
Molecular Profile as an Outcome Predictor in Glioblastoma along with MRI Features and Surgical Resection: A Scoping Review

 ,
,  ,
, 
Abstract
1. Introduction
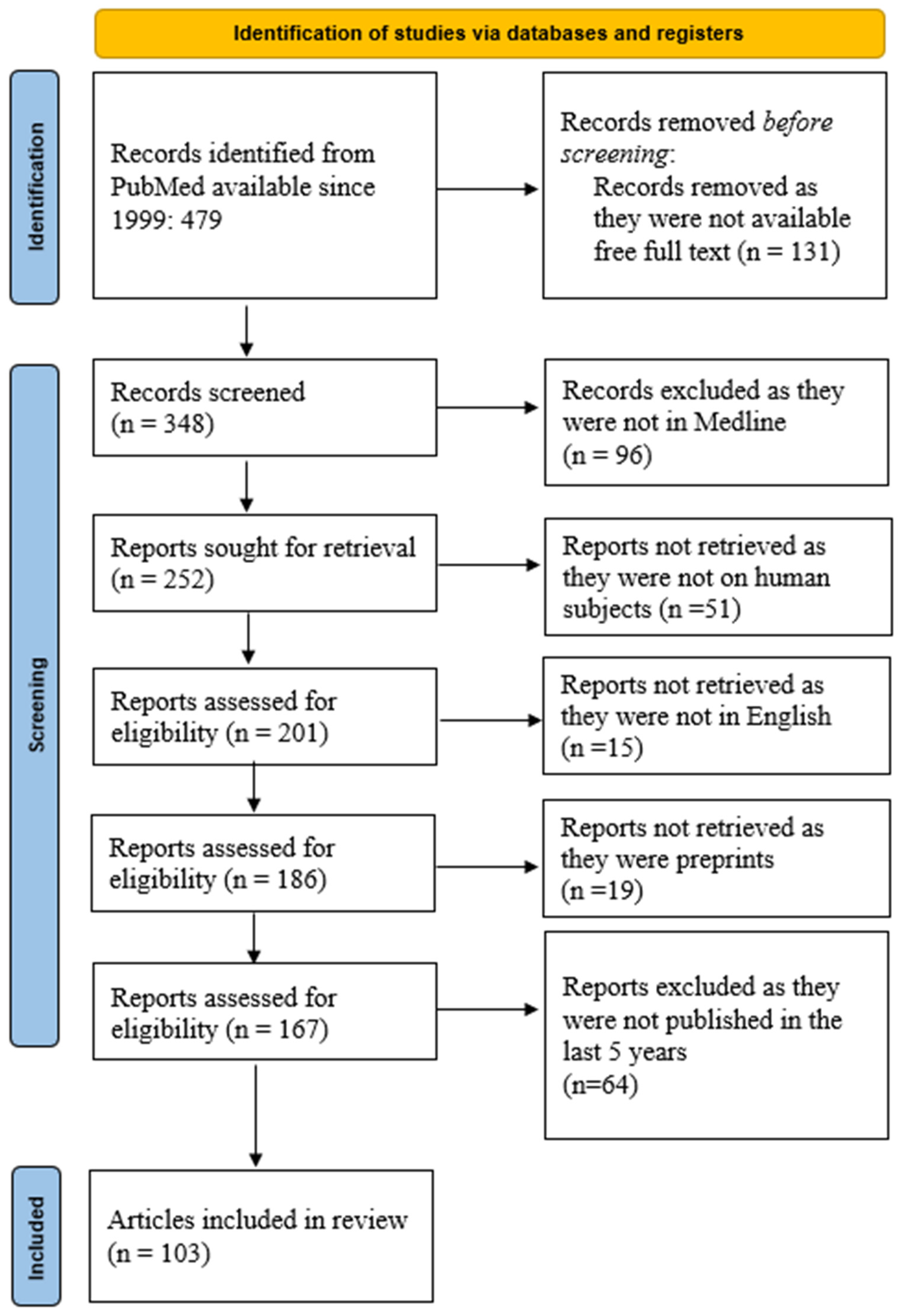
2. Material and Method
3. Results
3.1. IDH-Mutant 1/2 Grade 4 Astrocytoma (Previously Known as Secondary GBM) vs. GBM (IDH-Wildtype)
3.2. MGMT Promoter Methylation Status
3.3. Alterations of the Telomerase Reverse Transcriptase (TERT) Promoter
3.4. EGFR Mutations in Glioblastoma
4. Discussion
4.1. Clinical Aspects
4.2. Anatomical Aspects and MRI Features
- (a)
- Non-enhancing central area of necrosis: Intratumoral necrosis is usually a marker of aggressive and fast-growing tumors. As the tumor cell population increases and the tumor expands, the oxygen demand increases as well [84]. GBM is also characterized by microvascular proliferation. However, these vessels are often disorganized, structurally abnormal and, therefore, inefficient in providing an adequate blood supply [85]. In other words, the angiogenesis does not “keep up” with the tumor expansion. Lastly, tumor cells, especially those in GBM, have impaired or altered apoptosis mechanisms. As a result of these circumstances, cells at the center of the tumor suffer necrosis as a result of hypoxia and nutrient deprivation [85,86].
- (b)
- Peripheral ring enhancement: An essential imaging element of GBM, it can be used to assess the tumor both preoperatively and postoperatively. The aspect of the postoperative residual enhancement is used to quantify and define the extent of resection. However, it is worth noting that the absence of enhancement does not equate to the absence of tumor cells [87].
- (c)
- Peritumoral edema: Unlike the peritumoral edema characterizing meningiomas or metastases, which is pure vasogenic edema, in the case of GBM, the edema is tumor-infiltrated. These two types can be distinguished from each other on MRI using axial diffusivity and radial diffusivity [88].
4.3. Extent of Surgical Resection
- Gross total resection (GTR) is equivalent to the absence of enhancement on postoperative MRI.
- Near total resection (NTR) is equivalent to the existence of a rim enhancement on the resection cavity on postoperative MRI.
- Subtotal resection/partial resection (STR) implies the existence of a nodular enhancement on postoperative MRI.
- Partial resection is a resection of less than 95% of the tumor, based on contrast-enhanced MRI [98].
- Supramaximal resection (SMR) is the absence of any enhancement on postoperative MRI plus extending the resection into apparently normal brain tissue. Advocates of this concept argue that malignant cells extend well beyond MRI or visual macroscopic limits; therefore, removing additional brain tissue might increase survival [99]. There are recent studies backing up that SMR increases median survival without additional postoperative complications [100]. However, SMR is not always applicable. For tumors in the vicinity of eloquent areas, another concept is used, as follows:
- Maximum safe resection (MSR) consists of resecting as much brain tissue as possible without interfering with either the eloquent cortex or with the white matter tracks connected to it.
- Biopsy is where a small quantity of tissue is obtained for diagnostic rather than curative purposes.
- EOR—extent of resection (as a percentage).
4.4. Risk of Recurrence
5. Limitations and Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, N.F.; Ottaviani, D.; Tazare, J.; Gregson, J.; Kitchen, N.; Brandner, S.; Fersht, N.; Mulholland, P. Survival Outcomes and Prognostic Factors in Glioblastoma. Cancers 2022, 14, 3161. [Google Scholar] [CrossRef]
- Witthayanuwat, S.; Pesee, M.; Supaadirek, C.; Supakalin, N.; Thamronganantasakul, K.; Krusun, S. Survival Analysis of Glioblastoma Multiforme. Asian Pac. J. Cancer Prev. 2018, 19, 2613–2617. [Google Scholar] [CrossRef] [PubMed]
- Girardi, F.; Matz, M.; Stiller, C.; You, H.; Marcos Gragera, R.; Valkov, M.Y.; Bulliard, J.-L.; De, P.; Morrison, D.; Wanner, M.; et al. Global Survival Trends for Brain Tumors, by Histology: Analysis of Individual Records for 556,237 Adults Diagnosed in 59 Countries during 2000–2014 (CONCORD-3). Neuro Oncol. 2023, 25, 580–592. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Peruzzi, P.P.; Prabhu, V.C. Glioblastoma Multiforme. Available online: https://www.aans.org/patients/conditions-treatments/glioblastoma-multiforme/ (accessed on 5 September 2024).
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; Codon Publications: Singapore, 2017; pp. 143–153. [Google Scholar]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme–Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Tykocki, T.; Eltayeb, M. Ten-Year Survival in Glioblastoma. A Systematic Review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Hoppe, M.; Beiriger, J.; Kodavali, C.V.; Edwards, L.; Zinn, P.O. Letter: Glioblastoma Cell of Origin. Stem Cell Rev. Rep. 2022, 18, 691–693. [Google Scholar] [CrossRef]
- Claes, A.; Idema, A.J.; Wesseling, P. Diffuse Glioma Growth: A Guerilla War. Acta Neuropathol. 2007, 114, 443–458. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.-Y.; Kim, W.K.; Lee, J.-K.; Park, J.; et al. Human Glioblastoma Arises from Subventricular Zone Cells with Low-Level Driver Mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. The Definition of Primary and Secondary Glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef]
- Fei, X.; Wu, J.; Tian, H.; Jiang, D.; Chen, H.; Yan, K.; Wang, Y.; Zhao, Y.; Chen, H.; Xie, X.; et al. Glioma stem cells remodel immunotolerant microenvironment in GBM and are associated with therapeutic advancements. Cancer Biomark. 2024. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, H.; Yan, W.; Yang, P.; Bao, Z.; Zhang, C.; Jiang, T.; You, Y. Genetic and Clinical Characteristics of Primary and Secondary Glioblastoma Is Associated with Differential Molecular Subtype Distribution. Oncotarget 2015, 6, 7318–7324. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 Mutations Are Early Events in the Development of Astrocytomas and Oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [PubMed]
- Nobusawa, S.; Watanabe, T.; Kleihues, P.; Ohgaki, H. IDH1 Mutations as Molecular Signature and Predictive Factor of Secondary Glioblastomas. Clin. Cancer Res. 2009, 15, 6002–6007. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Simmons, M.L.; Lamborn, K.R.; Takahashi, M.; Chen, P.; Israel, M.A.; Berger, M.S.; Godfrey, T.; Nigro, J.; Prados, M.; Chang, S.; et al. Analysis of Complex Relationships between Age, P53, Epidermal Growth Factor Receptor, and Survival in Glioblastoma Patients. Cancer Res. 2001, 61, 1122–1128. [Google Scholar]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic Pathways to Glioblastoma: A Population-Based Study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Wesolowski, J.R.; Rajdev, P.; Mukherji, S.K. Temozolomide (Temodar). Am. J. Neuroradiol. 2010, 31, 1383–1384. [Google Scholar] [CrossRef]
- Villano, J.L.; Seery, T.E.; Bressler, L.R. Temozolomide in Malignant Gliomas: Current Use and Future Targets. Cancer Chemother. Pharmacol. 2009, 64, 647–655. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.-M.; Gallia, G.L.; et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Hickman, R.A.; Gedvilaite, E.; Ptashkin, R.; Reiner, A.S.; Cimera, R.; Nandakumar, S.; Price, A.; Vanderbilt, C.; Fahy, T.; Young, R.J.; et al. CDKN2A/B Mutations and Allele-Specific Alterations Stratify Survival Outcomes in IDH-Mutant Astrocytomas. Acta Neuropathol. 2023, 146, 845–847. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide Resistance in Glioblastoma Multiforme. Genes. Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Singer, L.S.; Feldman, A.Z.; Buerki, R.A.; Horbinski, C.M.; Lukas, R.V.; Stupp, R. The Impact of the Molecular Classification of Glioblastoma on the Interpretation of Therapeutic Clinical Trial Results. Chin. Clin. Oncol. 2021, 10, 38. [Google Scholar] [CrossRef]
- Galbraith, K.; Kumar, A.; Abdullah, K.G.; Walker, J.M.; Adams, S.H.; Prior, T.; Dimentberg, R.; Henderson, F.C.; Mirchia, K.; Sathe, A.A.; et al. Molecular Correlates of Long Survival in IDH-Wildtype Glioblastoma Cohorts. J. Neuropathol. Exp. Neurol. 2020, 79, 843–854. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef]
- Hertler, C.; Felsberg, J.; Gramatzki, D.; Le Rhun, E.; Clarke, J.; Soffietti, R.; Wick, W.; Chinot, O.; Ducray, F.; Roth, P.; et al. Long-Term Survival with IDH Wildtype Glioblastoma: First Results from the ETERNITY Brain Tumor Funders’ Collaborative Consortium (EORTC 1419). Eur. J. Cancer 2023, 189, 112913. [Google Scholar] [CrossRef]
- Nava, F.; Tramacere, I.; Fittipaldo, A.; Bruzzone, M.G.; DiMeco, F.; Fariselli, L.; Finocchiaro, G.; Pollo, B.; Salmaggi, A.; Silvani, A.; et al. Survival Effect of First- and Second-Line Treatments for Patients with Primary Glioblastoma: A Cohort Study from a Prospective Registry, 1997–2010. Neuro Oncol. 2014, 16, 719–727. [Google Scholar] [CrossRef]
- Krolicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Prolonged Survival in Secondary Glioblastoma Following Local Injection of Targeted Alpha Therapy with 213Bi-Substance P Analogue. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 Wild Type Anaplastic Astrocytomas Exhibit Worse Prognosis than IDH1-Mutated Glioblastomas, and IDH1 Mutation Status Accounts for the Unfavorable Prognostic Effect of Higher Age: Implications for Classification of Gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 Mutations in Gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Qi, S.; Yu, L.; Gui, S.; Ding, Y.; Han, H.; Zhang, X.; Wu, L.; Yao, F. IDH Mutations Predict Longer Survival and Response to Temozolomide in Secondary Glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef]
- Cruz, J.V.R.; Batista, C.; Afonso, B.d.H.; Alexandre-Moreira, M.S.; Dubois, L.G.; Pontes, B.; Moura Neto, V.; Mendes, F.d.A. Obstacles to Glioblastoma Treatment Two Decades after Temozolomide. Cancers 2022, 14, 3203. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Weller, M.; Tabatabai, G.; Kästner, B.; Felsberg, J.; Steinbach, J.P.; Wick, A.; Schnell, O.; Hau, P.; Herrlinger, U.; Sabel, M.C.; et al. MGMT Promoter Methylation Is a Strong Prognostic Biomarker for Benefit from Dose-Intensified Temozolomide Rechallenge in Progressive Glioblastoma: The DIRECTOR Trial. Clin. Cancer Res. 2015, 21, 2057–2064. [Google Scholar] [CrossRef]
- Molenaar, R.J.; Verbaan, D.; Lamba, S.; Zanon, C.; Jeuken, J.W.M.; Boots-Sprenger, S.H.E.; Wesseling, P.; Hulsebos, T.J.M.; Troost, D.; van Tilborg, A.A.; et al. The Combination of IDH1 Mutations and MGMT Methylation Status Predicts Survival in Glioblastoma Better than Either IDH1 or MGMT Alone. Neuro Oncol. 2014, 16, 1263–1273. [Google Scholar] [CrossRef]
- Brawanski, K.R.; Sprung, S.; Freyschlag, C.F.; Hoeftberger, R.; Ströbel, T.; Haybaeck, J.; Thomé, C.; Manzl, C.; Birkl-Toeglhofer, A.M. Influence of MMR, MGMT Promotor Methylation and Protein Expression on Overall and Progression-Free Survival in Primary Glioblastoma Patients Treated with Temozolomide. Int. J. Mol. Sci. 2023, 24, 6184. [Google Scholar] [CrossRef]
- Weller, M.; Stupp, R.; Reifenberger, G.; Brandes, A.A.; van den Bent, M.J.; Wick, W.; Hegi, M.E. MGMT Promoter Methylation in Malignant Gliomas: Ready for Personalized Medicine? Nat. Rev. Neurol. 2010, 6, 39–51. [Google Scholar] [CrossRef]
- Ortiz, R.; Perazzoli, G.; Cabeza, L.; Jiménez-Luna, C.; Luque, R.; Prados, J.; Melguizo, C. Temozolomide: An Updated Overview of Resistance Mechanisms, Nanotechnology Advances and Clinical Applications. Curr. Neuropharmacol. 2021, 19, 513–537. [Google Scholar] [CrossRef]
- Lang, F.; Liu, Y.; Chou, F.-J.; Yang, C. Genotoxic Therapy and Resistance Mechanism in Gliomas. Pharmacol. Ther. 2021, 228, 107922. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823–826. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing Repair and Tolerance of DNA Damage Caused by Alkylating Agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Yu, Y.; Grimmer, M.R.; Wahl, M.; Chang, S.M.; Costello, J.F. Temozolomide-Associated Hypermutation in Gliomas. Neuro Oncol. 2018, 20, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef]
- Mender, I.; Shay, J. Telomerase Repeated Amplification Protocol (TRAP). Bio Protoc. 2015, 5, e1657. [Google Scholar] [CrossRef]
- Lansdorp, P.M. Telomeres, Aging, and Cancer: The Big Picture. Blood 2022, 139, 813–821. [Google Scholar] [CrossRef]
- Maciejowski, J.; de Lange, T. Telomeres in Cancer: Tumour Suppression and Genome Instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Lorbeer, F.K.; Hockemeyer, D. TERT Promoter Mutations and Telomeres during Tumorigenesis. Curr. Opin. Genet. Dev. 2020, 60, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.C.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Nonoguchi, N.; Ohta, T.; Oh, J.-E.; Kim, Y.-H.; Kleihues, P.; Ohgaki, H. TERT Promoter Mutations in Primary and Secondary Glioblastomas. Acta Neuropathol. 2013, 126, 931–937. [Google Scholar] [CrossRef]
- Vuong, H.G.; Altibi, A.M.A.; Duong, U.N.P.; Ngo, H.T.T.; Pham, T.Q.; Chan, A.K.-Y.; Park, C.-K.; Fung, K.-M.; Hassell, L. TERT Promoter Mutation and Its Interaction with IDH Mutations in Glioma: Combined TERT Promoter and IDH Mutations Stratifies Lower-Grade Glioma into Distinct Survival Subgroups—A Meta-Analysis of Aggregate Data. Crit. Rev. Oncol. Hematol. 2017, 120, 1–9. [Google Scholar] [CrossRef]
- Terzi, N.K.; Yilmaz, I.; Oz, A.B. The Place and Prognostic Value of Tert Promoter Mutation in Molecular Classification in Grade Ii-Iii Glial Tumors and Primary Glioblastomas. Turk. J. Pathol. 2021, 38, 90–98. [Google Scholar] [CrossRef]
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.M.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult Infiltrating Gliomas with WHO 2016 Integrated Diagnosis: Additional Prognostic Roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. EGFR as a Clinical Marker in Glioblastomas and Other Gliomas. Int. J. Biol. Markers 2018, 33, 22–32. [Google Scholar] [CrossRef]
- Mizukami, T.; Izawa, N.; Nakajima, T.E.; Sunakawa, Y. Targeting EGFR and RAS/RAF Signaling in the Treatment of Metastatic Colorectal Cancer: From Current Treatment Strategies to Future Perspectives. Drugs 2019, 79, 633–645. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef]
- Montano, N.; Cenci, T.; Martini, M.; D’Alessandris, Q.G.; Pelacchi, F.; Ricci-Vitiani, L.; Maira, G.; De Maria, R.; Larocca, L.M.; Pallini, R. Expression of EGFRvIII in Glioblastoma: Prognostic Significance Revisited. Neoplasia 2011, 13, 1113–1121, IN3–IN6. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The Natural History of EGFR and EGFRvIII in Glioblastoma Patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.; Karajannis, M.; Harter, D. Glioblastoma Multiforme: State of the Art and Future Therapeutics. Surg. Neurol. Int. 2014, 5, 64. [Google Scholar] [CrossRef]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.; et al. Prognostic Value of Epidermal Growth Factor Receptor in Patients with Glioblastoma Multiforme1. Cancer Res. 2003, 63, 6962–6970. [Google Scholar]
- Amirpour, Z.; Bahari, A.; Nafisi, B.; Rahmani, K.; Taghipour Zahir, S. Prognosis and Survival Study in Patients with Glioblastoma Multiform and Its Relationship with EGFR Expression. Iran. J. Neurosurg. 2020, 6, 113–120. [Google Scholar] [CrossRef]
- Armocida, D.; Pesce, A.; Frati, A.; Santoro, A.; Salvati, M. EGFR Amplification Is a Real Independent Prognostic Impact Factor between Young Adults and Adults over 45yo with Wild-Type Glioblastoma? J. Neurooncol. 2020, 146, 275–284. [Google Scholar] [CrossRef]
- Hobbs, J.; Nikiforova, M.N.; Fardo, D.W.; Bortoluzzi, S.; Cieply, K.; Hamilton, R.L.; Horbinski, C. Paradoxical Relationship Between the Degree of EGFR Amplification and Outcome in Glioblastomas. Am. J. Surg. Pathol. 2012, 36, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.C.; Magge, R.S. Mechanisms of EGFR Resistance in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 8471. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef]
- Saeidi Borojeni, H.R.; Najafi, F.; Khosravi Shadmani, F.; Darabi, Z.; Darbandi, M.; Farhadi, K.; Saeidi Borojeni, S.; Maleki, S.; Naderi, M. Disability-Adjusted Life Years and Mortality Rate Attributed to Brain and Central Nervous System Cancer in the Middle East and North Africa Countries. Neuroepidemiology 2021, 55, 447–459. [Google Scholar] [CrossRef]
- Fan, Y.; Zhang, X.; Gao, C.; Jiang, S.; Wu, H.; Liu, Z.; Dou, T. Burden and Trends of Brain and Central Nervous System Cancer from 1990 to 2019 at the Global, Regional, and Country Levels. Arch. Public Health 2022, 80, 209. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.M.; Force, L.M.; Xu, R.; Compton, K.; Lu, D.; Henrikson, H.J.; Kocarnik, J.M.; Harvey, J.D.; Pennini, A.; Dean, F.E.; et al. The Global Burden of Adolescent and Young Adult Cancer in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Oncol. 2022, 23, 27–52. [Google Scholar] [CrossRef] [PubMed]
- Karnofsky, D.A.; Abelmann, W.H.; Craver, L.F.; Burchenal, J.H. The Use of the Nitrogen Mustards in the Palliative Treatment of Carcinoma. Cancer 1948, 1, 634–656. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, K.; Cui, Y.; Ren, X.; Li, M.; Zhang, G.; Yang, C.; Zhao, X.; Zhu, Q.; Lin, S. Differential Predictors and Clinical Implications Associated With Long-Term Survivors in IDH Wildtype and Mutant Glioblastoma. Front. Oncol. 2021, 11, 632663. [Google Scholar] [CrossRef] [PubMed]
- Chinot, O.; Barrie, M.; Calissi, B.; Fuentes, S.; Metellus, P.; Honore, S.; Boucard, C.; Loundou, A.; Figarella-Branger, D.; Dufour, H. Impact of Karnovsky Performance Status (KPS) on Outcome of Elderly Patients (Pts) with Glioblastoma (GBM) and Activity of Temozolomide (TMZ) as First Line Therapy: Retrospective Analysis of a Cohort. J. Clin. Oncol. 2006, 24, 1515. [Google Scholar] [CrossRef]
- Nikolov, V.; Stojanovic, M.; Kostic, A.; Radisavljevic, M.; Simonovic, N.; Jelenkovic, B.; Berilazic, L. Factors Affecting the Survival of Patients with Glioblastoma Multiforme. J. BUON 2018, 23, 173–178. [Google Scholar]
- Tejada-Solís, S.; Aldave-Orzaiz, G.; Pay-Valverde, E.; Marigil-Sánchez, M.; Idoate-Gastearena, M.A.; Díez-Valle, R. Prognostic Value of Ventricular Wall Fluorescence during 5-Aminolevulinic-Guided Surgery for Glioblastoma. Acta Neurochir. 2012, 154, 1997–2002. [Google Scholar] [CrossRef]
- Nestler, U.; Lutz, K.; Pichlmeier, U.; Stummer, W.; Franz, K.; Reulen, H.-J.; Bink, A. Anatomic Features of Glioblastoma and Their Potential Impact on Survival. Acta Neurochir. 2015, 157, 179–186. [Google Scholar] [CrossRef]
- Chaichana, K.L.; Cabrera-Aldana, E.E.; Jusue-Torres, I.; Wijesekera, O.; Olivi, A.; Rahman, M.; Quinones-Hinojosa, A. When Gross Total Resection of a Glioblastoma Is Possible, How Much Resection Should Be Achieved? World Neurosurg. 2014, 82, e257–e265. [Google Scholar] [CrossRef]
- Staub-Bartelt, F.; Rapp, M.; Sabel, M. Resection of Eloquent Located Brain Tumors by Mapping Only—A Feasibility Study. Brain Sci. 2023, 13, 1366. [Google Scholar] [CrossRef]
- Becker, A.; Sells, B.; Haque, S.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef] [PubMed]
- Shukla, G.; Alexander, G.S.; Bakas, S.; Nikam, R.; Talekar, K.; Palmer, J.D.; Shi, W. Advanced Magnetic Resonance Imaging in Glioblastoma: A Review. Chin. Clin. Oncol. 2017, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Orasanu, C.I.; Aschie, M.; Deacu, M.; Bosoteanu, M.; Vamesu, S.; Enciu, M.; Bălţătescu, G.I.; Cozaru, G.C.; Mitroi, A.F.; Voda, R.I. Implications of Cellular Immaturity in Necrosis and Microvascularization in Glioblastomas IDH-Wild-Type. Clin. Pr. 2022, 12, 1054–1068. [Google Scholar] [CrossRef]
- Krakstad, C.; Chekenya, M. Survival Signalling and Apoptosis Resistance in Glioblastomas: Opportunities for Targeted Therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chen, Z.; Chang, B.; Tu, M.; Li, S.; Wang, X.; Chen, M. Prediction of BRAF Mutation Status in Glioblastoma Multiforme by Preoperative Ring Enhancement Appearances on MRI. Front. Oncol. 2022, 12, 937345. [Google Scholar] [CrossRef] [PubMed]
- Min, Z.; Niu, C.; Rana, N.; Ji, H.; Zhang, M. Differentiation of Pure Vasogenic Edema and Tumor-Infiltrated Edema in Patients with Peritumoral Edema by Analyzing the Relationship of Axial and Radial Diffusivities on 3.0T MRI. Clin. Neurol. Neurosurg. 2013, 115, 1366–1370. [Google Scholar] [CrossRef]
- Sanghvi, D. Post-Treatment Imaging of High-Grade Gliomas. Indian. J. Radiol. Imaging 2015, 25, 102–108. [Google Scholar] [CrossRef]
- Macdonald, D.R.; Cascino, T.L.; Schold, S.C.; Cairncross, J.G. Response Criteria for Phase II Studies of Supratentorial Malignant Glioma. J. Clin. Oncol. 1990, 8, 1277–1280. [Google Scholar] [CrossRef]
- Taal, W.; Brandsma, D.; de Bruin, H.G.; Bromberg, J.E.; Swaak-Kragten, A.T.; Sillevis Smitt, P.A.E.; van Es, C.A.; van den Bent, M.J. Incidence of Early Pseudo-progression in a Cohort of Malignant Glioma Patients Treated with Chemoirradiation with Temozolomide. Cancer 2008, 113, 405–410. [Google Scholar] [CrossRef]
- Brandes, A.A.; Tosoni, A.; Franceschi, E.; Sotti, G.; Frezza, G.; Amistà, P.; Morandi, L.; Spagnolli, F.; Ermani, M. Recurrence Pattern After Temozolomide Concomitant With and Adjuvant to Radiotherapy in Newly Diagnosed Patients With Glioblastoma: Correlation With MGMT Promoter Methylation Status. J. Clin. Oncol. 2009, 27, 1275–1279. [Google Scholar] [CrossRef]
- Clarke, J.L.; Chang, S. Pseudoprogression and Pseudoresponse: Challenges in Brain Tumor Imaging. Curr. Neurol. Neurosci. Rep. 2009, 9, 241–246. [Google Scholar] [CrossRef]
- Bulik, M.; Kazda, T.; Slampa, P.; Jancalek, R. The Diagnostic Ability of Follow-Up Imaging Biomarkers after Treatment of Glioblastoma in the Temozolomide Era: Implications from Proton MR Spectroscopy and Apparent Diffusion Coefficient Mapping. BioMed Res. Int. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II Trial of Single-Agent Bevacizumab Followed by Bevacizumab Plus Irinotecan at Tumor Progression in Recurrent Glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Hygino da Cruz, L.C.; Rodriguez, I.; Domingues, R.C.; Gasparetto, E.L.; Sorensen, A.G. Pseudoprogression and Pseudoresponse: Imaging Challenges in the Assessment of Posttreatment Glioma. Am. J. Neuroradiol. 2011, 32, 1978–1985. [Google Scholar] [CrossRef]
- Tunthanathip, T.; Madteng, S. Factors Associated with the Extent of Resection of Glioblastoma. Precis. Cancer Med. 2020, 3, 12. [Google Scholar] [CrossRef]
- Mier-García, J.F.; Ospina-Santa, S.; Orozco-Mera, J.; Ma, R.; Plaha, P. Supramaximal versus Gross Total Resection in Glioblastoma, IDH Wild-Type and Astrocytoma, IDH-Mutant, Grade 4, Effect on Overall and Progression Free Survival: Systematic Review and Meta-Analysis. J. Neurooncol. 2023, 164, 31–41. [Google Scholar] [CrossRef]
- Wach, J.; Vychopen, M.; Kühnapfel, A.; Seidel, C.; Güresir, E. A Systematic Review and Meta-Analysis of Supramarginal Resection versus Gross Total Resection in Glioblastoma: Can We Enhance Progression-Free Survival Time and Preserve Postoperative Safety? Cancers 2023, 15, 1772. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Suki, D.; Hess, K.; Sawaya, R. The Influence of Maximum Safe Resection of Glioblastoma on Survival in 1229 Patients: Can We Do Better than Gross-Total Resection? J. Neurosurg. 2016, 124, 977–988. [Google Scholar] [CrossRef]
- Kreth, F.-W.; Thon, N.; Simon, M.; Westphal, M.; Schackert, G.; Nikkhah, G.; Hentschel, B.; Reifenberger, G.; Pietsch, T.; Weller, M.; et al. Gross Total but Not Incomplete Resection of Glioblastoma Prolongs Survival in the Era of Radiochemotherapy. Ann. Oncol. 2013, 24, 3117–3123. [Google Scholar] [CrossRef]
- Jusue-Torres, I.; Lee, J.; Germanwala, A.V.; Burns, T.C.; Parney, I.F. Effect of Extent of Resection on Survival of Patients with Glioblastoma, IDH–Wild-Type, WHO Grade 4 (WHO 2021): Systematic Review and Meta-Analysis. World Neurosurg. 2023, 171, e524–e532. [Google Scholar] [CrossRef] [PubMed]
- Polonara, G.; Aiudi, D.; Iacoangeli, A.; Raggi, A.; Ottaviani, M.M.; Antonini, R.; Iacoangeli, M.; Dobran, M. Glioblastoma: A Retrospective Analysis of the Role of the Maximal Surgical Resection on Overall Survival and Progression Free Survival. Biomedicines 2023, 11, 739. [Google Scholar] [CrossRef]
- Chaichana, K.L.; Zadnik, P.; Weingart, J.D.; Olivi, A.; Gallia, G.L.; Blakeley, J.; Lim, M.; Brem, H.; Quiñones-Hinojosa, A. Multiple Resections for Patients with Glioblastoma: Prolonging Survival. J. Neurosurg. 2013, 118, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Lamborn, K.R.; Chang, S.M.; Prados, M.D. Prognostic Factors for Survival of Patients with Glioblastoma: Recursive Partitioning Analysis. Neuro Oncol. 2004, 6, 227–235. [Google Scholar] [CrossRef]
- Fabian, D.; Guillermo Prieto Eibl, M.d.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef]
- Kirson, E.D.; Gurvich, Z.; Schneiderman, R.; Dekel, E.; Itzhaki, A.; Wasserman, Y.; Schatzberger, R.; Palti, Y. Disruption of Cancer Cell Replication by Alternating Electric Fields. Cancer Res. 2004, 64, 3288–3295. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma. JAMA 2017, 318, 2306. [Google Scholar] [CrossRef] [PubMed]
- Montemurro, N.; Pahwa, B.; Tayal, A.; Shukla, A.; De Jesus Encarnacion, M.; Ramirez, I.; Nurmukhametov, R.; Chavda, V.; De Carlo, A. Macrophages in Recurrent Glioblastoma as a Prognostic Factor in the Synergistic System of the Tumor Microenvironment. Neurol. Int. 2023, 15, 595–608. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Wang, Z.; Zhong, H.; Liang, X.; Ni, S. Targeting Tumor-Associated Macrophages for the Immunotherapy of Glioblastoma: Navigating the Clinical and Translational Landscape. Front. Immunol. 2022, 13, 1024921. [Google Scholar] [CrossRef]
- Florent, L.; Saby, C.; Slimano, F.; Morjani, H. BRAF V600-Mutated Metastatic Melanoma and Targeted Therapy Resistance: An Update of the Current Knowledge. Cancers 2023, 15, 2607. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.J.; Thornton, Z.A.; Saincher, S.S.; Yao, I.Y.; Dawson, S.; McGuinness, L.A.; Jones, H.E.; Jefferies, S.; Short, S.C.; Cheng, H.-Y.; et al. Prevalence of BRAF V600 in Glioma and Use of BRAF Inhibitors in Patients with BRAF V600 Mutation-Positive Glioma: Systematic Review. Neuro Oncol. 2022, 24, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.C.; Ronellenfitsch, M.W.; Lorenz, N.I.; Wagner, M.; Voss, M.; Capper, D.; Tzaridis, T.; Herrlinger, U.; Steinbach, J.P.; Stoffels, G.; et al. Dabrafenib in Patients with Recurrent, BRAF V600E Mutated Malignant Glioma and Leptomeningeal Disease. Oncol. Rep. 2017, 38, 3291–3296. [Google Scholar] [CrossRef]
- Arrieta, V.A.; Gould, A.; Kim, K.-S.; Habashy, K.J.; Dmello, C.; Vázquez-Cervantes, G.I.; Palacín-Aliana, I.; McManus, G.; Amidei, C.; Gomez, C.; et al. Ultrasound-Mediated Delivery of Doxorubicin to the Brain Results in Immune Modulation and Improved Responses to PD-1 Blockade in Gliomas. Nat. Commun. 2024, 15, 4698. [Google Scholar] [CrossRef]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological Analysis of Phase II Glioblastoma Dendritic Cell Vaccine (Audencel) Trial: Immune System Characteristics Influence Outcome and Audencel up-Regulates Th1-Related Immunovariables. Acta Neuropathol. Commun. 2018, 6, 135. [Google Scholar] [CrossRef]
- Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.F.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers 2018, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Heugenhauser, J.; Galijasevic, M.; Mangesius, S.; Goebel, G.; Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Stockhammer, G.; Iglseder, S.; et al. MRI Response Assessment in Glioblastoma Patients Treated with Dendritic-Cell-Based Immunotherapy. Cancers 2022, 14, 1579. [Google Scholar] [CrossRef]
- Taciuc, I.-A.; Dumitru, M.; Vrinceanu, D.; Gherghe, M.; Manole, F.; Marinescu, A.; Serboiu, C.; Neagos, A.; Costache, A. Applications and Challenges of Neural Networks in Otolaryngology (Review). Biomed. Rep. 2024, 20, 92. [Google Scholar] [CrossRef]

{kind=link}
| Study | Survival IDH-Wildtype | Survival IDH-Mutant ½ | Survival Benefit IDH-Mutant vs. Wildtype |
|---|---|---|---|
| Yan H. et al. (2009) [34] | 15 months | 30 months | 15 months (100%) |
| Hartmann C. et al. (2010) [33] | 12 months | 36 months | 24 months (200%) |
| Study | Unmethylated MGMT Promoter (Abundance of MGMT Enzyme) | Hypermethylated MGMT Promoter (Low Production of MGMT Enzyme) | Median Survival Benefit |
|---|---|---|---|
| Hegi M. et al. (2005) [37] | 12.2 months | 18.2 months | 6 months (~50%) |
| Weller et al. (2015) [38] | 7.9 months | 12.9 months | 5 months (~63%) |
| Molenaar R. et al. (2014) [39] * | 7.2 months | 14.3 months | 7.1 months (~98%) |
| Brawanski K. et al. (2023) [40] ** | 14 months | 11 months | 3 months (20%) |
| Study | Median Survival Time TERT Promoter Mutation GBM Patients | Median Survival Time No TERT Promoter Mutations GBM Patients |
|---|---|---|
| Nonoguchi N. et al. (2013) [56] | 9.3 months | 9.6 months |
| Pekmezci M. et al. [59] * | 13.2 months | 18.6 months |
| Study | Median Survival in Months GBMs with EGFR Amplification | Median Survival in Months GBMs with Absent EGFR Amplification |
|---|---|---|
| Shinojima, N et al. (2003) [66] | 14.4 months | 20.4 months |
| Amirpour Z. et al. * (2020) [67] | 20.6 months | 27.4 months |
| Armocida D. et al. ** (2019) [68] | 16 months | 21.7 months |
| Hobbs J. et al. (2012) [69] | 11 months (high amplification) 7.7 months (low amplification) | 7.9 months |
| Author | Median Survival GTR | Median Survival Non-GTR | Survival Benefit (Months) | Survival Benefit (Percentage) |
|---|---|---|---|---|
| Kreth F. et al. (2013) [102] | 17.1 months | 15.4 months | 5.4 | 46% |
| Li Y. et al. (2016) [101] | 15.6 | 9.8 | 5.8 | 60% |
| Jusue-Torres I. et al. (2023) [103] | 20 | 12 | 8 | 66% |
| Polonara G. et al. (2023) [104] | 16 | 14.2 | 1.8 | 12% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papacocea, S.I.; Vrinceanu, D.; Dumitru, M.; Manole, F.; Serboiu, C.; Papacocea, M.T. Molecular Profile as an Outcome Predictor in Glioblastoma along with MRI Features and Surgical Resection: A Scoping Review. Int. J. Mol. Sci. 2024, 25, 9714. https://doi.org/10.3390/ijms25179714
Papacocea SI, Vrinceanu D, Dumitru M, Manole F, Serboiu C, Papacocea MT. Molecular Profile as an Outcome Predictor in Glioblastoma along with MRI Features and Surgical Resection: A Scoping Review. International Journal of Molecular Sciences. 2024; 25(17):9714. https://doi.org/10.3390/ijms25179714
Chicago/Turabian StylePapacocea, Serban Iancu, Daniela Vrinceanu, Mihai Dumitru, Felicia Manole, Crenguta Serboiu, and Marius Toma Papacocea. 2024. "Molecular Profile as an Outcome Predictor in Glioblastoma along with MRI Features and Surgical Resection: A Scoping Review" International Journal of Molecular Sciences 25, no. 17: 9714. https://doi.org/10.3390/ijms25179714
APA StylePapacocea, S. I., Vrinceanu, D., Dumitru, M., Manole, F., Serboiu, C., & Papacocea, M. T. (2024). Molecular Profile as an Outcome Predictor in Glioblastoma along with MRI Features and Surgical Resection: A Scoping Review. International Journal of Molecular Sciences, 25(17), 9714. https://doi.org/10.3390/ijms25179714