
Insights from Structure-Based Simulations into the Persulfidation of Uridine Diphosphate-Glycosyltransferase71c5 Facilitating the Reversible Inactivation of Abscisic Acid

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
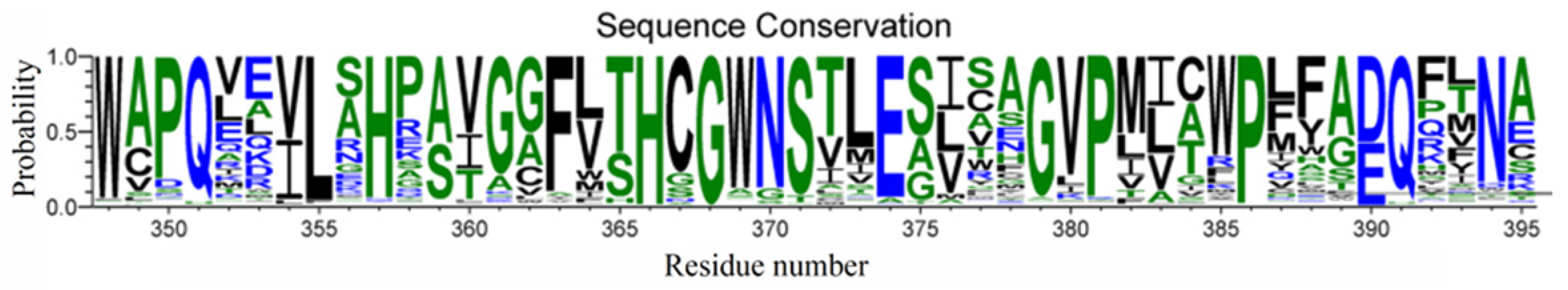
2.1. Identification of Persulfidation Sites in UGT71C5
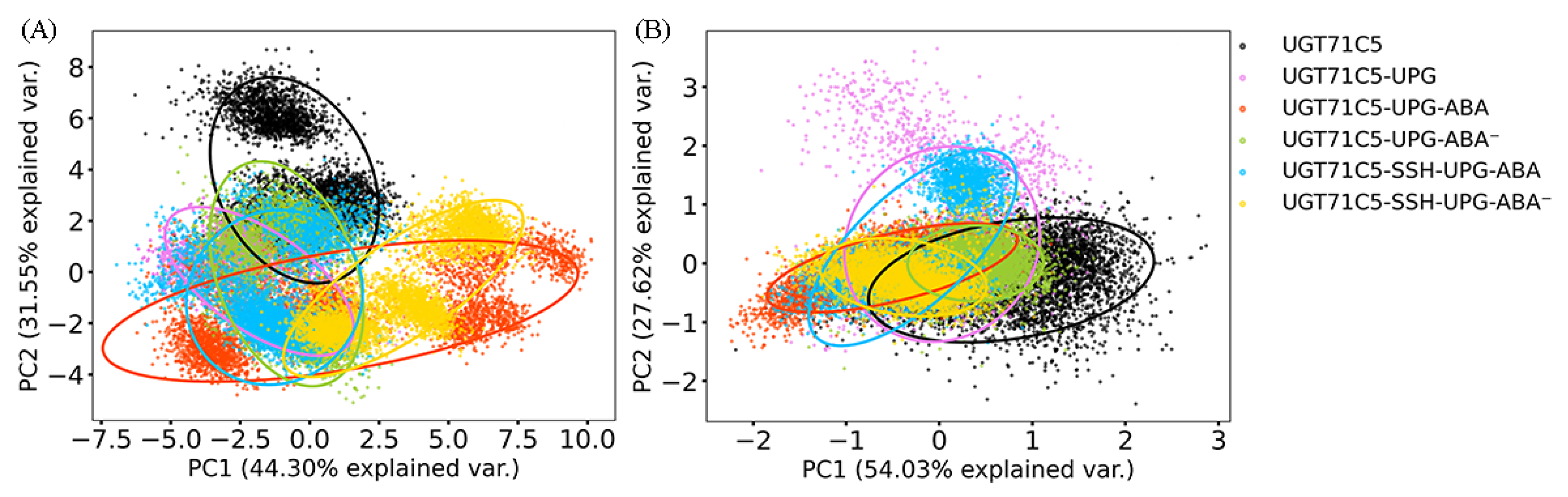
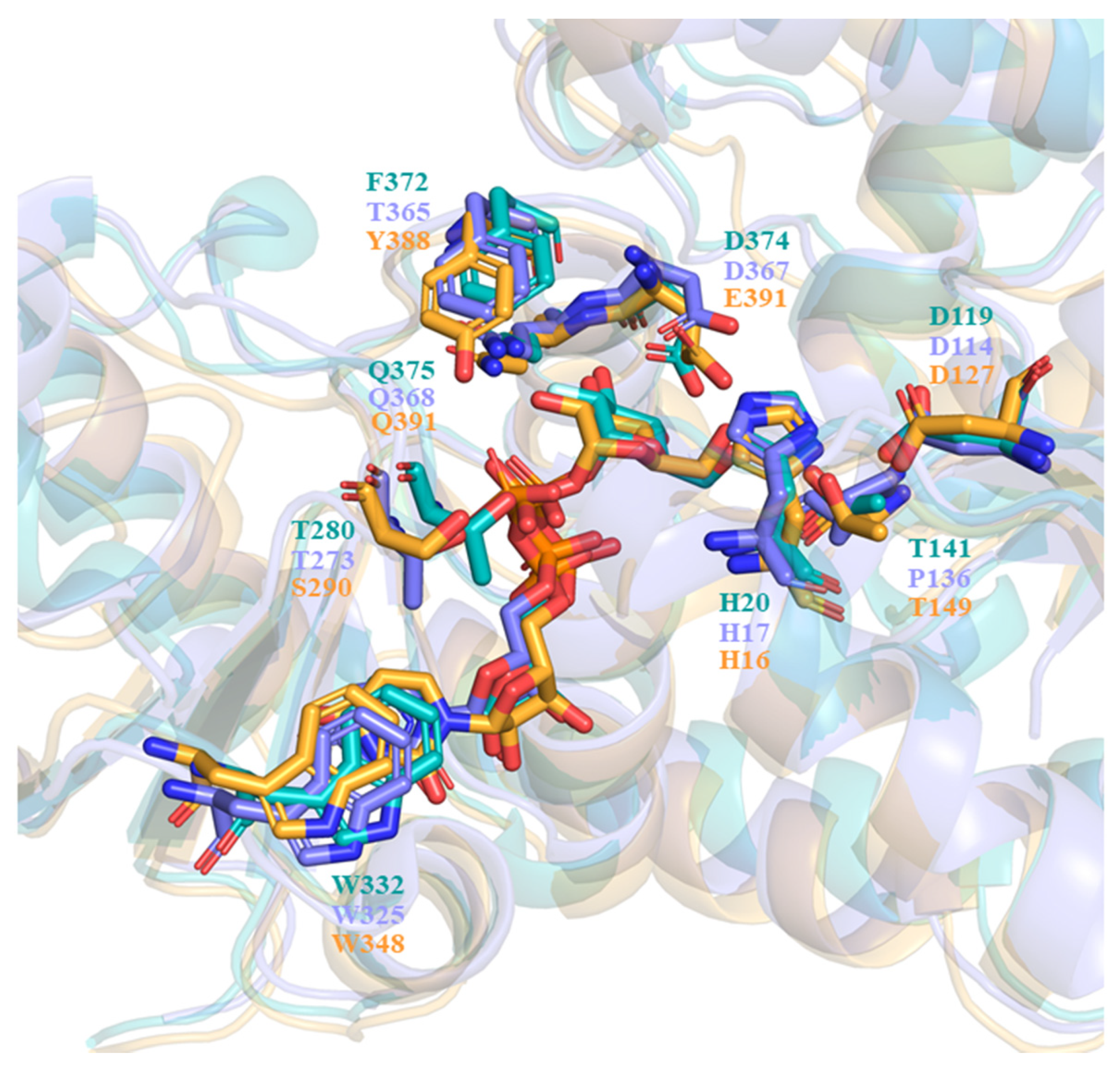
2.2. Structural Plasticity of UGT71C5 Associated with Substrate Binding and Persulfidation
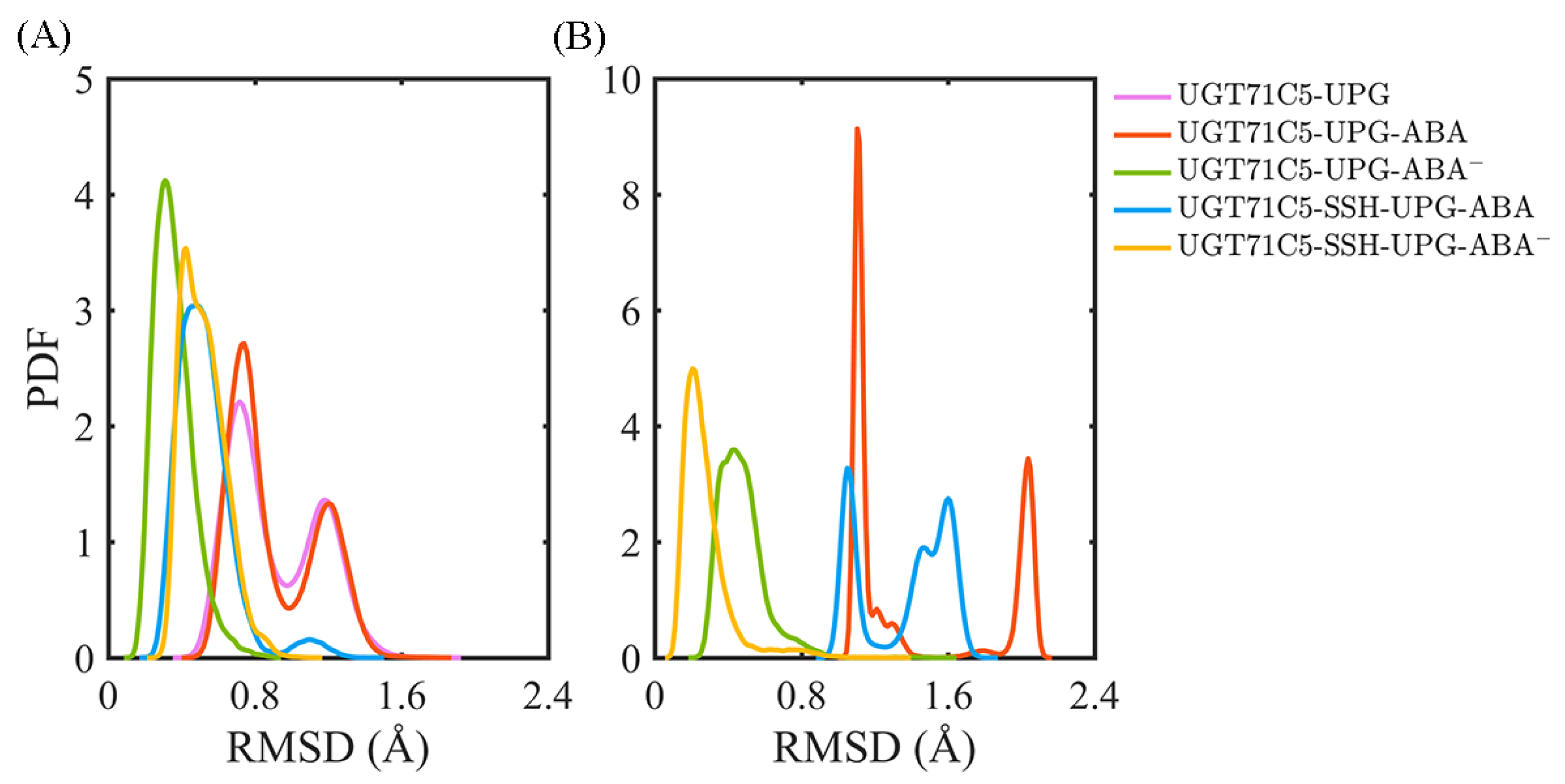
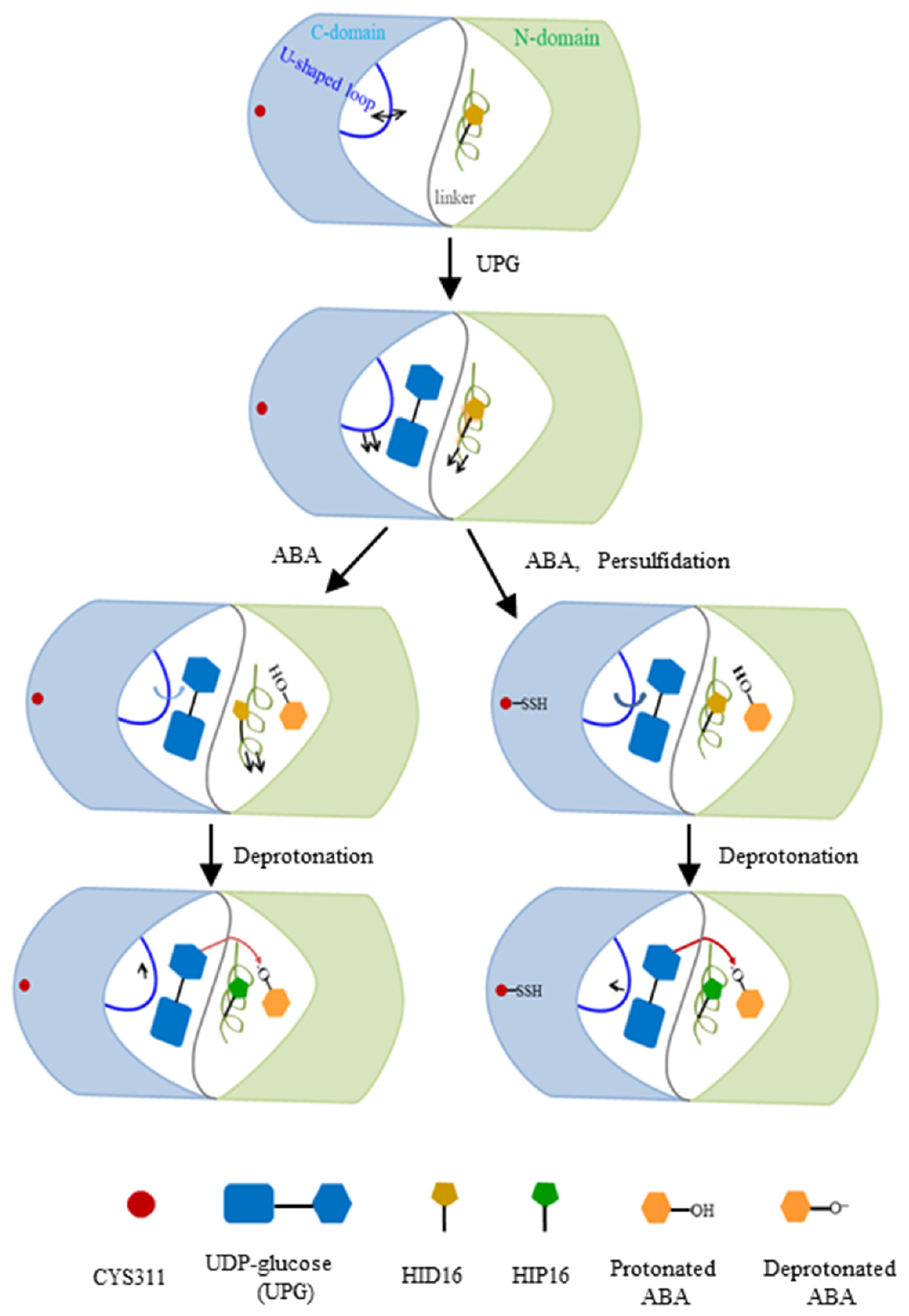
2.3. Substrate Binding vs. Persulfidation: Effects on the Linker and the U-Shaped Loop
2.4. Reduced Change in the Orientation of Deprotonated ABA in Persulfidated UGT71C5
2.5. A Dynamic Microenvironment for UPG Binding
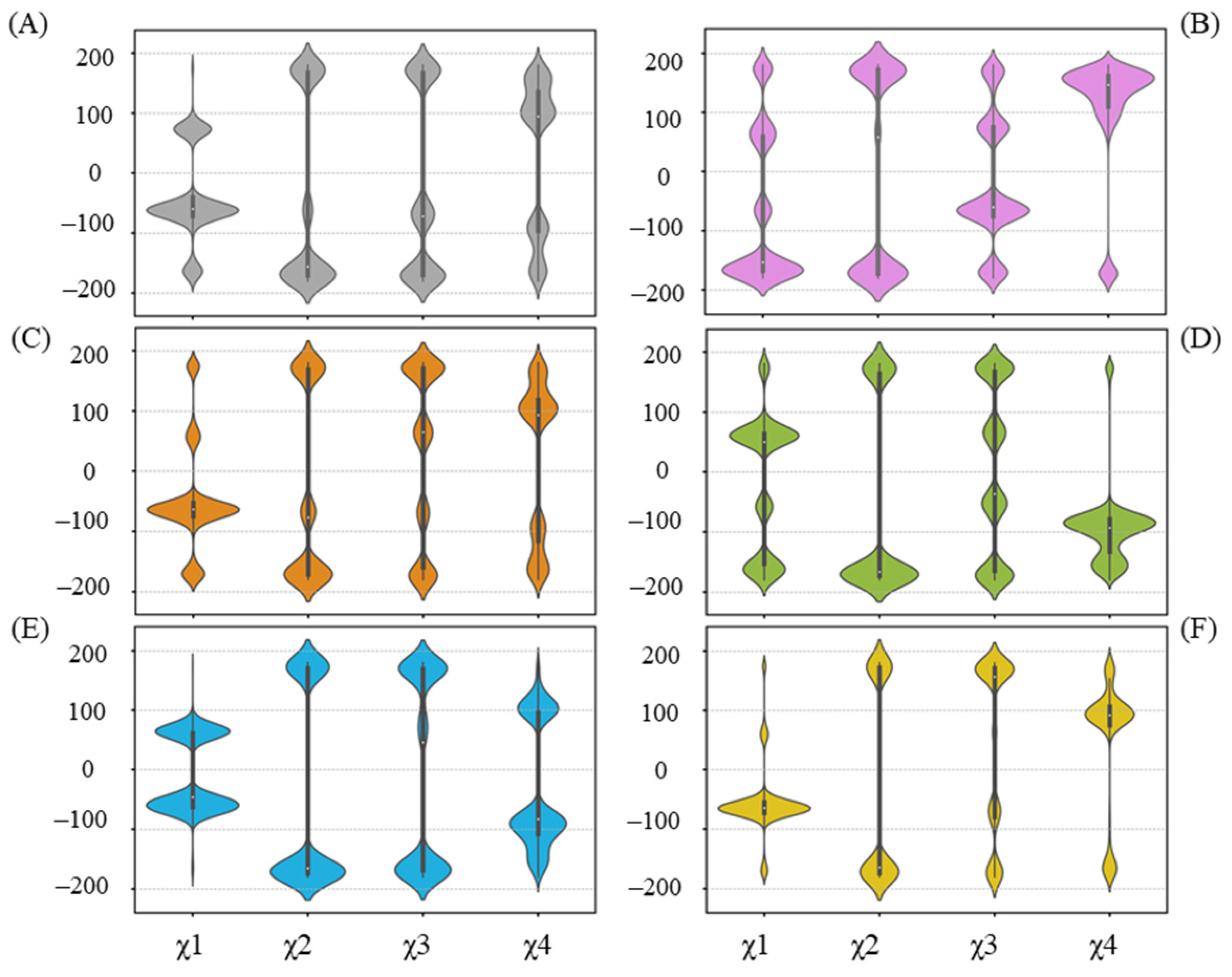
2.6. Alternative Conformations for Long Side-Chain Residues Highlight the Fluctuation in Donor Substrates
2.7. Preferable Configuration Condition for Glycosylating ABA Induced by Persulfidation
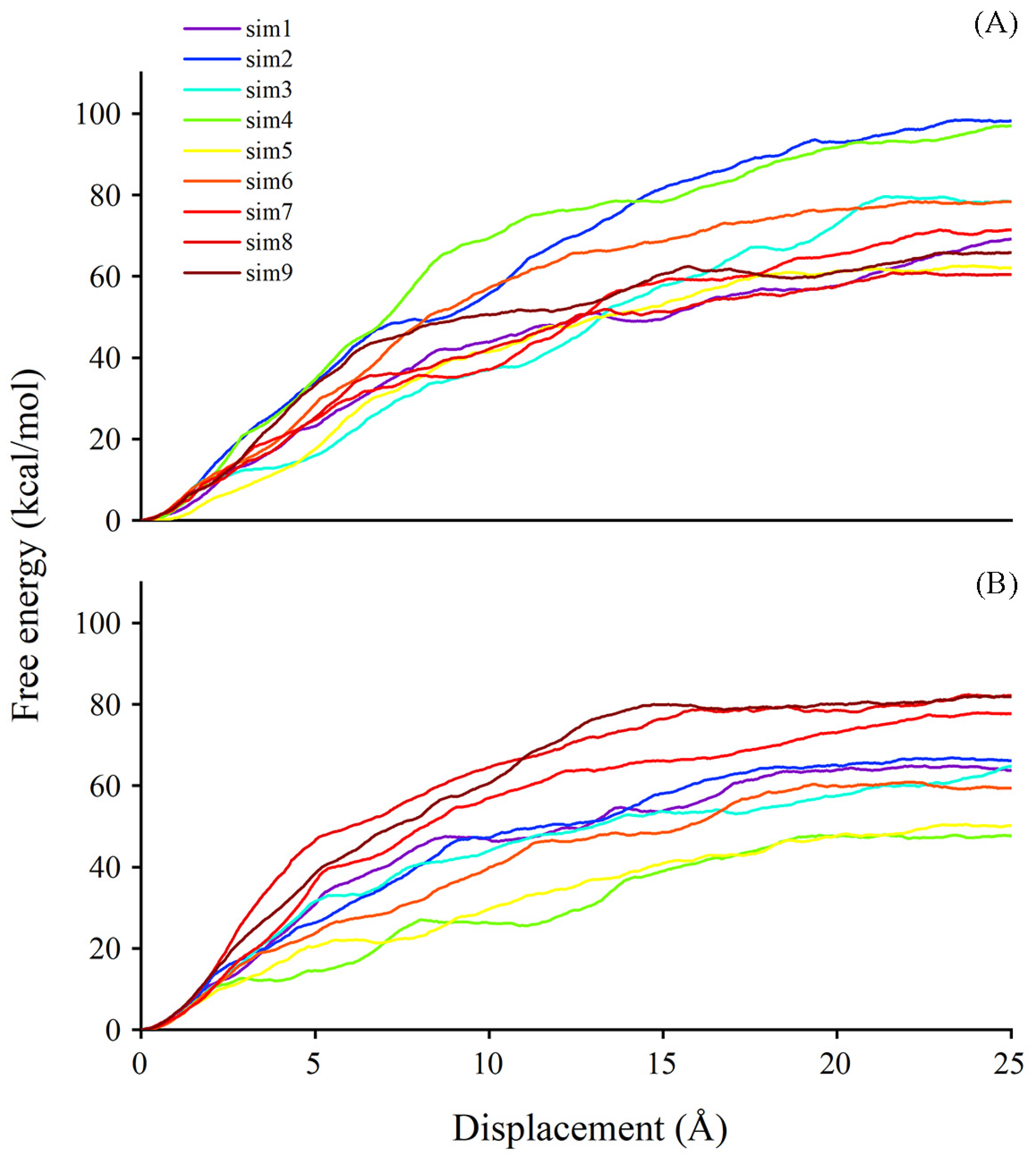
2.8. Better Performance of Acceptor Substrate Dissociation Exhibited in Persulfidated UGT71C5
3. Discussion
4. Materials and Methods
4.1. Recombinant Protein Expression and Site-Directed Mutagenesis
4.2. In Vitro Persulfidation Assay
4.3. System Setup
4.4. Conventional Molecular Dynamics (CMD) Simulation
4.5. Steered Molecular Dynamics (SMD) Simulation
4.6. Analysis of MD Trajectories
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilkinson, S.; Davies, W.J. ABA-based chemical signalling: The co-ordination of responses to stress in plants. Plant Cell Environ. 2002, 25, 195–210. [Google Scholar] [CrossRef]
- Chen, K.; Li, G.J.; Bressan, R.A.; Song, C.P.; Zhu, J.K.; Zhao, Y. Abscisic acid dynamics, signaling, and functions in plants. J. Integr. Plant Biol. 2020, 62, 25–54. [Google Scholar] [CrossRef] [PubMed]
- Marin, E.; Nussaume, L.; Quesada, A.; Gonneau, M.; Sotta, B.; Hugueney, P.; Frey, A.; Marion-Poll, A. Molecular identification of zeaxanthin epoxidase of Nicotiana plumbaginifolia, a gene involved in abscisic acid biosynthesis and corresponding to the ABA locus of Arabidopsis thaliana. EMBO J. 1996, 15, 2331–2342. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.H.; Tan, B.C.; Gage, D.A.; Zeevaart, J.A.; McCarty, D.R. Specific oxidative cleavage of carotenoids by VP14 of maize. Science 1997, 276, 1872–1874. [Google Scholar] [CrossRef]
- Xiong, L.; Lee, H.; Ishitani, M.; Zhu, J.K. Regulation of osmotic stress-responsive gene expression by the LOS6/ABA1 locus in Arabidopsis. J. Biol. Chem. 2002, 277, 8588–8596. [Google Scholar] [CrossRef]
- Seo, M.; Peeters, A.J.; Koiwai, H.; Oritani, T.; Marion-Poll, A.; Zeevaart, J.A.; Koornneef, M.; Kamiya, Y.; Koshiba, T. The Arabidopsis aldehyde oxidase 3 (AAO3) gene product catalyzes the final step in abscisic acid biosynthesis in leaves. Proc. Natl. Acad. Sci. USA 2000, 97, 12908–12913. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.H.; Endo, A.; Zhou, L.; Penney, J.; Chen, H.C.; Arroyo, A.; Leon, P.; Nambara, E.; Asami, T.; Seo, M.; et al. A unique short-chain dehydrogenase/reductase in Arabidopsis glucose signaling and abscisic acid biosynthesis and functions. Plant Cell 2002, 14, 2723–2743. [Google Scholar] [CrossRef]
- González-Guzmán, M.; Apostolova, N.; Bellés, J.M.; Barrero, J.M.; Piqueras, P.; Ponce, M.R.; Micol, J.L.; Serrano, R.; Rodríguez, P.L. The short-chain alcohol dehydrogenase ABA2 catalyzes the conversion of xanthoxin to abscisic aldehyde. Plant Cell 2002, 14, 1833–1846. [Google Scholar] [CrossRef]
- Kushiro, T.; Okamoto, M.; Nakabayashi, K.; Yamagishi, K.; Kitamura, S.; Asami, T.; Hirai, N.; Koshiba, T.; Kamiya, Y.; Nambara, E. The Arabidopsis cytochrome P450 CYP707A encodes ABA 8′-hydroxylases: Key enzymes in ABA catabolism. EMBO J. 2004, 23, 1647–1656. [Google Scholar] [CrossRef]
- Saito, S.; Hirai, N.; Matsumoto, C.; Ohigashi, H.; Ohta, D.; Sakata, K.; Mizutani, M. Arabidopsis CYP707As encode (+)-abscisic acid 8′-hydroxylase, a key enzyme in the oxidative catabolism of abscisic acid. Plant Physiol. 2004, 134, 1439–1449. [Google Scholar] [CrossRef]
- Lim, E.-K.; Doucet, C.; Hou, B.-K.; Jackson, R.; Abrams, S.; Bowles, D. Resolution of (+)-abscisic acid using an Arabidopsis glycosyltransferase. Tetrahedron-Asymmetry 2005, 16, 143–147. [Google Scholar] [CrossRef]
- Priest, D.M.; Ambrose, S.J.; Vaistij, F.E.; Elias, L.; Higgins, G.S.; Ross, A.R.; Abrams, S.R.; Bowles, D.J. Use of the glucosyltransferase UGT71B6 to disturb abscisic acid homeostasis in Arabidopsis thaliana. Plant J. 2006, 46, 492–502. [Google Scholar] [CrossRef]
- Lee, K.H.; Piao, H.L.; Kim, H.Y.; Choi, S.M.; Jiang, F.; Hartung, W.; Hwang, I.; Kwak, J.M.; Lee, I.J.; Hwang, I. Activation of glucosidase via stress-induced polymerization rapidly increases active pools of abscisic acid. Cell 2006, 126, 1109–1120. [Google Scholar] [CrossRef]
- Xu, Z.Y.; Lee, K.H.; Dong, T.; Jeong, J.C.; Jin, J.B.; Kanno, Y.; Kim, D.H.; Kim, S.Y.; Seo, M.; Bressan, R.A.; et al. A vacuolar β-glucosidase homolog that possesses glucose-conjugated abscisic acid hydrolyzing activity plays an important role in osmotic stress responses in Arabidopsis. Plant Cell 2012, 24, 2184–2199. [Google Scholar] [CrossRef]
- Lim, E.K.; Bowles, D.J. A class of plant glycosyltransferases involved in cellular homeostasis. EMBO J. 2004, 23, 2915–2922. [Google Scholar] [CrossRef]
- Bowles, D.; Isayenkova, J.; Lim, E.K.; Poppenberger, B. Glycosyltransferases: Managers of small molecules. Curr. Opin. Plant Biol. 2005, 8, 254–263. [Google Scholar] [CrossRef]
- Feldmann, K.A.; Goff, S.A. The First Plant Genome Sequence—Arabidopsis thaliana. Adv. Bot. Res. 2014, 69, 91–117. [Google Scholar]
- Dong, T.; Hwang, I. Contribution of ABA UDP-glucosyltransferases in coordination of ABA biosynthesis and catabolism for ABA homeostasis. Plant Signal. Behav. 2014, 9, e28888. [Google Scholar] [CrossRef]
- Liu, Z.; Yan, J.P.; Li, D.K.; Luo, Q.; Yan, Q.; Liu, Z.B.; Ye, L.M.; Wang, J.M.; Li, X.F.; Yang, Y. UDP-glucosyltransferase71c5, a major glucosyltransferase, mediates abscisic acid homeostasis in Arabidopsis. Plant Physiol. 2015, 167, 1659–1670. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef]
- Hoffmann, T.D.; Kurze, E.; Liao, J.; Hoffmann, T.; Song, C.; Schwab, W. Genome-wide identification of UDP-glycosyltransferases in the tea plant (Camellia sinensis) and their biochemical and physiological functions. Front. Plant Sci. 2023, 14, 1191625. [Google Scholar] [CrossRef]
- Wang, X. Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett. 2009, 583, 3303–3309. [Google Scholar] [CrossRef]
- Hughes, J.; Hughes, M.A. Multiple secondary plant product UDP-glucose glucosyltransferase genes expressed in cassava (Manihot esculenta Crantz) cotyledons. DNA Seq. 1994, 5, 41–49. [Google Scholar] [CrossRef]
- Paquette, S.; Møller, B.L.; Bak, S. On the origin of family 1 plant glycosyltransferases. Phytochemistry 2003, 62, 399–413. [Google Scholar] [CrossRef]
- Osmani, S.A.; Bak, S.; Møller, B.L. Substrate specificity of plant UDP-dependent glycosyltransferases predicted from crystal structures and homology modeling. Phytochemistry 2009, 70, 325–347. [Google Scholar] [CrossRef]
- Ozaki, S.; Imai, H.; Iwakiri, T.; Sato, T.; Shimoda, K.; Nakayama, T.; Hamada, H. Regioselective glucosidation of trans-resveratrol in Escherichia coli expressing glucosyltransferase from Phytolacca americana. Biotechnol. Lett. 2012, 34, 475–481. [Google Scholar] [CrossRef]
- Maharjan, R.; Fukuda, Y.; Nakayama, T.; Nakayama, T.; Hamada, H.; Ozaki, S.I.; Inoue, T. Structural basis for substrate recognition in the Phytolacca americana glycosyltransferase PaGT3. Acta Crystallogr. D Struct. Biol. 2022, 78, 379–389. [Google Scholar] [CrossRef]
- Offen, W.; Martinez-Fleites, C.; Yang, M.; Kiat-Lim, E.; Davis, B.G.; Tarling, C.A.; Ford, C.M.; Bowles, D.J.; Davies, G.J. Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification. EMBO J. 2006, 25, 1396–1405. [Google Scholar] [CrossRef]
- Breton, C.; Fournel-Gigleux, S.; Palcic, M.M. Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 2012, 22, 540–549. [Google Scholar] [CrossRef]
- Hiromoto, T.; Honjo, E.; Noda, N.; Tamada, T.; Kazuma, K.; Suzuki, M.; Blaber, M.; Kuroki, R. Structural basis for acceptor-substrate recognition of UDP-glucose: Anthocyanidin 3-O-glucosyltransferase from Clitoria ternatea. Protein Sci. 2015, 24, 395–407. [Google Scholar] [CrossRef]
- Calderwood, A.; Kopriva, S. Hydrogen sulfide in plants: From dissipation of excess sulfur to signaling molecule. Nitric Oxide 2014, 41, 72–78. [Google Scholar] [CrossRef]
- Jin, Z.; Pei, Y. Physiological Implications of Hydrogen Sulfide in Plants: Pleasant Exploration behind Its Unpleasant Odour. Oxid. Med. Cell. Longev. 2015, 2015, 397502. [Google Scholar] [CrossRef]
- Aroca, Á.; Serna, A.; Gotor, C.; Romero, L.C. S-sulfhydration: A cysteine posttranslational modification in plant systems. Plant Physiol. 2015, 168, 334–342. [Google Scholar] [CrossRef]
- Aroca, A.; Schneider, M.; Scheibe, R.; Gotor, C.; Romero, L.C. Hydrogen Sulfide Regulates the Cytosolic/Nuclear Partitioning of Glyceraldehyde-3-Phosphate Dehydrogenase by Enhancing its Nuclear Localization. Plant Cell Physiol. 2017, 58, 983–992. [Google Scholar] [CrossRef]
- Huang, J.; Xie, Y. Hydrogen Sulfide Signaling in Plants. Antioxid. Redox Signal. 2023, 39, 40–58. [Google Scholar] [CrossRef]
- Jin, Z.; Xue, S.; Luo, Y.; Tian, B.; Fang, H.; Li, H.; Pei, Y. Hydrogen sulfide interacting with abscisic acid in stomatal regulation responses to drought stress in Arabidopsis. Plant Physiol. Biochem. 2013, 62, 41–46. [Google Scholar] [CrossRef]
- Scuffi, D.; Álvarez, C.; Laspina, N.; Gotor, C.; Lamattina, L.; García-Mata, C. Hydrogen sulfide generated by L-cysteine desulfhydrase acts upstream of nitric oxide to modulate abscisic acid-dependent stomatal closure. Plant Physiol. 2014, 166, 2065–2076. [Google Scholar] [CrossRef]
- Papanatsiou, M.; Scuffi, D.; Blatt, M.R.; García-Mata, C. Hydrogen sulfide regulates inward-rectifying K+ channels in conjunction with stomatal closure. Plant Physiol. 2015, 168, 29–35. [Google Scholar] [CrossRef]
- Chen, S.; Jia, H.; Wang, X.; Shi, C.; Wang, X.; Ma, P.; Wang, J.; Ren, M.; Li, J. Hydrogen Sulfide Positively Regulates Abscisic Acid Signaling through Persulfidation of SnRK2.6 in Guard Cells. Mol. Plant 2020, 13, 732–744. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, J.; Zhou, M.; Zhou, H.; Cui, B.; Gotor, C.; Romero, L.C.; Fu, L.; Yang, J.; Foyer, C.H.; et al. Persulfidation-based Modification of Cysteine Desulfhydrase and the NADPH Oxidase RBOHD Controls Guard Cell Abscisic Acid Signaling. Plant Cell 2020, 32, 1000–1017. [Google Scholar] [CrossRef]
- Laureano-Marín, A.M.; Aroca, Á.; Pérez-Pérez, M.E.; Yruela, I.; Jurado-Flores, A.; Moreno, I.; Crespo, J.L.; Romero, L.C.; Gotor, C. Abscisic Acid-Triggered Persulfidation of the Cys Protease ATG4 Mediates Regulation of Autophagy by Sulfide. Plant Cell 2020, 32, 3902–3920. [Google Scholar] [CrossRef]
- Li, S.; Yu, K.; Wu, G.; Zhang, Q.; Wang, P.; Zheng, J.; Liu, Z.X.; Wang, J.; Gao, X.; Cheng, H. pCysMod: Prediction of Multiple Cysteine Modifications Based on Deep Learning Framework. Front. Cell Dev. Biol. 2021, 9, 617366. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Tsai, C.J.; Haliloğlu, T.; Nussinov, R. Dynamic allostery: Linkers are not merely flexible. Structure 2011, 19, 907–917. [Google Scholar] [CrossRef]
- Patel, D.K.; Menon, D.V.; Patel, D.H.; Dave, G. Linkers: A synergistic way for the synthesis of chimeric proteins. Protein Expr. Purif. 2022, 191, 106012. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.K. Plant glycosyltransferases: Their potential as novel biocatalysts. Chemistry 2005, 11, 5486–5494. [Google Scholar] [CrossRef] [PubMed]
- Hansen, E.H.; Osmani, S.A.; Kristensen, C.; Møller, B.L.; Hansen, J. Substrate specificities of family 1 UGTs gained by domain swapping. Phytochemistry 2009, 70, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Black, G.W. Structural, functional, and mutagenesis studies of UDP-glycosyltransferases. Adv. Protein Chem. Struct. Biol. 2012, 87, 87–115. [Google Scholar]
- Shao, H.; He, X.; Achnine, L.; Blount, J.W.; Dixon, R.A.; Wang, X. Crystal structures of a multifunctional triterpene/flavonoid glycosyltransferase from Medicago truncatula. Plant Cell 2005, 17, 3141–3154. [Google Scholar] [CrossRef]
- Brazier-Hicks, M.; Offen, W.A.; Gershater, M.C.; Revett, T.J.; Lim, E.K.; Bowles, D.J.; Davies, G.J.; Edwards, R. Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc. Natl. Acad. Sci. USA 2007, 104, 20238–20243. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Li, M.; Wu, T.; Wang, S.; Duan, T.; Huang, S.; Xie, Y. The Modulation of Sucrose Nonfermenting 1-Related Protein Kinase 2.6 State by Persulfidation and Phosphorylation: Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2023, 24, 11512. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., 3rd. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Harvey, M.J.; De Fabritiis, G. An Implementation of the Smooth Particle Mesh Ewald Method on GPU Hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. [Google Scholar] [CrossRef]
- Kalikka, J.; Akola, J. Steered molecular dynamics simulations of ligand-receptor interaction in lipocalins. Eur. Biophys. J. 2011, 40, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shen, J.; Luo, X.; Cheng, F.; Xu, Y.; Chen, K.; Arnold, E.; Ding, J.; Jiang, H. Steered molecular dynamics simulation on the binding of NNRTI to HIV-1 RT. Biophys. J. 2003, 84, 3547–3563. [Google Scholar] [CrossRef]
- Zhang, Z.; Santos, A.P.; Zhou, Q.; Liang, L.; Wang, Q.; Wu, T.; Franzen, S. Steered molecular dynamics study of inhibitor binding in the internal binding site in dehaloperoxidase-hemoglobin. Biophys. Chem. 2016, 211, 28–38. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Poornam, G.P.; Matsumoto, A.; Ishida, H.; Hayward, S. A method for the analysis of domain movements in large biomolecular complexes. Proteins 2009, 76, 201–212. [Google Scholar] [CrossRef]
- Prompers, J.J.; Brüschweiler, R. General framework for studying the dynamics of folded and nonfolded proteins by NMR relaxation spectroscopy and MD simulation. J. Am. Chem. Soc. 2002, 124, 4522–4534. [Google Scholar] [CrossRef]
- Sander, J.; Ester, M.; Kriegel, H.-P.; Xu, X. Density-Based Clustering in Spatial Databases: The Algorithm GDBSCAN and Its Applications. Data Min. Knowl. Discov. 1998, 2, 169–194. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, M.; Xiao, L.; Sun, K.; Qiu, T.; Lai, S.; Chen, G.; Geng, L.; Huang, S.; Xie, Y. Insights from Structure-Based Simulations into the Persulfidation of Uridine Diphosphate-Glycosyltransferase71c5 Facilitating the Reversible Inactivation of Abscisic Acid. Int. J. Mol. Sci. 2024, 25, 9679. https://doi.org/10.3390/ijms25179679
Li M, Xiao L, Sun K, Qiu T, Lai S, Chen G, Geng L, Huang S, Xie Y. Insights from Structure-Based Simulations into the Persulfidation of Uridine Diphosphate-Glycosyltransferase71c5 Facilitating the Reversible Inactivation of Abscisic Acid. International Journal of Molecular Sciences. 2024; 25(17):9679. https://doi.org/10.3390/ijms25179679
Chicago/Turabian StyleLi, Miaomiao, Lihui Xiao, Ke Sun, Taotao Qiu, Sisong Lai, Guojing Chen, Lingxi Geng, Siqi Huang, and Yanjie Xie. 2024. "Insights from Structure-Based Simulations into the Persulfidation of Uridine Diphosphate-Glycosyltransferase71c5 Facilitating the Reversible Inactivation of Abscisic Acid" International Journal of Molecular Sciences 25, no. 17: 9679. https://doi.org/10.3390/ijms25179679
APA StyleLi, M., Xiao, L., Sun, K., Qiu, T., Lai, S., Chen, G., Geng, L., Huang, S., & Xie, Y. (2024). Insights from Structure-Based Simulations into the Persulfidation of Uridine Diphosphate-Glycosyltransferase71c5 Facilitating the Reversible Inactivation of Abscisic Acid. International Journal of Molecular Sciences, 25(17), 9679. https://doi.org/10.3390/ijms25179679