 Deficiency in Smooth Muscle Cells Triggers Cell Reprogramming and Aggravates Aneurysm Development. JCI Insight. 10 de enero de 2023; 8. Available online: https://insight.jci.org/articles/view/162987 (accessed on 1 April 2024).
- Koitabashi, N.; Arai, M.; Niwano, K.; Watanabe, A.; Endoh, M.; Suguta, M.; Yokoyama, T.; Tada, H.; Toyama, T.; Adachi, H.; et al. Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur. J. Hear. Fail. 2008, 10, 373–379. [Google Scholar] [CrossRef]
- Wang, R.; Xu, Y.-J.; Liu, X.-S.; Zeng, D.-X.; Xiang, M. Knockdown of connective tissue growth factor by plasmid-based short hairpin RNA prevented pulmonary vascular remodeling in cigarette smoke-exposed rats. Arch. Biochem. Biophys. 2011, 508, 93–100. [Google Scholar] [CrossRef]
- Szabó, Z.; Magga, J.; Alakoski, T.; Ulvila, J.; Piuhola, J.; Vainio, L.; Kivirikko, K.I.; Vuolteenaho, O.; Ruskoaho, H.; Lipson, K.E.; et al. Connective Tissue Growth Factor Inhibition Attenuates Left Ventricular Remodeling and Dysfunction in Pressure Overload–Induced Heart Failure. Hypertension 2014, 63, 1235–1240. [Google Scholar] [CrossRef]
- Panek, A.N.; Posch, M.G.; Alenina, N.; Ghadge, S.K.; Erdmann, B.; Popova, E.; Perrot, A.; Geier, C.; Morano, R.D.I.; Bader, M.; et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS ONE 2009, 4, e6743. [Google Scholar] [CrossRef]
- Ahmed, M.S.; Gravning, J.; Martinov, V.N.; von Lueder, T.G.; Edvardsen, T.; Czibik, G.; Moe, I.T.; Vinge, L.E.; Øie, E.; Valen, G.; et al. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1291–H1302. [Google Scholar] [CrossRef]
- Gravning, J.; Ahmed, M.S.; von Lueder, T.G.; Edvardsen, T.; Attramadal, H. CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload. Int. J. Cardiol. 2013, 168, 2049–2056. [Google Scholar] [CrossRef]
- Kaasbøll, O.J.; Moe, I.T.; Ahmed, M.S.; Stang, E.; Hagelin, E.M.V.; Attramadal, H. CTGF/CCN2 Postconditioning Increases Tolerance of Murine Hearts towards Ischemia-Reperfusion Injury. PLoS ONE 2016, 11, e0149000. [Google Scholar] [CrossRef]
- Larsen, J.H.; Stubbe, J.; Beck, H.C.; Overgaard, M.; Lindegaard, C.A.; Hansen, D.R.; Goldschmeding, R.; Rodriguez-Díez, R.R.; Ruiz-Ortega, M.; Pyke, C.; et al. Ccn2 deficiency causes smooth muscle cell de-differentiation and severe atherosclerosis in hyperlipidemic mice. bioRxiv. 2023. [Google Scholar] [CrossRef]
- Pillai, V.B.; Bindu, S.; Sharp, W.; Fang, Y.H.; Kim, G.; Gupta, M.; Samant, S.; Gupta, M.P. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H962–H972. [Google Scholar] [CrossRef]
- Syukri, A.; Budu; Hatta, M.; Amir, M.; Rohman, M.S.; Mappangara, I.; Kaelan, C.; Wahyuni, S.; Bukhari, A.; Junita, A.R.; et al. Doxorubicin induced immune abnormalities and inflammatory responses via HMGB1, HIF1-α and VEGF pathway in progressive of cardiovascular damage. Ann. Med. Surg. 2022, 76, 103501. [Google Scholar] [CrossRef]
- Wenningmann, N.; Knapp, M.; Ande, A.; Vaidya, T.R.; Ait-Oudhia, S. Insights into Doxorubicin-induced Cardiotoxicity: Molecular Mechanisms, Preventive Strategies, and Early Monitoring. Mol. Pharmacol. 2019, 96, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wan, W.; Fan, M. HMOX1 silencing prevents doxorubicin-induced cardiomyocyte injury, mitochondrial dysfunction, and ferroptosis by downregulating CTGF. Gen. Thorac. Cardiovasc. Surg. 2023, 71, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Dorn, L.E.; Petrosino, J.M.; Wright, P.; Accornero, F. CTGF/CCN2 is an autocrine regulator of cardiac fibrosis. J. Mol. Cell. Cardiol. 2018, 121, 205–211. [Google Scholar] [CrossRef]
- Fontes, M.S.; Kessler, E.L.; van Stuijvenberg, L.; Brans, M.A.; Falke, L.L.; Kok, B.; Leask, A.; van Rijen, H.V.; Vos, M.A.; Goldschmeding, R.; et al. CTGF knockout does not affect cardiac hypertrophy and fibrosis formation upon chronic pressure overload. J. Mol. Cell Cardiol. 2015, 88, 82–90. [Google Scholar] [CrossRef]
- Biondo, L.A.; Junior, E.A.L.; Souza, C.O.; Cruz, M.M.; Cunha, R.D.C.; Alonso-Vale, M.I.; Oyama, L.M.; Nascimento, C.M.O.; Pimentel, G.D.; dos Santos, R.V.T.; et al. Impact of Doxorubicin Treatment on the Physiological Functions of White Adipose Tissue. PLoS ONE 2016, 11, e0151548. [Google Scholar] [CrossRef]
- Ohlig, J.; Henninger, C.; Zander, S.; Merx, M.; Kelm, M.; Fritz, G. Rac1-mediated cardiac damage causes diastolic dysfunction in a mouse model of subacute doxorubicin-induced cardiotoxicity. Arch. Toxicol. 2018, 92, 441–453. [Google Scholar] [CrossRef]
- Porteiro, B.; Fondevila, M.F.; Buque, X.; Gonzalez-Rellan, M.J.; Fernandez, U.; Mora, A.; Beiroa, D.; Senra, A.; Gallego, R.; Fernø, J.; et al. Pharmacological stimulation of p53 with low-dose doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis. Mol. Metab. 2018, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Parry, T.L.; Brown, D.I.; Mota, R.I.; Huang, W.; Beak, J.Y.; Sola, M.; Zhou, C.; Hicks, S.T.; Caughey, M.C.; et al. Doxorubicin Exposure Causes Subacute Cardiac Atrophy Dependent on the Striated Muscle–Specific Ubiquitin Ligase MuRF1. Circ. Hear. Fail. 2019, 12, e005234. [Google Scholar] [CrossRef]
- Henninger, C.; Huelsenbeck, S.; Wenzel, P.; Brand, M.; Huelsenbeck, J.; Schad, A.; Fritz, G. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacol. Res. 2015, 91, 47–56. [Google Scholar] [CrossRef]
- Podyacheva, E.Y.; Kushnareva, E.A.; Karpov, A.A.; Toropova, Y.G. Analysis of Models of Doxorubicin-Induced Cardiomyopathy in Rats and Mice. A Modern View from the Perspective of the Pathophysiologist and the Clinician. Front. Pharmacol. 2021, 12, 670479. [Google Scholar] [CrossRef]
- Rodrigues-Diez, R.R.; Garcia-Redondo, A.B.; Orejudo, M.; Rodrigues-Diez, R.; Briones, A.M.; Bosch-Panadero, E.; Kery, G.; Pato, J.; Ortiz, A.; Salaices, M.; et al. The C-terminal module IV of connective tissue growth factor, through EGFR/Nox1 signaling, activates the NF-κB pathway and proinflammatory factors in vascular smooth muscle cells. Antioxid. Redox Signal. 2015, 22, 29–47. [Google Scholar] [CrossRef]
- Valentijn, F.A.; Knoppert, S.N.; Pissas, G.; Rodrigues-Diez, R.R.; Marquez-Exposito, L.; Broekhuizen, R.; Mokry, M.; Kester, L.A.; Falke, L.L.; Goldschmeding, R.; et al. CCN2 Aggravates the Immediate Oxidative Stress–DNA Damage Response following Renal Ischemia–Reperfusion Injury. Antioxidants 2021, 10, 2020. [Google Scholar] [CrossRef]
- Chen, M.M.; Lam, A.; Abraham, J.A.; Schreiner, G.F.; Joly, A.H. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: A potential role in heart fibrosis. J. Mol. Cell Cardiol. 2000, 32, 1805–1819. [Google Scholar] [CrossRef] [PubMed]
- Hall-Glenn, F.; De Young, R.A.; Huang, B.L.; van Handel, B.; Hofmann, J.J.; Chen, T.T.; Choi, A.; Ong, J.R.; Benya, P.D.; Mikkola, H.; et al. CCN2/connective tissue growth factor is essential for pericyte adhesion and endothelial basement membrane formation during angiogenesis. PLoS ONE 2012, 7, e30562. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Gender-based differences in mechanisms of protection in myocardial ischemia–reperfusion injury. Cardiovasc. Res. 2007, 75, 478–486. [Google Scholar] [CrossRef]
- Gao, S.; Ho, D.; Vatner, D.E.; Vatner, S.F. Echocardiography in Mice. Curr. Protoc. Mouse Biol. 2011, 1, 71–83. [Google Scholar] [CrossRef]
- Jiménez-Castilla, L.; Opazo-Ríos, L.; Marin-Royo, G.; Orejudo, M.; Rodrigues-Diez, R.; Ballesteros-Martínez, C.; Soto-Catalán, M.; Caro-Ordieres, T.; Artaiz, I.; Suarez-Cortés, T.; et al. The Synthetic Flavonoid Hidrosmin Improves Endothelial Dysfunction and Atherosclerotic Lesions in Diabetic Mice. Antioxidants 2022, 11, 2499. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejera-Muñoz, A.; Cortés, M.; Rodriguez-Rodriguez, A.; Tejedor-Santamaria, L.; Marchant, V.; Rayego-Mateos, S.; Gimeno-Longas, M.J.; Leask, A.; Nguyen, T.Q.; Martín, M.; et al. Ccn2 Deletion Reduces Cardiac Dysfunction, Oxidative Markers, and Fibrosis Induced by Doxorubicin Administration in Mice. Int. J. Mol. Sci. 2024, 25, 9617. https://doi.org/10.3390/ijms25179617
Tejera-Muñoz A, Cortés M, Rodriguez-Rodriguez A, Tejedor-Santamaria L, Marchant V, Rayego-Mateos S, Gimeno-Longas MJ, Leask A, Nguyen TQ, Martín M, et al. Ccn2 Deletion Reduces Cardiac Dysfunction, Oxidative Markers, and Fibrosis Induced by Doxorubicin Administration in Mice. International Journal of Molecular Sciences. 2024; 25(17):9617. https://doi.org/10.3390/ijms25179617
Chicago/Turabian StyleTejera-Muñoz, Antonio, Marcelino Cortés, Alianet Rodriguez-Rodriguez, Lucia Tejedor-Santamaria, Vanessa Marchant, Sandra Rayego-Mateos, Maria José Gimeno-Longas, Andrew Leask, Tri Q. Nguyen, María Martín, and et al. 2024. "Ccn2 Deletion Reduces Cardiac Dysfunction, Oxidative Markers, and Fibrosis Induced by Doxorubicin Administration in Mice" International Journal of Molecular Sciences 25, no. 17: 9617. https://doi.org/10.3390/ijms25179617