
Thiostrepton as a Potential Therapeutic Agent for Hepatocellular Carcinoma

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. TST Is Identified as a Potential Agent against HCC
2.2. TST Inhibits the Migration of HCC Cells
2.3. TST Arrests the Cell Cycle and Induces Cell Apoptosis of HCC Cells
2.4. TST Impairs the Mitochondria in HCC Cells
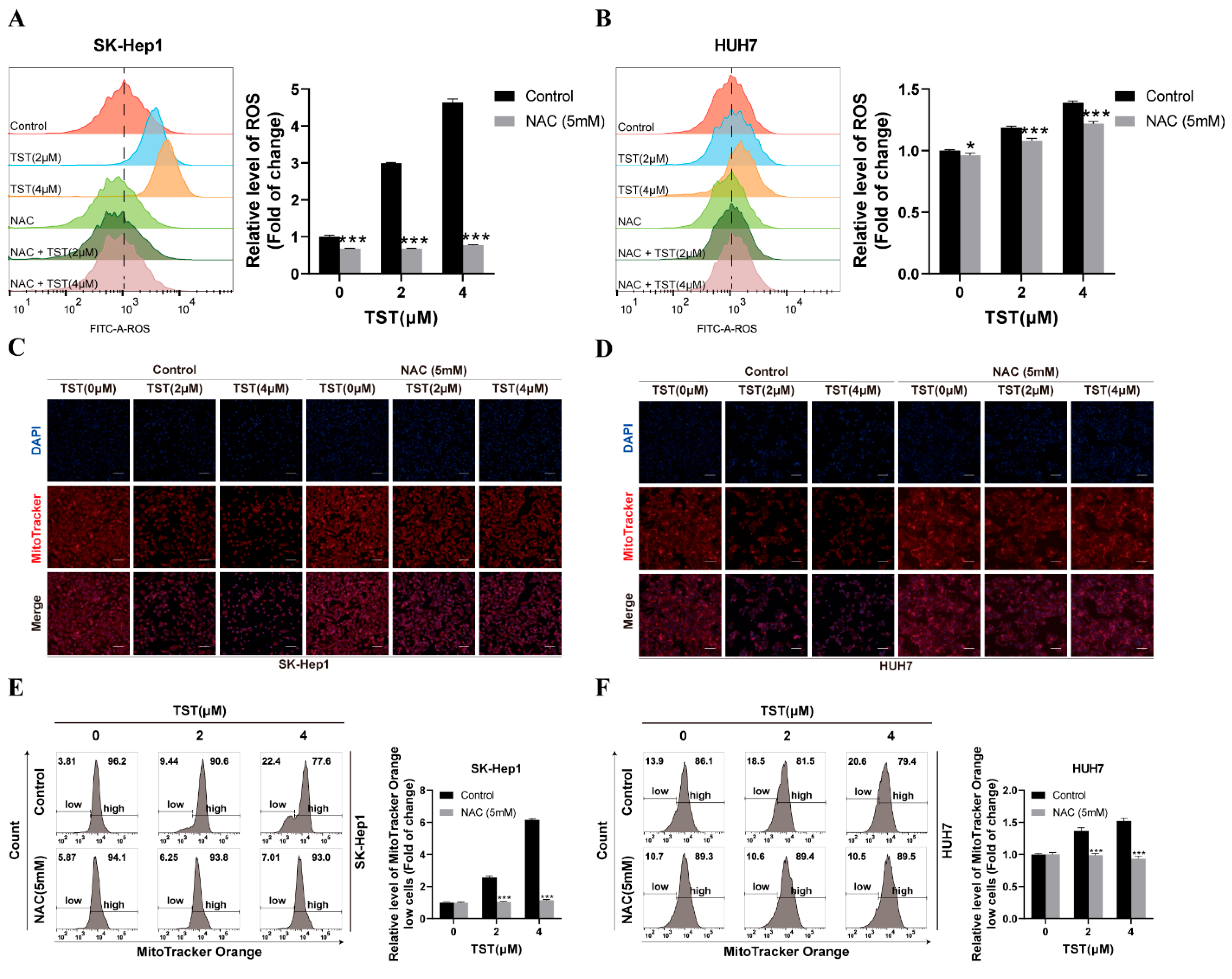
2.5. Mitochondrial Damage in HCC Cells Triggered by TST Relies on ROS
2.6. TST Suppresses the Growth of HCC Cells in an ROS-Dependent Manner
2.7. Mitophagy Blockade Enhances the Inhibitory Activity of TST against HCC Cells
3. Discussion and Conclusions
4. Material and Methods
4.1. PRISM Dataset and Drug Screening
4.2. Reagent and Plasmids
4.3. Cell Culture
4.4. Cell Viability and Colony Formation Assay
4.5. Drug Combination Analysis
4.6. Cell Cycle Analysis
4.7. Apoptosis Assay
4.8. Western Blot Assay
4.9. Immunofluorescence Assay
4.10. RNA Interference
4.11. Cell Scratch Assay
4.12. Determination of ROS
4.13. Determination of Mitochondrial Mass
4.14. MMP Measurement
4.15. TEM Assay
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bruix, J.; Han, K.H.; Gores, G.; Llovet, J.M.; Mazzaferro, V. Liver cancer: Approaching a personalized care. J. Hepatol. 2015, 62, S144–S156. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Q.; Liang, N.; Xue, H.; Yang, T.; Chen, X.; Qiu, Z.; Zeng, C.; Sun, T.; Yuan, W.; et al. Oncogenic driver genes and tumor microenvironment determine the type of liver cancer. Cell Death Dis. 2020, 11, 313. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.; Meyer, T.; Sapisochin, G.; Salem, R.; Saborowski, A. Hepatocellular carcinoma. Lancet 2022, 400, 1345–1362. [Google Scholar] [CrossRef] [PubMed]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in liver cancer and possible treatment approaches. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188314. [Google Scholar] [CrossRef]
- Bruix, J.; Reig, M.; Sherman, M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016, 150, 835–853. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Mei, S.; Zhang, K. A Multi-Label Learning Framework for Drug Repurposing. Pharmaceutics 2019, 11, 466. [Google Scholar] [CrossRef]
- Singhal, S.; Mehta, J.; Desikan, R.; Ayers, D.; Roberson, P.; Eddlemon, P.; Munshi, N.; Anaissie, E.; Wilson, C.; Dhodapkar, M.; et al. Antitumor activity of thalidomide in refractory multiple myeloma. N. Engl. J. Med. 1999, 341, 1565–1571. [Google Scholar] [CrossRef]
- Hurle, M.R.; Yang, L.; Xie, Q.; Rajpal, D.K.; Sanseau, P.; Agarwal, P. Computational drug repositioning: From data to therapeutics. Clin. Pharmacol. Ther. 2013, 93, 335–341. [Google Scholar] [CrossRef]
- Issa, N.T.; Stathias, V.; Schurer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. Semin. Cancer Biol. 2021, 68, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Wu, H.; Wang, T.; Liu, Y.; Li, X.; Xu, S.; Wu, C.; Zou, H.; Cao, M.; Jin, G.; Lang, J.; et al. Mitophagy promotes sorafenib resistance through hypoxia-inducible ATAD3A dependent Axis. J. Exp. Clin. Cancer Res. 2020, 39, 274. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Luo, L.; Guo, C.Y.; Goto, S.; Urata, Y.; Shao, J.H.; Li, T.S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, G.; Zheng, Y.; Shen, H.M.; Hu, X.; Ming, Q.L.; Huang, C.; Li, P.; Gao, N. A novel autophagy/mitophagy inhibitor liensinine sensitizes breast cancer cells to chemotherapy through DNM1L-mediated mitochondrial fission. Autophagy 2015, 11, 1259–1279. [Google Scholar] [CrossRef]
- Cai, X.; Xiao, W.; Shen, J.; Lian, H.; Lu, Y.; Liu, X.; Gu, J. Thiostrepton and miR-216b synergistically promote osteosarcoma cell cytotoxicity and apoptosis by targeting FoxM1. Oncol. Lett. 2020, 20, 391. [Google Scholar] [CrossRef]
- Liu, S.X.; Zhou, Y.; Zhao, L.; Zhou, L.S.; Sun, J.; Liu, G.J.; Du, Y.S.; Zhou, Y.N. Thiostrepton confers protection against reactive oxygen species-related apoptosis by restraining FOXM1-triggerred development of gastric cancer. Free Radic. Biol. Med. 2022, 193, 385–404. [Google Scholar] [CrossRef]
- Kongsema, M.; Wongkhieo, S.; Khongkow, M.; Lam, E.W.F.; Boonnoy, P.; Vongsangnak, W.; Wong-Ekkabut, J. Molecular mechanism of Forkhead box M1 inhibition by thiostrepton in breast cancer cells. Oncol. Rep. 2019, 42, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Kuttikrishnan, S.; Prabhu, K.S.; Khan, A.Q.; Alali, F.Q.; Ahmad, A.; Uddin, S. Thiostrepton inhibits growth and induces apoptosis by targeting FoxM1/SKP2/MTH1 axis in B-precursor acute lymphoblastic leukemia cells. Leuk. Lymphoma 2021, 62, 3170–3180. [Google Scholar] [CrossRef]
- Kwok, J.M.M.; Myatt, S.S.; Marson, C.M.; Coombes, R.C.; Constantinidou, D.; Lam, E.W.F. Thiostrepton selectively targets breast cancer cells through inhibition of forkhead box M1 expression. Mol. Cancer Ther. 2008, 7, 2022–2032. [Google Scholar] [CrossRef]
- Newick, K.; Cunniff, B.; Preston, K.; Held, P.; Arbiser, J.; Pass, H.; Mossman, B.; Shukla, A.; Heintz, N. Peroxiredoxin 3 is a redox-dependent target of thiostrepton in malignant mesothelioma cells. PLoS ONE 2012, 7, e39404. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, J.; Xie, J.; Niu, Y.; Ye, S.; Wan, Q.; Ye, Q. Detection and identification of peroxiredoxin 3 as a biomarker in hepatocellular carcinoma by a proteomic approach. Int. J. Mol. Med. 2012, 29, 832–840. [Google Scholar] [CrossRef]
- Bird, K.E.; Xander, C.; Murcia, S.; Schmalstig, A.A.; Wang, X.; Emanuele, M.J.; Braunstein, M.; Bowers, A.A. Thiopeptides Induce Proteasome-Independent Activation of Cellular Mitophagy. ACS Chem. Biol. 2020, 15, 2164–2174. [Google Scholar] [CrossRef]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, W.; Humeau, J.; Chen, G.; Liu, P.; Pol, J.; Zhang, Z.; Kepp, O.; Kroemer, G. Autophagy induction by thiostrepton improves the efficacy of immunogenic chemotherapy. J. ImmunoTher. Cancer 2020, 8, e000462. [Google Scholar] [CrossRef]
- Su, G.; Wang, D.; Yang, Q.; Kong, L.; Ju, X.; Yang, Q.; Zhu, Y.; Zhang, S.; Li, Y. Cepharanthine suppresses APC-mutant colorectal cancers by down-regulating the expression of β-catenin. Nat. Prod. Bioprospect. 2024, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Qin, T.; An, T.; Wu, H.; Xu, G.; Xiang, J.; Lei, K.; Zhang, S.; Xia, J.; Su, G.; et al. Novel PORCN inhibitor WHN-88 targets Wnt/beta-catenin pathway and prevents the growth of Wnt-driven cancers. Eur. J. Pharmacol. 2023, 945, 175628. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Fan, D.; Kong, L.; Yang, Q.; Zhu, Y.; Zhang, S.; Su, G.; Li, Y. Antimicrobial Peptide Brevinin-1RL1 from Frog Skin Secretion Induces Apoptosis and Necrosis of Tumor Cells. Molecules 2021, 26, 2059. [Google Scholar] [CrossRef]
- Wang, Y.; Su, G.F.; Huang, Z.X.; Wang, Z.G.; Zhou, P.J.; Fan, J.L.; Wang, Y.F. Cepharanthine hydrochloride induces mitophagy targeting GPR30 in hepatocellular carcinoma (HCC). Expert. Opin. Ther. Targets 2020, 24, 389–402. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, G.; Yang, Q.; Zhou, H.; Huang, Y.; Nie, S.; Wang, D.; Ma, G.; Zhang, S.; Kong, L.; Zou, C.; et al. Thiostrepton as a Potential Therapeutic Agent for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2024, 25, 9717. https://doi.org/10.3390/ijms25179717
Su G, Yang Q, Zhou H, Huang Y, Nie S, Wang D, Ma G, Zhang S, Kong L, Zou C, et al. Thiostrepton as a Potential Therapeutic Agent for Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2024; 25(17):9717. https://doi.org/10.3390/ijms25179717
Chicago/Turabian StyleSu, Guifeng, Qianqing Yang, Heyang Zhou, Ying Huang, Shiyun Nie, Dan Wang, Guangchao Ma, Shaohua Zhang, Lingmei Kong, Chenggang Zou, and et al. 2024. "Thiostrepton as a Potential Therapeutic Agent for Hepatocellular Carcinoma" International Journal of Molecular Sciences 25, no. 17: 9717. https://doi.org/10.3390/ijms25179717