
Alzheimer’s Disease Pathology and Assistive Nanotheranostic Approaches for Its Therapeutic Interventions

 ,
,  ,
,  , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
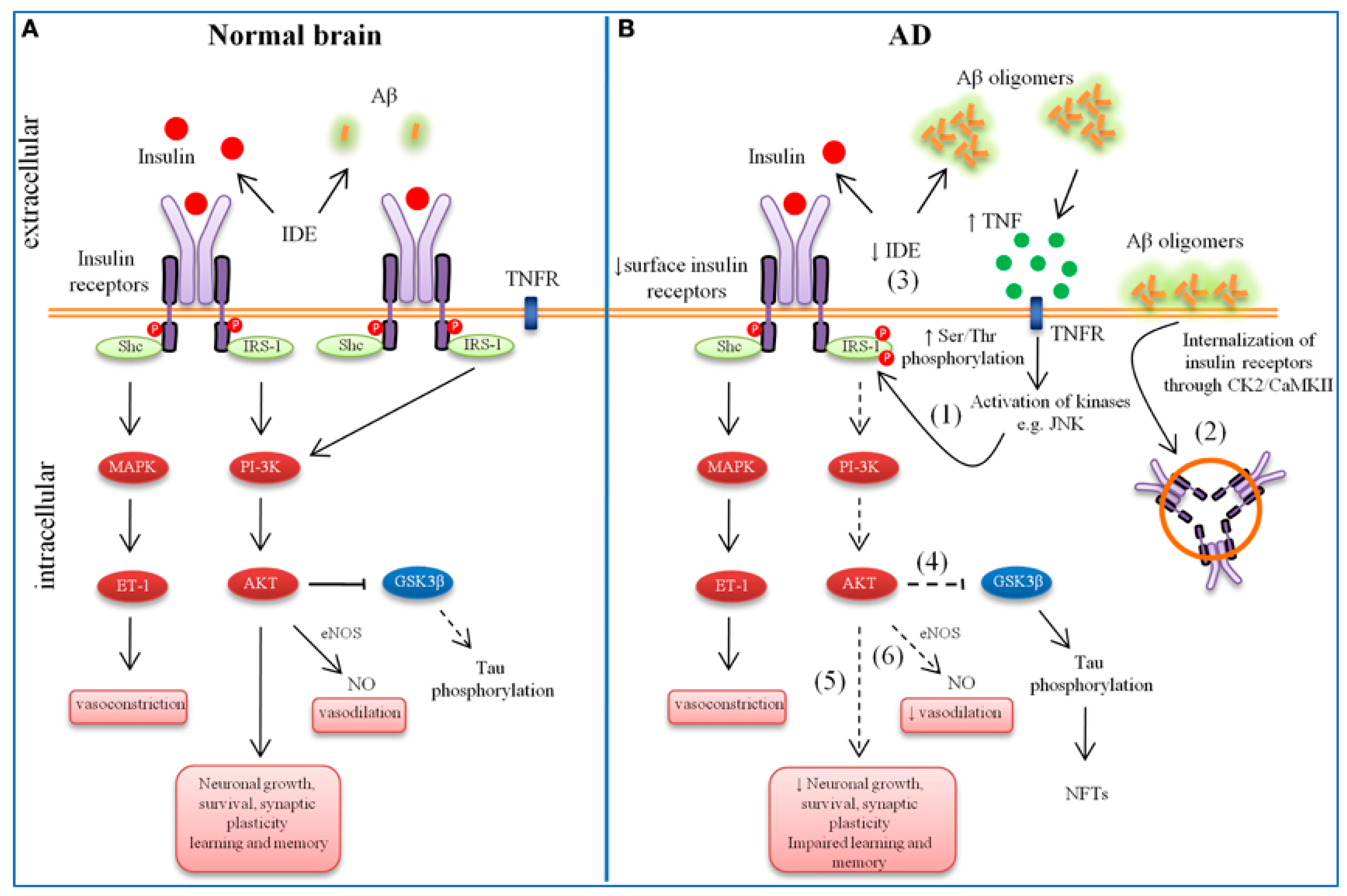
2. Role of Aβ in AD
3. Role of Tau and Dysregulated Phosphorylation in AD
4. Synaptic Loss in AD
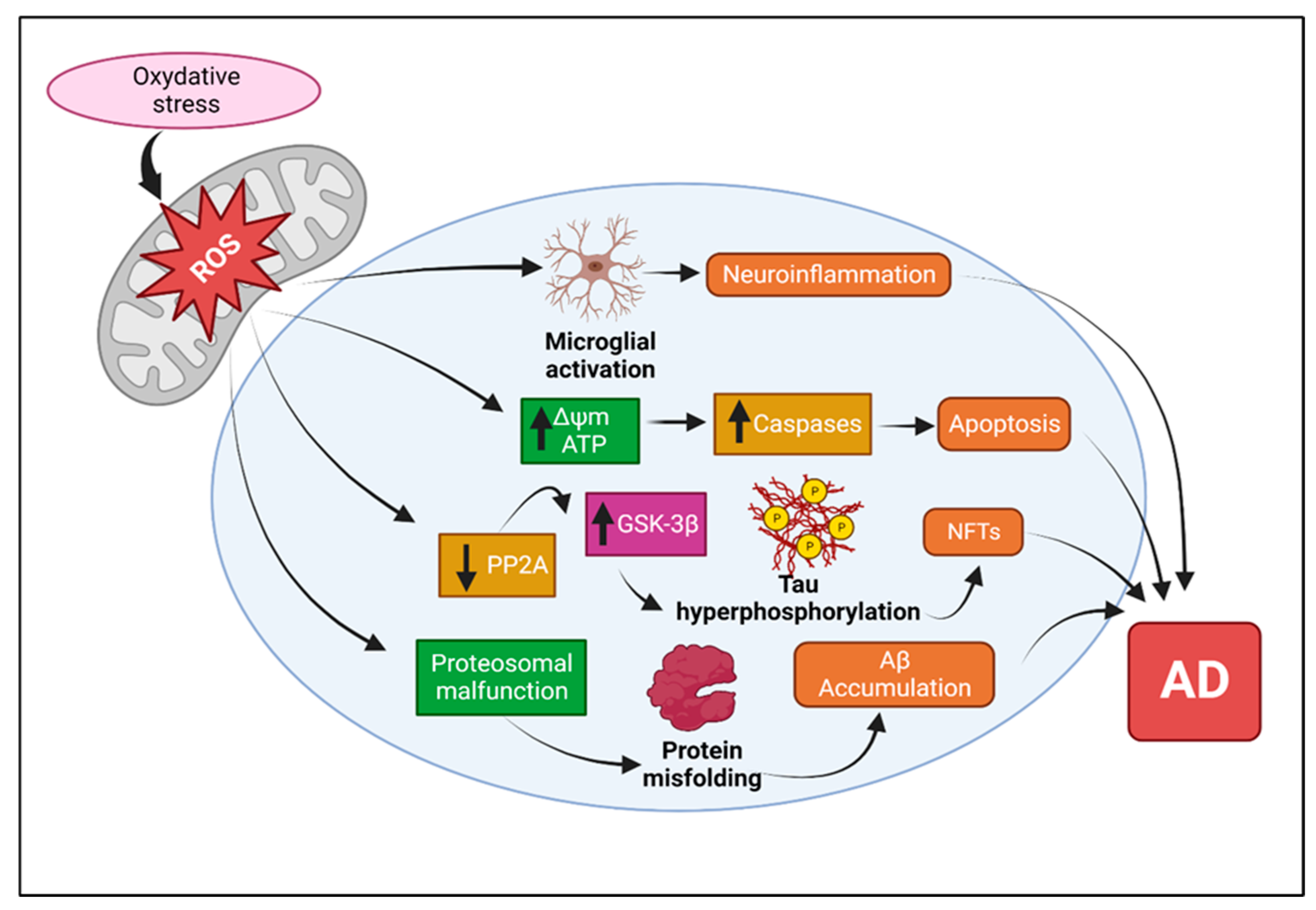
4.1. Mitochondrial Dysfunction and Defects in AD
4.1.1. Role in the Loss of Neuronal Plasticity and Synaptic Plasticity
4.1.2. Mitochondrial Dynamics in Axonal Transport
4.1.3. Mitochondrial Biogenesis
4.1.4. Mitochondrial Functions
4.1.5. Effects of Aβ and Tau on Mitochondrial Functions
4.2. Endoplasmic Reticulum Stress
5. Hypometabolism of Glucose in AD
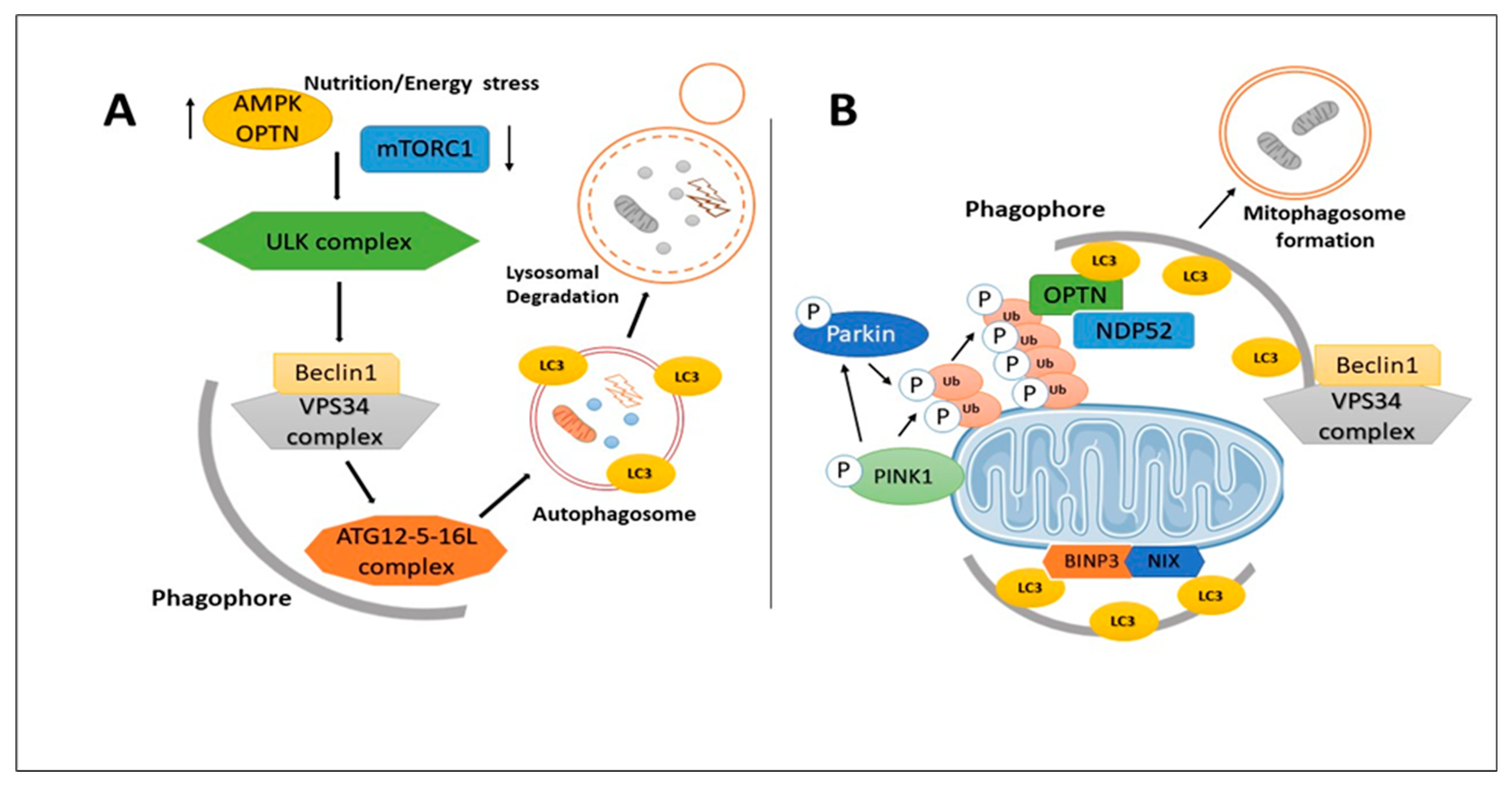
6. Mitophagy and Autophagy Dysregulation in AD
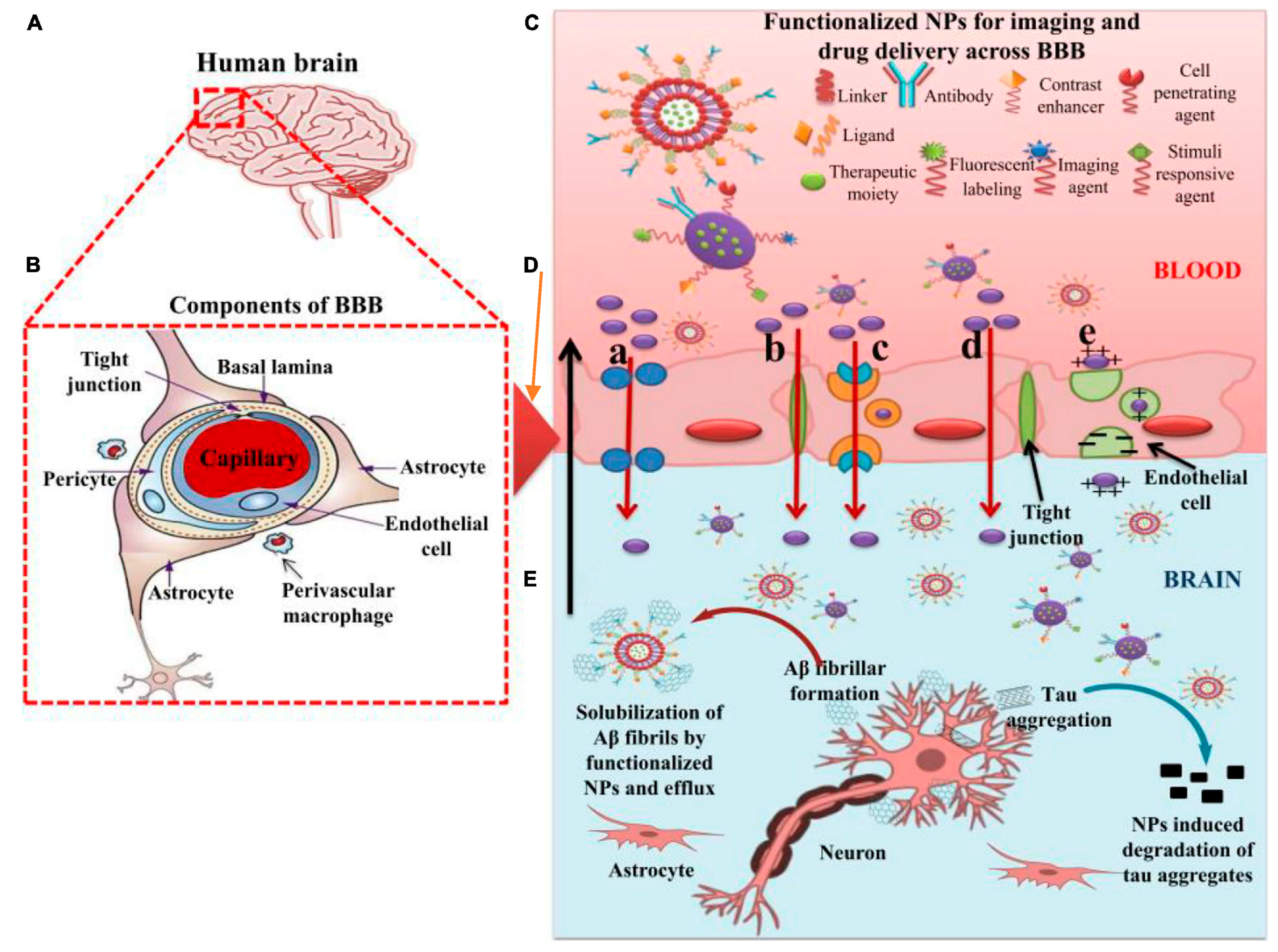
7. Nano-Based Theranostics in AD
8. Nanotechnological Diagnostic Tools
8.1. In Vivo Diagnosis
8.1.1. Nanoparticles in AD Diagnosis Using MRI
8.1.2. Optical Imaging
8.2. In Vitro Diagnosis
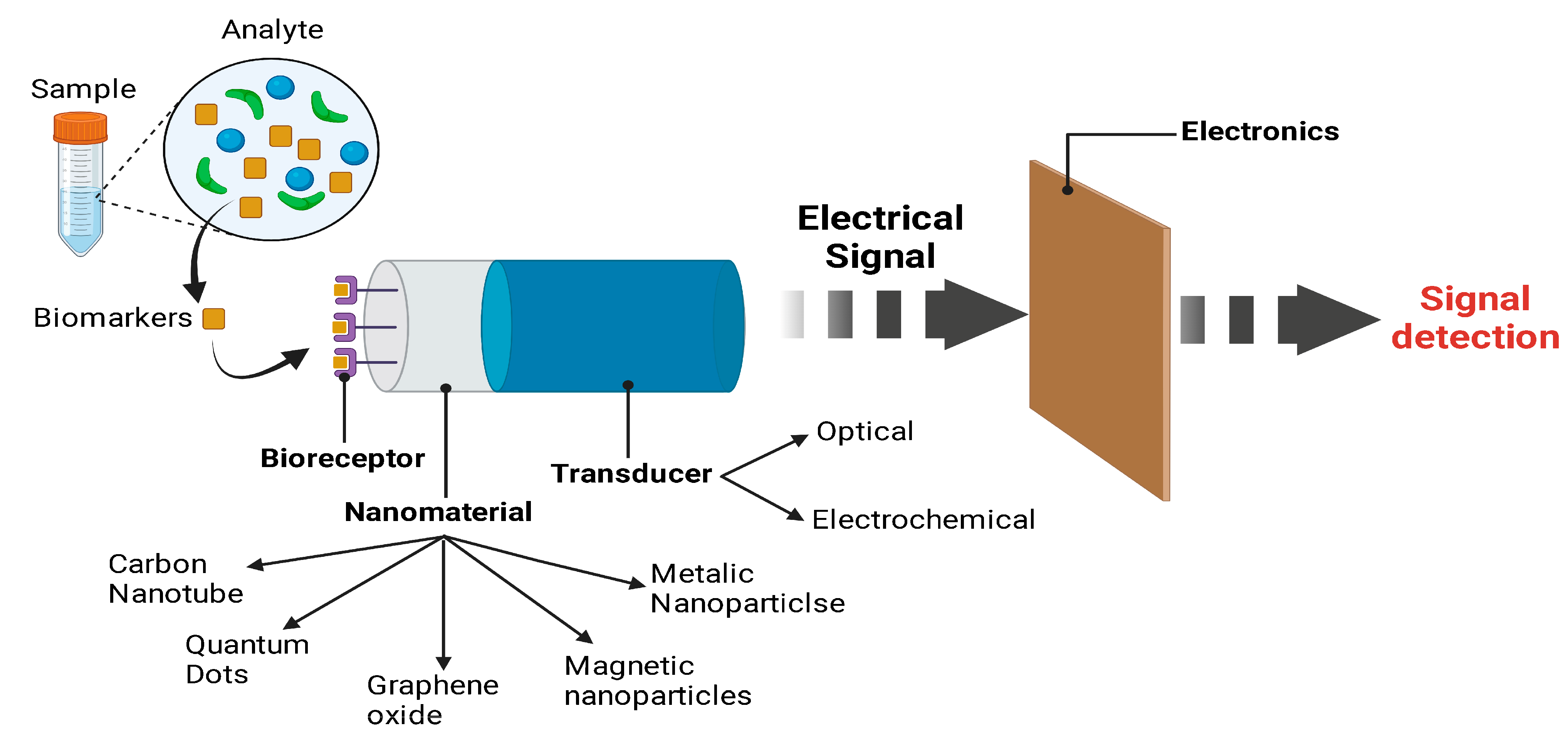
8.2.1. Biosensor or ELISA-Based Detections
8.2.2. Electrochemical Biosensors
8.2.3. Optical Biosensors
9. Therapeutic Interventions for AD
- In Situ PEG Coating
- “Don’t-Eat-Me” Signal Peptides
- Enhanced Brain Delivery
9.1. Biogenic Nanotherapeutics
9.1.1. Exosomes
9.1.2. Liposomes and Lipid Nanoparticles
9.1.3. Biopolymers and Nanoformulations
9.1.4. Phytocompound-Conjugated Systems
9.2. Metallic and Inorganic Nanosystems
9.2.1. Carbon Nanotubes (CNTs)
9.2.2. Dendrimers
9.2.3. Quantum Dots (QDs)
9.2.4. Metallic Nanoparticles and LSPR-Based NPs
10. Clinical Trials
11. Challenges Associated with Nanotheranostic Approaches
11.1. Pre-Targeting with Biotinylated PECAM-1 Antibody
11.2. Specific Binding with Avidin-Functionalized Nanoparticles
12. Future Scopes and Prospects
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bagga, S.; Kumar, M. Current Status of Alzheimer’s Disease and Pathological Mechanisms Investigating the Therapeutic Molecular Targets. Curr. Mol. Med. 2022, 23, 492–508. [Google Scholar] [CrossRef]
- Jeyaraman, M.; Rajendran, R.L.; Muthu, S.; Jeyaraman, N.; Sharma, S.; Jha, S.K.; Muthukanagaraj, P.; Hong, C.M.; da Fonseca, L.F.; Lana, J.F.S.D.; et al. An update on stem cell and stem cell-derived extracellular vesicle-based therapy in the management of Alzheimer’s disease. Heliyon 2023, 9, e17808. [Google Scholar] [CrossRef] [PubMed]
- Onkar, A.; Khan, F.; Goenka, A.; Rajendran, R.L.; Dmello, C.; Hong, C.M.; Mubin, N.; Gangadaran, P.; Ahn, B.C. Smart Nanoscale Extracellular Vesicles in the Brain: Unveiling their Biology, Diagnostic Potential, and Therapeutic Applications. ACS Appl. Mater. Interfaces 2024, 16, 6709–6742. [Google Scholar] [CrossRef]
- Petralla, S.; Colavitta, M.F.; Barrantes, F.J. Therapeutic Strategies Aimed at Improving Neuroplasticity in Alzheimer Disease. Pharmaceutics 2023, 15, 2052. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Luo, H.; Ruan, Y.; Mai, Y.; Liu, C.; Chen, J.; Yang, S.; Xuan, A.; Liu, J. Identification of candidate genes associated with clinical onset of Alzheimer’s disease. Front. Neurosci. 2022, 16, 1060111. [Google Scholar] [CrossRef]
- Mohd Murshid, N.; Aminullah Lubis, F.; Makpol, S. Epigenetic Changes and Its Intervention in Age-Related Neurodegenerative Diseases. Cell Mol. Neurobiol. 2022, 42, 577–595. [Google Scholar] [CrossRef]
- Ozben, T.; Ozben, S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin. Biochem. 2019, 72, 87–89. [Google Scholar] [CrossRef]
- Som Chaudhury, S.; Nandi, M.; Kumar, K.; Ruidas, B.; Sur, T.K.; Prasad, P.; Chakrabarti, S.; De, P.; Sil, J.; Das Mukhopadhyay, C.; et al. Rodent Model Preclinical Assessment of PEGylated Block Copolymer Targeting Cognition and Oxidative Stress Insults of Alzheimer’s Disease. Mol. Neurobiol. 2023, 60, 2036–2050. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Bai, F. The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front. Neurosci. 2018, 12, 320523. [Google Scholar] [CrossRef]
- Barbu, E.; Molnàr, É.; Tsibouklis, J.; Górecki, D.C. The potential for nanoparticle-based drug delivery to the brain: Overcoming the blood–brain barrier. Expert Opin. Drug Deliv. 2009, 6, 553–565. [Google Scholar] [CrossRef]
- Karthivashan, G.; Ganesan, P.; Park, S.Y.; Kim, J.S.; Choi, D.K. Therapeutic strategies and nano-drug delivery applications in management of ageing Alzheimer’s disease. Drug Deliv. 2018, 25, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Harilal, S.; Jose, J.; Parambi, D.G.T.; Kumar, R.; Mathew, G.E.; Uddin, M.S.; Kim, H.; Mathew, B. Advancements in nanotherapeutics for Alzheimer’s disease: Current perspectives. J. Pharm. Pharmacol. 2019, 71, 1370–1383. [Google Scholar] [CrossRef]
- Liu, R.; Luo, C.; Pang, Z.; Zhang, J.; Ruan, S.; Wu, M.; Wang, L.; Sun, T.; Li, N.; Han, L.; et al. Advances of nanoparticles as drug delivery systems for disease diagnosis and treatment. Chin. Chem. Lett. 2023, 34, 107518. [Google Scholar] [CrossRef]
- Chandrakala, V.; Aruna, V.; Angajala, G. Review on metal nanoparticles as nanocarriers: Current challenges and perspectives in drug delivery systems. Emergent Mater. 2022, 5, 1593–1615. [Google Scholar] [CrossRef] [PubMed]
- Binda, A.; Murano, C.; Rivolta, I. Innovative therapies and nanomedicine applications for the treatment of alzheimer’s disease: A. state-of-the-art (2017–2020). Int. J. Nanomed. 2020, 15, 6113–6135. [Google Scholar] [CrossRef]
- Nazıroğlu, M.; Muhamad, S.; Pecze, L. Nanoparticles as potential clinical therapeutic agents in Alzheimer’s disease: Focus on selenium nanoparticles. Expert Rev. Clin. Pharmacol. 2017, 10, 773–782. [Google Scholar] [CrossRef]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 2015, 9, 140046. [Google Scholar] [CrossRef]
- Stark, W.J.; Stoessel, P.R.; Wohlleben, W.; Hafner, A. Industrial applications of nanoparticles. Chem. Soc. Rev. 2015, 44, 5793–5805. [Google Scholar] [CrossRef] [PubMed]
- Oberdörster, G.; Sharp, Z.; Atudorei, V.; Elder, A.; Gelein, R.; Kreyling, W.; Cox, C. Translocation of Inhaled Ultrafine Particles to the Brain. Inhal. Toxicol. 2008, 16, 437–445. [Google Scholar] [CrossRef]
- Zolnik, B.S.; González-Fernández, Á.; Sadrieh, N.; Dobrovolskaia, M.A. Minireview: Nanoparticles and the Immune System. Endocrinology 2010, 151, 458–465. [Google Scholar] [CrossRef]
- Martín-Rapun, R.; De Matteis, L.; Ambrosone, A.; Garcia-Embid, S.; Gutierrez, L.; M de la Fuente, J. Targeted Nanoparticles for the Treatment of Alzheimer’s Disease. Curr. Pharm. Des. 2017, 23, 1927–1952. [Google Scholar] [CrossRef]
- Kononenko, V.; Narat, M.; Drobne, D. Nanoparticle interaction with the immune system. Arch. Ind. Hyg. Toxicol. 2015, 66, 97–108. [Google Scholar] [CrossRef]
- Amor, S.; Peferoen, L.A.N.; Vogel, D.Y.S.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hardie, J.; Zhang, X.; Rotello, V.M. Effects of engineered nanoparticles on the innate immune system. Semin. Immunol. 2017, 34, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xie, W.; Zhu, X.; Xu, M.; Liu, J. Sulfur Nanoparticles with Novel Morphologies Coupled with Brain-Targeting Peptides RVG as a New Type of Inhibitor against Metal-Induced Aβ Aggregation. ACS Chem. Neurosci. 2018, 9, 749–761. [Google Scholar] [CrossRef]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef]
- Sengul, A.B.; Asmatulu, E. Toxicity of metal and metal oxide nanoparticles: A review. Environ. Chem. Lett. 2020, 18, 1659–1683. [Google Scholar] [CrossRef]
- Annangi, B.; Bach, J.; Vales, G.; Rubio, L.; Marcos, R.; Hernández, A. Long-term exposures to low doses of cobalt nanoparticles induce cell transformation enhanced by oxidative damage. Nanotoxicology 2015, 9, 138–147. [Google Scholar] [CrossRef]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-amyloid Peptides and Amyloid Plaques in Alzheimer’s Disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M.A. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ikonomovic, M.D.; Counts, S.E.; Perez, S.E.; Malek-Ahmadi, M.; Scheff, S.W.; Ginsberg, S.D. Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behav. Brain Res. 2016, 311, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- O’Day, D.H. Calmodulin and Amyloid Beta as Coregulators of Critical Events during the Onset and Progression of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 1393. [Google Scholar] [CrossRef]
- Roda, A.; Serra-Mir, G.; Montoliu-Gaya, L.; Tiessler, L.; Villegas, S. Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1666. [Google Scholar] [CrossRef]
- Plotkin, S.S.; Cashman, N.R. Passive immunotherapies targeting Aβ and tau in Alzheimer’s disease. Neurobiol. Dis. 2020, 144, 105010. [Google Scholar] [CrossRef]
- Budvytyte, R.; Valincius, G. The interactions of amyloid β aggregates with phospholipid membranes and the implications for neurodegeneration. Biochem. Soc. Trans. 2023, 51, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Chévez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and Proteolytic Processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Honda, K.; Saito, Y.; Saito, H.; Toyoda, M.; Abe, R.; Saito, T.; Saido, T.C.; Michikawa, M.; Taru, H.; Sobu, Y.; et al. Accumulation of amyloid-β in the brain of mouse models of Alzheimer’s disease is modified by altered gene expression in the presence of human apoE isoforms during aging. Neurobiol. Aging 2023, 123, 63–74. [Google Scholar] [CrossRef]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, Å.K.; Blennow, K.; Hansson, O. Cerebrospinal Fluid Levels ofβ-Amyloid 1-2012, 42, but Not of Tau, Are Fully Changed Already 5 to 10 Years Before the Onset of Alzheimer Dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef]
- Ferreira, A.; Busciglio, J.; Cáceres, A. Microtubule formation and neurite growth in cerebellar macroneurons which develop in vitro: Evidence for the involvement of the microtubule-associated proteins, MAP-1a, HMW-MAP2 and Tau. Dev. Brain Res. 1989, 49, 215–228. [Google Scholar] [CrossRef]
- Takei, Y.; Teng, J.; Harada, A.; Hirokawa, N. Defects in Axonal Elongation and Neuronal Migration in Mice with Disrupted tau and map1b Genes. J. Cell Biol. 2000, 150, 989–1000. [Google Scholar] [CrossRef]
- Perea, J.R.; López, E.; Diéz-Ballesteros, J.C. Extracellular monomeric tau is internalized by astrocytes. Front. Neurosci. 2019, 13, 444353. [Google Scholar] [CrossRef] [PubMed]
- Boyko, S.; Surewicz, W.K. Tau liquid–liquid phase separation in neurodegenerative diseases. Trends Cell Biol. 2022, 32, 611–623. [Google Scholar] [CrossRef]
- Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. [Google Scholar] [CrossRef]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau with Cognition in Preclinical Alzheimer Disease: A. Longitudinal Study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C.; Wang, Y. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimer’s Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; De Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal Interaction of Oligomeric Amyloid-β with Phosphorylated Tau: Implications to Synaptic Dysfunction and Neuronal Damage. J. Alzheimer’s Dis. 2013, 36, 285–295. [Google Scholar] [CrossRef]
- Pascoal, T.A.; Mathotaarachchi, S.; Shin, M.; Benedet, A.L.; Mohades, S.; Wang, S.; Beaudry, T.; Kang, M.S.; Soucy, J.P.; Labbe, A.; et al. Synergistic interaction between amyloid and tau predicts the progression to dementia. Alzheimer’s Dement. 2017, 13, 644–653. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- DeKosky, S.T.; Scheff, S.W.; Styren, S.D. Structural Correlates of Cognition in Dementia: Quantification and Assessment of Synapse Change. Neurodegeneration 1996, 5, 417–421. [Google Scholar] [CrossRef]
- Almeida, C.G.; Tampellini, D.; Takahashi, R.H.; Greengard, P.; Lin, M.T.; Snyder, E.M.; Gouras, G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005, 20, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Amyloid-β-Derived Diffusible Ligands Cause Impaired Axonal Transport of Mitochondria in Neurons. Neurodegener. Dis. 2010, 7, 56–59. [Google Scholar] [CrossRef]
- Rossi, M.J.; Pekkurnaz, G. Powerhouse of the mind: Mitochondrial plasticity at the synapse. Curr. Opin. Neurobiol. 2019, 57, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Mesulam, M.M. Neuroplasticity Failure in Alzheimer’s Disease: Bridging the Gap between Plaques and Tangles. Neuron 1999, 24, 521–529. [Google Scholar] [CrossRef]
- Kamenetz, F.; Tomita, T.; Hsieh, H. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Scheff, S.W.; Scott, S.A.; DeKosky, S.T. Quantitation of synaptic density in the septal nuclei of young and aged Fischer 344 rats. Neurobiol. Aging 1991, 12, 3–12. [Google Scholar] [CrossRef]
- Gylys, K.H.; Fein, J.A.; Yang, F.; Wiley, D.J.; Miller, C.A.; Cole, G.M. Synaptic Changes in Alzheimer’s Disease: Increased Amyloid-β and Gliosis in Surviving Terminals Is Accompanied by Decreased PSD-95 Fluorescence. Am. J. Pathol. 2004, 165, 1809–1817. [Google Scholar] [CrossRef]
- Tampellini, D.; Gouras, G.K. Synapses, synaptic activity and intraneuronal Aβ in Alzheimer’s disease. Front. Aging Neurosci. 2010, 2, 1330. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 507–513. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Calkins, M.J.; Manczak, M.; Mao, P. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 2011, 20, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Lewis, T.L.; Hirabayashi, Y. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [PubMed]
- Bray, N. Many makes of mitochondria. Nat. Rev. Neurosci. 2019, 20, 645. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Drerup, C.M. Axonal Transport and Mitochondrial Function in Neurons. Front. Cell Neurosci. 2019, 13, 475553. [Google Scholar] [CrossRef]
- Son, G.; Han, J. Roles of mitochondria in neuronal development. BMB Rep. 2018, 51, 549. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332. [Google Scholar] [CrossRef]
- Bi, R.; Zhang, W.; Zhang, D.F. Genetic association of the cytochrome c oxidase-related genes with Alzheimer’s disease in Han Chinese. Neuropsychopharmacology 2018, 43, 2264–2276. [Google Scholar] [CrossRef]
- Quntanilla, R.A.; Tapia-Monsalves, C. The Role of Mitochondrial Impairment in Alzheimer’s Disease Neurodegeneration: The Tau Connection. Curr. Neuropharmacol. 2020, 18, 1076–1091. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Ishii, A.; Kamano, N.; Watamura, N.; Saito, T.; Ohshima, T.; Yokosuka, M.; Saido, T.C. Endoplasmic reticulum stress responses in mouse models of Alzheimer’s disease: Overexpression paradigm versus knockin paradigm. J. Biol. Chem. 2018, 293, 3118–3125. [Google Scholar] [CrossRef]
- Peng, Y.; Gao, P.; Shi, L. Central and peripheral metabolic defects contribute to the pathogenesis of Alzheimer’s disease: Targeting mitochondria for diagnosis and prevention. Antioxid. Redox Signal 2020, 32, 1188–1236. [Google Scholar] [CrossRef]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and its involvement in Alzheimer’s disease: A review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef]
- Malkov, A.; Popova, I.; Ivanov, A.; Jang, S.S.; Yoon, S.Y.; Osypov, A.; Huang, Y.; Zilberter, Y.; Zilberter, M. Aβ initiates brain hypometabolism, network dysfunction and behavioral abnormalities via NOX2-induced oxidative stress in mice. Commun. Biol. 2021, 4, 1054. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Kim, S.; Nam, Y. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850. [Google Scholar] [CrossRef]
- Kalra, R.S.; Kandimalla, R.; Binukumar, K.B. Editorial: Apoptosis, autophagy, and mitophagy dysfunction in Alzheimer’s disease: Evolving emergence and mechanisms. Front. Mol. Neurosci. 2022, 15, 1049914. [Google Scholar] [CrossRef]
- Zhao, N.; Zhang, X.; Li, B.; Wang, J.; Zhang, C.; Xu, B. Treadmill Exercise Improves PINK1/Parkin-Mediated Mitophagy Activity Against Alzheimer’s Disease Pathologies by Upregulated SIRT1-FOXO1/3 Axis in APP/PS1 Mice. Mol. Neurobiol. 2023, 60, 277–291. [Google Scholar] [CrossRef]
- Travaglio, M.; Michopoulos, F.; Yu, Y. Increased cysteine metabolism in PINK1 models of Parkinson’s disease. DMM Dis. Models Mech. 2023, 16, dmm049727. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Bagchi, A. Study of the Effects of Nicotine and Caffeine for the Treatment of Parkinson’s Disease. Appl. Biochem. Biotechnol. 2023, 195, 639–654. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Sterky, F.H.; Lee, S.; Wibom, R. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12937–12942. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Hemachandra Reddy, P. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-López, E.; Schuhmacher, A.J. Transportation of Single-Domain Antibodies through the Blood–Brain Barrier. Biomolecules 2021, 11, 1131. [Google Scholar] [CrossRef]
- Chopra, H.; Bibi, S.; Singh, I.; Kamal, M.A.; Islam, F.; Alhumaydhi, F.A.; Emran, T.B.; Cavalu, S. Nanomedicines in the Management of Alzheimer’s Disease: Current View and Future Prospects. Front. Aging Neurosci. 2022, 14, 879114. [Google Scholar] [CrossRef]
- Cavalu, S.; Fritea, L.; Brocks, M. Novel Hybrid Composites Based on PVA/SeTiO2 Nanoparticles and Natural Hydroxyapatite for Orthopedic Applications: Correlations between Structural, Morphological and Biocompatibility Properties. Materials 2020, 13, 2077. [Google Scholar] [CrossRef]
- Wang, L.; Bharti; Kumar, R.; Pavlov, P.F.; Winblad, B. Small molecule therapeutics for tauopathy in Alzheimer’s disease: Walking on the path of most resistance. Eur. J. Med. Chem. 2021, 209, 112915. [Google Scholar] [CrossRef]
- Harini, K.; Koyeli, G.; Vijaya, A.A. Nano-mediated Strategies for Metal Ion–Induced Neurodegenerative Disorders: Focus on Alzheimer’s and Parkinson’s Diseases. Curr. Pharmacol. Rep. 2022, 8, 450–463. [Google Scholar] [CrossRef]
- Dan, S.; Sharma, D.; Rastogi, K. Therapeutic and Diagnostic Applications of Nanocomposites in the Treatment Alzheimer’s Disease Studies. Review 2022, 12, 940–960. [Google Scholar] [CrossRef]
- Song, Y.; Zhu, X.Y.; Zhang, X.M.; Xiong, H. Targeted Mitochondrial Epigenetics: A New Direction in Alzheimer’s Disease Treatment. Int. J. Mol. Sci. 2022, 23, 9703. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.U.; Kim, S.; Sim, S.J. SERS-based Nanoplasmonic Exosome Analysis: Enabling Liquid Biopsy for Cancer Diagnosis and Monitoring Progression. Biochip J. 2020, 14, 231–241. [Google Scholar] [CrossRef]
- Iranifar, E.; Seresht, B.M.; Momeni, F. Exosomes and microRNAs: New potential therapeutic candidates in Alzheimer disease therapy. J. Cell Physiol. 2019, 234, 2296–2305. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Ji, X.; Lv, R. Targetting exosomes as a new biomarker and therapeutic approach for Alzheimer’s disease. Clin. Interv. Aging 2020, 15, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Lee, J.U.; Jeon, M.J.; Kim, S.; Sim, S.J. Detection of multiplex exosomal miRNAs for clinically accurate diagnosis of Alzheimer’s disease using label-free plasmonic biosensor based on DNA-Assembled advanced plasmonic architecture. Biosens. Bioelectron. 2022, 199, 113864. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Sajadimajd, S.; Jafari, S.; Izadi, Z.; Sarvari, S.; Sharifi, M.; Falahati, M.; Moakedi, F.; Muganda, W.C.A.; Müller, M.; et al. Novel therapeutic strategies for Alzheimer’s disease: Implications from cell-based therapy and nanotherapy. Nanomedicine 2020, 24, 102149. [Google Scholar] [CrossRef]
- Boyuklieva, R.; Pilicheva, B. Micro- and Nanosized Carriers for Nose-to-Brain Drug Delivery in Neurodegenerative Disorders. Biomedicines 2022, 10, 1706. [Google Scholar] [CrossRef]
- Neganova, M.E.; Aleksandrova, Y.R.; Sukocheva, O.A.; Klochkov, S.G. Benefits and limitations of nanomedicine treatment of brain cancers and age-dependent neurodegenerative disorders. Semin. Cancer Biol. 2022, 86, 805–833. [Google Scholar] [CrossRef]
- Cui, N.; Lu, H.; Li, M. Magnetic nanoparticles associated PEG/PLGA block copolymer targeted with anti-transferrin receptor antibodies for Alzheimer’s disease. J. Biomed. Nanotechnol. 2018, 14, 1017–1024. [Google Scholar] [CrossRef]
- Singh, S.; Raina, D.; Rishipathak, D.; Babu, K.R.; Khurana, R.; Gupta, Y.; Garg, K.; Rehan, F.; Gupta, S.M. Quantum dots in the biomedical world: A smart advanced nanocarrier for multiple venues application. Arch. Pharm. 2022, 355, 2200299. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Nguyen, T.D.; Nguyen, T.K.O. Advances in developing therapeutic strategies for Alzheimer’s disease. Biomed. Pharmacother. 2021, 139, 111623. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sachdeva, B.; Sachdeva, P.; Chaudhary, V.; Rani, G.M.; Sinha, J.K. Graphene quantum dots as a potential diagnostic and therapeutic tool for the management of Alzheimer’s disease. Carbon Lett. 2022, 32, 1381–1394. [Google Scholar] [CrossRef]
- Ribeiro, T.d.C.; Sábio, R.M.; Luiz, M.T. Curcumin-Loaded Mesoporous Silica Nanoparticles Dispersed in Thermo-Responsive Hydrogel as Potential Alzheimer Disease Therapy. Pharmaceutics 2022, 14, 1976. [Google Scholar] [CrossRef]
- Sohail, I.; Bhatti, I.A.; Ashar, A.; Sarim, F.M.; Mohsin, M.; Naveed, R.; Yasir, M.; Iqbal, M.; Nazir, A. Polyamidoamine (PAMAM) dendrimers synthesis, characterization and adsorptive removal of nickel ions from aqueous solution. J. Mater. Res. Technol. 2020, 9, 498–506. [Google Scholar] [CrossRef]
- Seo, M.W.; Park, T.E. Recent advances with liposomes as drug carriers for treatment of neurodegenerative diseases. Biomed. Eng. Lett. 2021, 11, 211–216. [Google Scholar] [CrossRef]
- Rabiee, N.; Ahmadi, S.; Afshari, R. Polymeric Nanoparticles for Nasal Drug Delivery to the Brain: Relevance to Alzheimer’s Disease. Adv. Ther. 2021, 4, 2000076. [Google Scholar] [CrossRef]
- Poudel, P.; Park, S. Recent Advances in the Treatment of Alzheimer’s Disease Using Nanoparticle-Based Drug Delivery Systems. Pharmaceutics 2022, 14, 835. [Google Scholar] [CrossRef]
- Agraharam, G.; Saravanan, N.; Girigoswami, A.; Girigoswami, K. Future of Alzheimer’s Disease: Nanotechnology-Based Diagnostics and Therapeutic Approach. BioNanoScience 2022, 12, 1002–1017. [Google Scholar] [CrossRef]
- Brazaca, L.C.; Sampaio, I.; Zucolotto, V.; Janegitz, B.C. Applications of biosensors in Alzheimer’s disease diagnosis. Talanta 2020, 210, 120644. [Google Scholar] [CrossRef]
- Carneiro, P.; Morais, S.; Pereira, M.C. Nanomaterials towards Biosensing of Alzheimer’s Disease Biomarkers. Nanomaterials 2019, 9, 1663. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Jo, D.G.; Cho, M.; Lee, Y. Monitoring of early diagnosis of Alzheimer’s disease using the cellular prion protein and poly(pyrrole-2-carboxylic acid) modified electrode. Biosens. Bioelectron. 2018, 113, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Xu, N.; Yang, X.; Ling, G.; Zhang, P. The roles of gold nanoparticles in the detection of amyloid-β peptide for Alzheimer’s disease. Colloid Interface Sci. Commun. 2022, 46, 100579. [Google Scholar] [CrossRef]
- Bilal, M.; Barani, M.; Sabir, F. Nanomaterials for the treatment and diagnosis of Alzheimer’s disease: An overview. NanoImpact 2020, 20, 100251. [Google Scholar] [CrossRef]
- Trino, L.D.; Albano, L.G.S.; Granato, D.C. ZIF-8 Metal-Organic Framework Electrochemical Biosensor for the Detection of Protein-Protein Interaction. Chem. Mater. 2021, 33, 1293–1306. [Google Scholar] [CrossRef]
- Shui, B.; Tao, D.; Cheng, J. A novel electrochemical aptamer–antibody sandwich assay for the detection of tau-381 in human serum. Analyst 2018, 143, 3549–3554. [Google Scholar] [CrossRef]
- Špringer, T.; Hemmerová, E.; Finocchiaro, G.; Krištofiková, Z.; Vyhnálek, M.; Homola, J. Surface plasmon resonance biosensor for the detection of tau-amyloid β complex. Sens. Actuator B Chem. 2020, 316, 128146. [Google Scholar] [CrossRef]
- Khan, Z.A.; Park, S. AuNPs- Aβ-Ni-HRP sandwich assay: A new sensitive colorimetric method for the detection of Aβ 1-40. Talanta 2022, 237, 122946. [Google Scholar] [CrossRef]
- Li, L.; He, R.; Yan, H. Nanotechnology for the diagnosis and treatment of Alzheimer’s disease: A bibliometric analysis. Nano Today 2022, 47, 101654. [Google Scholar] [CrossRef]
- Kim, K.; Lee, C.H.; Park, C.B. Chemical sensing platforms for detecting trace-level Alzheimer’s core biomarkers. Chem. Soc. Rev. 2020, 49, 5446–5472. [Google Scholar] [CrossRef] [PubMed]
- Chitvoranund, N.; Jiemsirilers, S.; Kashima, D.P. Effects of Surface Treatment on Adhesion of Silver Film on Glass Substrate Fabricated by Electroless Plating. Adv. Mat. Res. 2013, 664, 566–573. [Google Scholar] [CrossRef]
- Khun, K.; Ibupoto, Z.H.; AlSalhi, M.S. Fabrication of Well-Aligned ZnO Nanorods Using a Composite Seed Layer of ZnO Nanoparticles and Chitosan Polymer. Materials 2013, 6, 4361–4374. [Google Scholar] [CrossRef]
- Akhtar, N.; Metkar, S.K.; Girigoswami, A.; Girigoswami, K. ZnO nanoflower based sensitive nano-biosensor for amyloid detection. Mater. Sci. Eng. C 2017, 78, 960–968. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular Mechanism of Thioflavin-T Binding to Amyloid Fibrils. Biochim. Biophys. Acta 2010, 1804, 1405. [Google Scholar] [CrossRef] [PubMed]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. J. Prev. Alzheimer’s Dis. 2021, 8, 371–386. [Google Scholar] [CrossRef]
- Dirisala, A.; Uchida, S.; Toh, K.; Li, J.; Osawa, S.; Tockary, T.A.; Liu, X.; Abbasi, S.; Hayashi, K.; Mochida, Y.; et al. Transient stealth coating of liver sinusoidal wall by anchoring two-armed PEG for retargeting nanomedicines. Sci. Adv. 2020, 6, eabb8133. [Google Scholar] [CrossRef]
- Liu, H.; Lv, H.; Duan, X.; Du, Y.; Tang, Y.; Xu, W. Advancements in Macrophage-Targeted Drug Delivery for Effective Disease Management. Int. J. Nanomed. 2023, 18, 6915–6940. [Google Scholar] [CrossRef]
- Khan, N.H.; Mir, M.; Ngowi, E.E.; Zafar, U.; Khakwani, M.M.A.K.; Khattak, S.; Zhai, Y.-K.; Jiang, E.-S.; Zheng, M.; Duan, S.-F.; et al. Nanomedicine: A Promising Way to Manage Alzheimer’s Disease. Front. Bioeng. Biotechnol. 2021, 9, 630055. [Google Scholar] [CrossRef]
- Patil, S.; Chandrasekaran, R. Biogenic nanoparticles: A comprehensive perspective in synthesis, characterization, application and its challenges. J. Genet. Eng. Biotechnol. 2020, 18, 67. [Google Scholar] [CrossRef]
- Ke, W.; Afonin, K.A. Exosomes as natural delivery carriers for programmable therapeutic nucleic acid nanoparticles (NANPs). Adv. Drug. Deliv. Rev. 2021, 176, 113835. [Google Scholar] [CrossRef]
- Yang, Z.Z.; Zhang, Y.Q.; Wang, Z.Z. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int. J. Pharm. 2013, 452, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Dong, H.; Cao, H.; Ji, X.; Luan, S.; Liu, J. Exosomes in Pathogenesis, Diagnosis, and Treatment of Alzheimer’s Disease. Med. Sci. Monit. 2019, 25, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Wu, P.; Zhou, X. Extracellular Vesicles: Novel Roles in Neurological Disorders. Stem Cells Int. 2021, 2021, 6640836. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Guo, L.; Jiang, Y. Brain delivery of quercetin-loaded exosomes improved cognitive function in AD mice by inhibiting phosphorylated tau-mediated neurofibrillary tangles. Drug Deliv. 2020, 27, 745–755. [Google Scholar] [CrossRef]
- Beltrán-Gracia, E.; López-Camacho, A.; Higuera-Ciapara, I. Nanomedicine review: Clinical developments in liposomal applications. Cancer Nanotechnol. 2019, 10, 11. [Google Scholar] [CrossRef]
- Agrawal, M.; Ajazuddin; Tripathi, D.K. Recent advancements in liposomes targeting strategies to cross blood-brain barrier (BBB) for the treatment of Alzheimer’s disease. J. Control. Release 2017, 260, 61–77. [Google Scholar] [CrossRef]
- Kong, L.; Li, X.T.; Ni, Y.N. Transferrin-modified osthole PEGylated liposomes travel the blood-brain barrier and mitigate alzheimer’s disease-related pathology in APP/PS-1 mice. Int. J. Nanomed. 2020, 15, 2841–2858. [Google Scholar] [CrossRef]
- Ji, B.; Wang, M.; Gao, D. Combining nanoscale magnetic nimodipine liposomes with magnetic resonance image for Parkinson’s disease targeting therapy. Nanomedicine 2017, 12, 237–253. [Google Scholar] [CrossRef]
- Papadia, K.; Markoutsa, E.; Mourtas, S. Multifunctional LUV liposomes decorated for BBB and amyloid targeting. A. In vitro proof-of-concept. Eur. J. Pharm. Sci. 2017, 101, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, B.Q.; Li, Y.H.; Jiang, Q.Q.; Cong, W.H.; Chen, K.J.; Wen, X.M.; Wu, Z.Z. Lactoferrin modification of berberine nanoliposomes enhances the neuroprotective effects in a mouse model of Alzheimer’s disease. Neural Regen. Res. 2023, 18, 226. [Google Scholar] [CrossRef]
- Yadav, H.K.S.; Almokdad, A.A.; Shaluf, S.I.M.; Debe, M.S. Polymer-Based Nanomaterials for Drug-Delivery Carriers. Nanocarriers Drug Deliv. 2018, 531–556. [Google Scholar] [CrossRef]
- Montegiove, N.; Calzoni, E.; Emiliani, C.; Cesaretti, A. Biopolymer Nanoparticles for Nose-to-Brain Drug Delivery: A New Promising Approach for the Treatment of Neurological Diseases. J. Funct. Biomater. 2022, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Mazahir, F.; Yadav, A.K. Recent Trends in Nano-Particulate Carriers for the Diagnosis and Treatment of Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2022, 22, 477–499. [Google Scholar] [CrossRef]
- Ailioaie, L.M.; Ailioaie, C.; Litscher, G. Photobiomodulation in Alzheimer’s Disease-A Complementary Method to State-of-the-Art Pharmaceutical Formulations and Nanomedicine? Pharmaceutics 2023, 15, 916. [Google Scholar] [CrossRef]
- Xie, J.; Gonzalez-Carter, D.; Tockary, T.A.; Nakamura, N.; Xue, Y.; Nakakido, M.; Akiba, H.; Dirisala, A.; Liu, X.; Toh, K.; et al. Dual-sensitive nanomicelles enhancing systemic delivery of therapeutically active antibodies specifically into the brain. ACS Nano 2020, 14, 6729–6742. [Google Scholar] [CrossRef]
- Abbasi, S.; Uchida, S.; Toh, K.; Tockary, T.A.; Dirisala, A.; Hayashi, K.; Fukushima, S.; Kataoka, K. Co-encapsulation of Cas9 mRNA and guide RNA in polyplex micelles enables genome editing in mouse brain. J. Control. Release 2021, 332, 260–268. [Google Scholar] [CrossRef]
- Keller, B.L.; Lohmann, C.A.; Kyeremateng, S.O.; Fricker, G. Synthesis and Characterization of Biodegradable Poly(butyl cyanoacrylate) for Drug Delivery Applications. Polymers 2022, 14, 998. [Google Scholar] [CrossRef]
- Mathew, A.; Fukuda, T.; Nagaoka, Y. Curcumin Loaded-PLGA Nanoparticles Conjugated with Tet-1 Peptide for Potential Use in Alzheimer’s Disease. PLoS ONE 2012, 7, e32616. [Google Scholar] [CrossRef]
- Bukhari, S.N.A.; Yogesh, R. An Overview of Tetramethylpyrazine (Ligustrazine) and its Derivatives as Potent Anti-Alzheimer’s Disease Agents. Lett. Drug. Des. Discov. 2022, 19, 565–578. [Google Scholar] [CrossRef]
- Hernandez, C.; Shukla, S. Liposome based drug delivery as a potential treatment option for Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1190. [Google Scholar] [CrossRef]
- Kanojia, N.; Thapa, K.; Kaur, G. Update on Therapeutic potential of emerging nanoformulations of phytocompounds in Alzheimer’s and Parkinson’s disease. J. Drug. Deliv. Sci. Technol. 2023, 79, 104074. [Google Scholar] [CrossRef]
- Fasae, K.D.; Abolaji, A.O.; Faloye, T.R. Metallobiology and therapeutic chelation of biometals (copper, zinc and iron) in Alzheimer’s disease: Limitations, and current and future perspectives. J. Trace Elem. Med. Biol. 2021, 67, 126779. [Google Scholar] [CrossRef]
- Mishra, J.; Kumar, B.; Pandey, M. Carbon Nano Tubes: Novel Drug Delivery System in Amelioration of Alzheimer’s Disease. Comb. Chem. High Throughput Screen 2020, 24, 1528–1543. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, Y.; Yang, Y. Pharmacological and toxicological target organelles and safe use of single-walled carbon nanotubes as drug carriers in treating Alzheimer disease. Nanomedicine 2010, 6, 427–441. [Google Scholar] [CrossRef]
- Lyu, Z.; Ding, L.; Huang, A.Y.T.; Kao, C.L.; Peng, L. Poly(amidoamine) dendrimers: Covalent and supramolecular synthesis. Mater. Today Chem. 2019, 13, 34–48. [Google Scholar] [CrossRef]
- Aurelia Chis, A.; Dobrea, C.; Morgovan, C.; Arseniu, A.M.; Rus, L.L.; Butuca, A.; Juncan, A.M.; Totan, M.; Vonica-Tincu, A.L.; Cormos, G.; et al. Applications and Limitations of Dendrimers in Biomedicine. Molecules 2020, 25, 3982. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, T.; Chen, Q. Biomimetic Dendrimer–Peptide Conjugates for Early Multi-Target Therapy of Alzheimer’s Disease by Inflammatory Microenvironment Modulation. Adv. Mater. 2021, 33, 2100746. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Pang, Z. Dendrimer-Based Drug Delivery Systems for Brain Targeting. Biomolecules 2019, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Arotiba, O.; Owino, J.; Songa, E.; Hendricks, N.; Waryo, T.; Jahed, N.; Baker, P.; Iwuoha, E. An Electrochemical DNA Biosensor Developed on a Nanocomposite Platform of Gold and Poly(propyleneimine) Dendrimer. Sensors 2008, 8, 6791–6809. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.A.; Henry, J.E.; Good, T.A. Attenuation of β-amyloid-induced toxicity by sialic-acid-conjugated dendrimers: Role of sialic acid attachment. Brain Res. 2007, 1161, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Palan, F.; Chatterjee, B. Dendrimers in the context of targeting central nervous system disorders. J. Drug. Deliv. Sci. Technol. 2022, 73, 103474. [Google Scholar] [CrossRef]
- Pezzini, I.; Mattoli, V.; Ciofani, G. Mitochondria and neurodegenerative diseases: The promising role of nanotechnology in targeted drug delivery. Expert Opin. Drug Deliv. 461 2016, 14, 513–523. [Google Scholar] [CrossRef]
- Ren, C.; Li, D.; Zhou, Q.; Hu, X. Mitochondria-targeted TPP-MoS2 with dual enzyme activity provides efficient neuroprotection through M1/M2 microglial polarization in an Alzheimer’s disease model. Biomaterials 2020, 232, 119752. [Google Scholar] [CrossRef]
- Feng, J.; Li, F.; Li, X. An amplification label of core–shell CdSe@CdS QD sensitized GO for a signal-on photoelectrochemical immunosensor for amyloid β-protein. J. Mater. Chem. B 2019, 7, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Villalva, M.D.; Agarwal, V.; Ulanova, M. Quantum dots as a theranostic approach in Alzheimer’s disease: A systematic review. Nanomedicine 2021, 16, 1595–1611. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, R.; Zhang, D. Neuroprotective effects of maize tetrapeptide-anchored gold nanoparticles in Alzheimer’s disease. Colloids Surf. B Biointerfaces 2021, 200, 111584. [Google Scholar] [CrossRef] [PubMed]
- Combes, A.; Camosseto, V.; N’Guessan, P. BAD-LAMP controls TLR9 trafficking and signalling in human plasmacytoid dendritic cells. Nat. Commun. 2017, 8, 913. [Google Scholar] [CrossRef]
- Guo, X.; Lie, Q.; Liu, Y. Multifunctional Selenium Quantum Dots for the Treatment of Alzheimer’s Disease by Reducing Aβ-Neurotoxicity and Oxidative Stress and Alleviate Neuroinflammation. ACS Appl. Mater. Interfaces 2021, 13, 30261–30273. [Google Scholar] [CrossRef]
- Birla, H.; Minocha, T.; Kumar, G. Role of Oxidative Stress and Metal Toxicity in the Progression of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 552. [Google Scholar] [CrossRef]
- Fakhri, S.; Abdian, S.; Zarneshan, S.N. Nanoparticles in Combating Neuronal Dysregulated Signaling Pathways: Recent Approaches to the Nanoformulations of Phytochemicals and Synthetic Drugs against Neurodegenerative Diseases. Int. J. Nanomed. 2022, 17, 299. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Yang, L.; Liu, Y. Sialic acid (SA)-modified selenium nanoparticles coated with a high blood-brain barrier permeability peptide-B6 peptide for potential use in Alzheimer’s disease. Acta Biomater. 2015, 25, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Suganthy, N.; Sri Ramkumar, V.; Pugazhendhi, A. Biogenic synthesis of gold nanoparticles from Terminalia arjuna bark extract: Assessment of safety aspects and neuroprotective potential via antioxidant, anticholinesterase, and antiamyloidogenic effects. Environ. Sci. Pollut. Res. Int. 2018, 25, 10418–10433. [Google Scholar] [CrossRef]
- Gupta, D.; Boora, A.; Thakur, A.; Gupta, T.K. Green and sustainable synthesis of nanomaterials: Recent advancements and limitations. Environ. Res. 2023, 231, 116316. [Google Scholar] [CrossRef]
- Ayaz, M.; Ovais, M.; Ahmad, I. Biosynthesized metal nanoparticles as potential Alzheimer’s disease therapeutics. Metal Nanoparticles Drug Deliv. Diagn. Appl. 2020, 31–42. [Google Scholar] [CrossRef]
- Zhu, X.; Vo, C.; Taylor, M.; Smith, B.R. Non-spherical micro- and nanoparticles in nanomedicine. Mater. Horiz. 2019, 6, 1094–1121. [Google Scholar] [CrossRef]
- Chen, P.; Wang, H.; He, M. Size-dependent cytotoxicity study of ZnO nanoparticles in HepG2 cells. Ecotoxicol. Environ. Saf. 2019, 171, 337–346. [Google Scholar] [CrossRef]
- Farah, F.H. Nanocarriers AS Delivery Systems FOR Therapeutics Agents. Int. J. Pharm. Sci. Res. 2019, 10, 3487. [Google Scholar] [CrossRef]
- Chaicherd, S.; Killingsworth, M.C.; Pissuwan, D. Toxicity of gold nanoparticles in a commercial dietary supplement drink on connective tissue fibroblast cells. SN Appl. Sci. 2019, 1, 336. [Google Scholar] [CrossRef]
- Carro, C.E.; Pilozzi, A.R.; Huang, X. Nanoneurotoxicity and Potential Nanotheranostics for Alzheimer’s Disease. EC Pharmacol. Toxicol. 2019, 7, 1. [Google Scholar]
- Shi, Y.; Pilozzi, A.R.; Huang, X. Exposure of CuO Nanoparticles Contributes to Cellular Apoptosis, Redox Stress, and Alzheimer’s Aβ Amyloidosis. Int. J. Environ. Res. Public Health 2020, 17, 1005. [Google Scholar] [CrossRef]
- Egbuna, C.; Parmar, V.K.; Jeevanandam, J.; Ezzat, S.M.; Patrick-Iwuanyanwu, K.C.; Adetunji, C.O.; Khan, J.; Onyeike, E.N.; Uche, C.Z.; Akram, M.; et al. Toxicity of Nanoparticles in Biomedical Application: Nanotoxicology. J. Toxicol. 2021, 2021, 9954443. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Liu, H.; Zhang, S.; Liu, X.; Hayat, T.; Alsaedi, A.; Wang, X. Mechanism of toxic effects of Nano-ZnO on cell cycle of zebrafish (Danio rerio). Chemosphere 2019, 229, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.W.; Cambre, M.; Lee, H.J. The Toxicity of Nanoparticles Depends on Multiple Molecular and Physicochemical Mechanisms. Int. J. Mol. Sci. 2017, 18, 2702. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Ma, N.J.; Zhou, H.; Wang, Q.; Zhang, H.; Wang, P.; Hou, H.; Wen, H.; Li, L. Zinc oxide nanoparticles-induced epigenetic change and G2/M arrest are associated with apoptosis in human epidermal keratinocytes. Int. J. Nanomed. 2016, 11, 3859–3874. [Google Scholar] [CrossRef]
- Jaragh-Alhadad, L.A.; Falahati, M. Copper oxide nanoparticles promote amyloid-β-triggered neurotoxicity through formation of oligomeric species as a prelude to Alzheimer’s diseases. Int. J. Biol. Macromol. 2022, 207, 121–129. [Google Scholar] [CrossRef]
- Wimmer, I.; Tietz, S.; Nishihara, H.; Deutsch, U.; Sallusto, F.; Gosselet, F.; Lyck, R.; Muller, W.A.; Lassmann, H.; Engelhardt, B. PECAM-1 stabilizes blood-brain barrier integrity and favors paracellular T-cell diapedesis across the blood-brain barrier during neuroinflammation. Front. Immunol. 2019, 10, 711. [Google Scholar] [CrossRef]
- Soares, S.; Sousa, J.; Pais, A.; Vitorino, C. Nanomedicine: Principles, properties, and regulatory issues. Front. Chem. 2018, 6, 356901. [Google Scholar] [CrossRef]
- Min, H.S.; Kim, H.J.; Naito, M.; Ogura, S.; Toh, K.; Hayashi, K.; Kim, B.S.; Fukushima, S.; Anraku, Y.; Miyata, K.; et al. Systemic Brain Delivery of Antisense Oligonucleotides across the Blood–Brain Barrier with a Glucose-Coated Polymeric Nanocarrier. Angew. Chem. 2020, 59, 8173–8180. [Google Scholar] [CrossRef]
- Ling, T.S.; Chandrasegaran, S.; Xuan, L.Z.; Suan, T.L.; Elaine, E.; Nathan, D.V.; Chai, Y.H.; Gunasekaran, B.; Salvamani, S. The Potential Benefits of Nanotechnology in Treating Alzheimer’s Disease. Biomed. Res. Int. 2021, 2021, 5550938. [Google Scholar] [CrossRef]
- Fabrizio, C.; Termine, A.; Caltagirone, C.; Sancesario, G. Artificial Intelligence for Alzheimer’s Disease: Promise or Challenge? Diagnostics 2021, 11, 1473. [Google Scholar] [CrossRef] [PubMed]
- Silva-Spínola, A.; Baldeiras, I.; Arrais, J.P.; Santana, I. The Road to Personalized Medicine in Alzheimer’s Disease: The Use of Artificial Intelligence. Biomedicines 2022, 10, 315. [Google Scholar] [CrossRef] [PubMed]
- Olaniyan, O.T.; Adetunji, C.O.; Adeyomoye, O.; Dare, A.; Adeniyi, M.J.; Enoch, A. Cognitive therapy for brain diseases using deep learning models. Artif. Intell. Neurol. Disord. 2023, 171–184. [Google Scholar] [CrossRef]
- Yadav, A.; Pandey, D.; Ashraf, G.M. Rachana Peptide Based Therapy for Neurological Disorders. Curr. Protein. Pept. Sci. 2021, 22, 656–665. [Google Scholar] [CrossRef]
- Vassilakopoulou, V.; Karachaliou, C.E.; Evangelou, A.; Zikos, C.; Livaniou, E. Peptide-Based Vaccines for Neurodegenerative Diseases: Recent Endeavors and Future Perspectives. Vaccines 2021, 9, 1278. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef]
- Chopade, P.; Chopade, N.; Zhao, Z.; Mitragotri, S.; Liao, R.; Chandran Suja, V. Alzheimer’s and Parkinson’s disease therapies in the clinic. Bioeng. Transl. Med. 2023, 8, e10367. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, R. The application of nanotechnology in treatment of Alzheimer’s disease. Front. Bioeng. Biotechnol. 2022, 10, 1042986. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specific Nanosystem Name | Type | Strategy | Size | Conjugated Drugs | Target | Main Function | References |
|---|---|---|---|---|---|---|---|
| Carbon Nanotubes | Inorganic NPs | In vivo/in vitro | 1–100 nm | Berberine | BBB transcytosis, Cholinergic systems | Reduces Aβ accumulation | [98] |
| Lipid carriers | Organic NPs | Ex vivo | 211.4 ± 3.54 nm | Curcumin | Amyloid cascades | Reduces Aβ burden | [99] |
| In vivo/in vitro | Curcumin and nerve growth | BBB transcytosis, Amyloid cascades and tau hyperphosphorylation | Reduces Aβ plaque deposition and lowers AChE activity inside the hippocampus of AD rats | [99] | |||
| Polymeric NPs | Organic NPs | In vivo/in vitro | 161.3 ± 4.7 nm | RVG29 peptide and tau tangles BACE1- AS shRNA gene As | Amyloid cascades | Primarily suppresses Aβ plaque burden and reduces phosphorylated-tau-tangles formation | [100] |
| Metal NPs | Inorganic NPs | 1–100 nm | Anthocyanin | Amyloid cascades and tau Hyperphosphorylation | Anthocyanin-loaded PEG-AuNPs can exhibit neuroprotective potential in comparison to their free form by regulating the p-PI3K/p-Akt/p-GSK3b pathway, inhibition of tau hyperphosphorylation and amyloid cascades formation in AD mice model | [98] | |
| CLPFFD peptide | Amyloid cascades | PEGlyation of AuNPS, effective stabilization of the NPs through masking its negative charge and by facilitating BBB transport improved functionalization with CLPFFD peptide and enhancement of their selective binding toward amyloid fibrils | [98] | ||||
| Magnetic NPs | Inorganic NPs | In vitro | <70 nm | Anti-transferrin monoclonal antibody (OX-26) | Amyloid cascades | Hindrance formation of extracellular accretion of Aβ aggregates | [101] |
| Iron oxide | Amyloid cascades | The larger the concentration of NPs, the more will fibrillation in a magnetic field, whereas a smaller concentration downregulates it Surprisingly, negatively charged or uncharged nanoparticles show better fibrillation suppression | [98] | ||||
| Quantum dots | Inorganic NPs | In vitro | 1–100 nm | QD-biphenyl ethers | Amyloid cascades | Aβ fibril formation inhibition | [98] |
| In vitro | Graphene quantum dots | Amyloid cascades | Aβ aggregation inhibition | [102] | |||
| Liposomal | 10–100 nm | Curcumin derivative | Aβ, Cholinergic dysfunction | Represented as a carrier molecule | [103] | ||
| Peptide Aβ | BBB transcytosis, Amyloid cascades | [103] | |||||
| AuNPs | 1–150 nm | Anthocyanin | Amyloid cascades and tau hyperphosphorylation | Affects different biological activities related to AD | [104] | ||
| CLPFFD peptide | Amyloid cascades | [103] | |||||
| Mesoporous silica NPs (MSN) | 2–50 nm | Rivastigmine hydrogen tartrate | Neuronal cell death/Cholinergic systems | Acts as carrier molecules | [105] | ||
| Metal chelator 5-chloro-4-hydroxy-7-iodoquinoline | BBB transcytosis, Amyloid cascades | [103] | |||||
| Carbon dots | 1–10 nm | Tunable zero-dimension | Acetylcholinesterase enzyme, Amyloid cascades | Take part in theranostic | [103] | ||
| Dendrimers | Organic NPs | 1–10 nm | o-phenylene diamine | Amyloid cascades | Functions as carrier molecules and different chemical loading abilities can be carried to different brain parts | [106] | |
| Nanoliposome (1,2-distearoyl-sn-glycero-3-phosphocholine; cholesterol) | 110 ± 6 nm | Curcumin | Aβ fibril formation retardation | [103] | |||
| Liposome (Shirasu porous glass + cholesterol) | 102 ± 2 nm | Modulate tau phosphorylation and glycogen synthase kinase 3 activities | Reduced Aβ clearance | [107] | |||
| Retro-Inverso peptide inhibitor nanoparticles | 131 ± 43 nm | Inhibitors of aggregation of the Alzheimer’s Aβ peptide | Reduced Aβ clearance | [103] | |||
| Iron oxide | 250–350 nm | Inhibitors of aggregation of the Alzheimer’s Aβ peptide | Inhibited formation of Aβ oligomers and fibrils in vitro | [103] | |||
| H2O2-responsive therapy | Interfered with Aβ aggregation and neurotoxicity | ||||||
| Z-DEVD-FMK (caspase-3 inhibitor) | Decreased infarct volume, neurological deficit, and caspase-3 activity | ||||||
| Chitosan | 650 ± 2 nm | Z-DEVD-FMK and bFGF | Low infarct volume; improved motor function | [108] | |||
| Cationic Bovine Serum Albumin | 114 ± 14 nm | Tanshinone IIA | Low infarct volume, neurological function deficit, neutrophil infiltration, and ultimately neuronal apoptosis | [103] | |||
| Lipidic (Squalene) | 120 nm | Adenosine | Lower infarct volume; improved neurological deficit scores | [109] | |||
| Polylactic acid | 118.3 ± 7.8 nm | nanoparticles angiopep-2-conjugated, 125 NAP (NAPVSIPQ)-loaded (NAP: neuroprotective peptide) | Increased drug uptake inside the brain, impairment in ameliorated learning, cholinergic disruption, and functional loss of hippocampal neurons | [103] | |||
| Poly(butylcyanoacrylate) | 250 ± 30 nm | Nerve growth factor | Reversed scopolamine-induced amnesia and improvement in recognition and memory | [108] | |||
| Carboxyl-conjugated AuNPs (negatively charged) | 250 ± 30 nm | Negatively charged AuNPs | Disrupted the Aβ fibrillation and fragmented the fibrils already that were formed | [103] |
| Trial ID | Intervention | Clinical Importance |
|---|---|---|
| NCT03806478 | Intranasal Nanoparticles of APH-1105 | Phase 2 study assessing the safety, tolerability, and efficacy of intranasal delivery of APH-1105 for treating mild to moderate AD in adults. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dey, A.; Ghosh, S.; Rajendran, R.L.; Bhuniya, T.; Das, P.; Bhattacharjee, B.; Das, S.; Mahajan, A.A.; Samant, A.; Krishnan, A.; et al. Alzheimer’s Disease Pathology and Assistive Nanotheranostic Approaches for Its Therapeutic Interventions. Int. J. Mol. Sci. 2024, 25, 9690. https://doi.org/10.3390/ijms25179690
Dey A, Ghosh S, Rajendran RL, Bhuniya T, Das P, Bhattacharjee B, Das S, Mahajan AA, Samant A, Krishnan A, et al. Alzheimer’s Disease Pathology and Assistive Nanotheranostic Approaches for Its Therapeutic Interventions. International Journal of Molecular Sciences. 2024; 25(17):9690. https://doi.org/10.3390/ijms25179690
Chicago/Turabian StyleDey, Anuvab, Subhrojyoti Ghosh, Ramya Lakshmi Rajendran, Tiyasa Bhuniya, Purbasha Das, Bidyabati Bhattacharjee, Sagnik Das, Atharva Anand Mahajan, Anushka Samant, Anand Krishnan, and et al. 2024. "Alzheimer’s Disease Pathology and Assistive Nanotheranostic Approaches for Its Therapeutic Interventions" International Journal of Molecular Sciences 25, no. 17: 9690. https://doi.org/10.3390/ijms25179690
APA StyleDey, A., Ghosh, S., Rajendran, R. L., Bhuniya, T., Das, P., Bhattacharjee, B., Das, S., Mahajan, A. A., Samant, A., Krishnan, A., Ahn, B.-C., & Gangadaran, P. (2024). Alzheimer’s Disease Pathology and Assistive Nanotheranostic Approaches for Its Therapeutic Interventions. International Journal of Molecular Sciences, 25(17), 9690. https://doi.org/10.3390/ijms25179690