
Utilizing Molecular Dynamics Simulations, Machine Learning, Cryo-EM, and NMR Spectroscopy to Predict and Validate Protein Dynamics
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Techniques for Studying Protein Dynamics
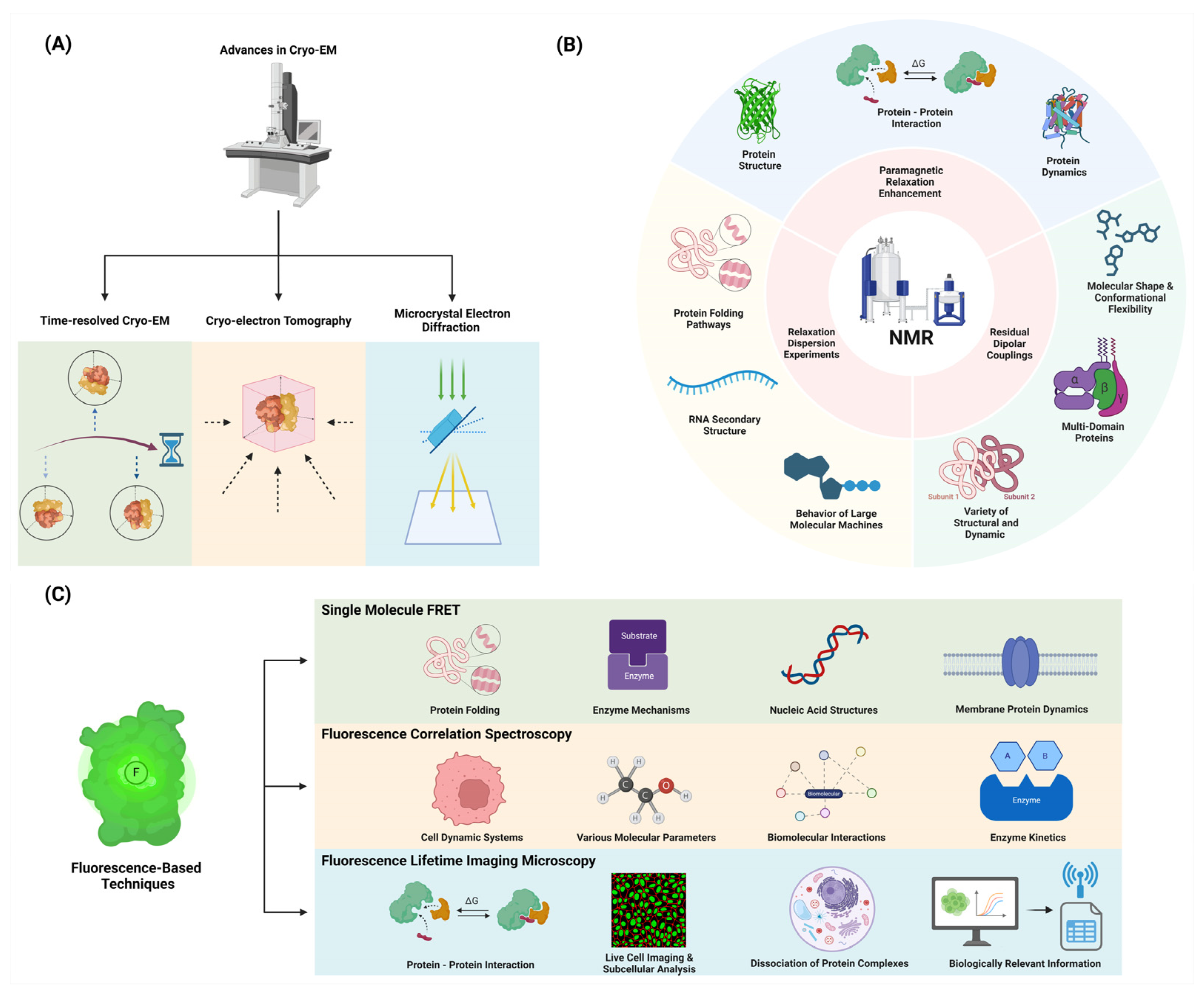
2.1. Innovations in Cryo-EM and X-ray Crystallography for Dynamics Studies
2.1.1. Time-Resolved Cryo-EM: Capturing Protein Motions at Different Time Points
2.1.2. Cryo-Electron Tomography: Visualizing Proteins in Their Cellular Context
2.1.3. Time-Resolved X-ray Crystallography: Studying Room Temperature and Computational Modeling
2.1.4. Microcrystal Electron Diffraction (MicroED): Studying Small Protein Dynamics
2.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.2.1. Relaxation Dispersion Experiments: Detecting and Characterizing Excited States
2.2.2. Paramagnetic Relaxation Enhancement (PRE): Probing Long-Range Interactions
2.2.3. Residual Dipolar Couplings (RDCs): Characterizing Domain Orientations and Flexibility
2.3. Fluorescence-Based Techniques
2.3.1. Single-Molecule FRET: Probing Conformational Changes in Individual Molecules
2.3.2. Fluorescence Correlation Spectroscopy (FCS): Analyzing Diffusion and Binding Kinetics
2.3.3. Fluorescence Lifetime Imaging Microscopy (FLIM): Mapping Protein Interactions in Cells
3. Computational Approaches to Protein Dynamics
3.1. Molecular Dynamics Simulations
3.1.1. Fundamentals of Molecular Dynamics (MD) Simulations: Force Fields and Integration Algorithms
3.1.2. Long-Timescale Simulations: Accessing Biologically Relevant Timescales (Ms–S)
3.1.3. Enhanced Sampling Techniques: Exploring Rare Events and Conformational Transitions
3.1.4. Coarse-Grained Models: Simulating Large Systems and Complex Assemblies
3.2. Machine Learning and AI in Protein Dynamics
3.2.1. Deep Learning for Feature Extraction: Identifying Relevant Collective Variables
3.2.2. Generative Models: Predicting Protein Conformations and Dynamics
3.2.3. Reinforcement Learning: Optimizing Sampling Strategies in MD Simulations
4. Applications and Insights from Protein Dynamics Studies
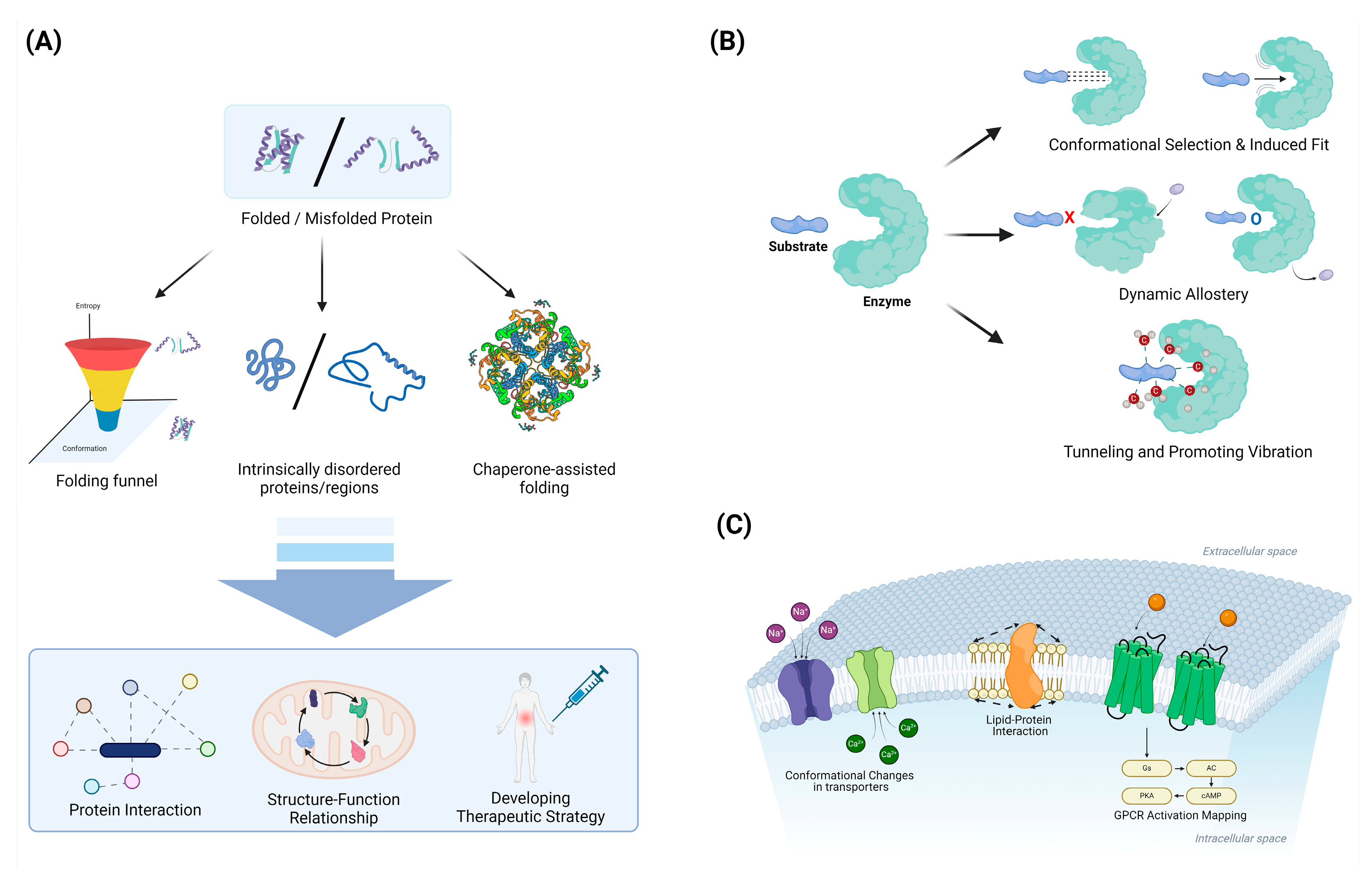
4.1. Protein Folding and Misfolding
4.1.1. Folding Funnels and Energy Landscapes: Characterizing the Thermodynamics and Kinetics of Folding
4.1.2. Intrinsically Disordered Proteins: Recognizing the Functional Importance of Structural Flexibility
4.1.3. Chaperone-Assisted Folding: Elucidating the Role of Cellular Machinery in Protein Folding
4.2. Enzyme Catalysis and Allostery
4.2.1. Conformational Selection vs. Induced Fit: Understanding Substrate Binding Mechanisms
4.2.2. Dynamic Allostery: Recognizing the Importance of Entropy in Allosteric Regulation
4.2.3. Tunneling and Promoting Vibrations: Exploring Quantum Effects in Enzyme Catalysis
4.3. Membrane Protein Dynamics
4.3.1. Lipid–Protein Interactions: Characterizing the Influence of the Membrane Environment
4.3.2. Conformational Changes in Transporters: Elucidating Alternating Access Mechanisms
4.3.3. GPCR Activation: Mapping the Energy Landscape of Receptor Activation
5. Integrating Complementary Techniques for Comprehensive Understanding of Protein Dynamics
5.1. Cryo-EM and MD Simulations: Synergy in Structural and Dynamic Studies
5.2. NMR Spectroscopy and Fluorescence Techniques: Complementary Insights into Protein Dynamics
5.3. Computational and Experimental Integration: A Holistic Approach to Protein Dynamics
5.4. Applications in Drug Discovery and Protein Engineering
6. Future Directions and Challenges
6.1. Limitations of Experimental Techniques for Studying Protein Dynamics
6.2. Limitations of Molecular Dynamics Simulations
6.3. Integration of Multi-Scale Approaches: Combining Atomistic Simulations with Coarse-Grained Models and Experimental Data to Bridge Timescales and Length Scales
6.4. In-Cell Dynamics: Developing Methods to Study Protein Motions in Their Native Cellular Environment
6.5. AI-Driven Discovery: Leveraging Machine Learning to Predict Functional Motions and Design Proteins with Specific Dynamic Properties
6.6. Dynamics in Complex Assemblies: Extending Our Understanding to Large Macromolecular Complexes and Cellular Machines
6.7. Linking Dynamics to Function: Developing Quantitative Frameworks to Relate Protein Motions to Biological Function and Disease States
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nam, K.; Wolf-Watz, M. Protein dynamics: The future is bright and complicated! Struct. Dyn. 2023, 10, 014301. [Google Scholar] [CrossRef]
- Jayaraman, V.; Toledo-Patino, S.; Noda-Garcia, L.; Laurino, P. Mechanisms of protein evolution. Protein Sci. 2022, 31, e4362. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef]
- Klyshko, E.; Kim, J.S.; McGough, L.; Valeeva, V.; Lee, E.; Ranganathan, R.; Rauscher, S. Functional protein dynamics in a crystal. Nat. Commun. 2024, 15, 3244. [Google Scholar] [CrossRef]
- Roca-Martinez, J.; Lazar, T.; Gavalda-Garcia, J.; Bickel, D.; Pancsa, R.; Dixit, B.; Tzavella, K.; Ramasamy, P.; Sanchez-Fornaris, M.; Grau, I.; et al. Challenges in describing the conformation and dynamics of proteins with ambiguous behavior. Front. Mol. Biosci. 2022, 9, 959956. [Google Scholar] [CrossRef]
- Ghosh, D.; Biswas, A.; Radhakrishna, M. Advanced computational approaches to understand protein aggregation. Biophys. Rev. 2024, 5, 021302. [Google Scholar] [CrossRef]
- Chua, E.Y.D.; Mendez, J.H.; Rapp, M.; Ilca, S.L.; Tan, Y.Z.; Maruthi, K.; Kuang, H.; Zimanyi, C.M.; Cheng, A.; Eng, E.T.; et al. Better, Faster, Cheaper: Recent Advances in Cryo-Electron Microscopy. Annu. Rev. Biochem. 2022, 91, 1–32. [Google Scholar] [CrossRef]
- Krieger, J.M.; Sorzano, C.O.S.; Carazo, J.M.; Bahar, I. Protein dynamics developments for the large scale and cryoEM: Case study of ProDy 2.0. Acta Crystallogr. D Struct. Biol. 2022, 78, 399–409. [Google Scholar] [CrossRef]
- Maeots, M.E.; Enchev, R.I. Structural dynamics: Review of time-resolved cryo-EM. Acta Crystallogr. D Struct. Biol. 2022, 78, 927–935. [Google Scholar] [CrossRef]
- Bongiovanni, G.; Harder, O.F.; Voss, J.M.; Drabbels, M.; Lorenz, U.J. Near-atomic resolution reconstructions from in situ revitrified cryo samples. Acta Crystallogr. D Struct. Biol. 2023, 79, 473–478. [Google Scholar] [CrossRef]
- Srajer, V.; Royer, W.E., Jr. Time-resolved x-ray crystallography of heme proteins. Methods Enzym. 2008, 437, 379–395. [Google Scholar] [CrossRef]
- Hekstra, D.R. Emerging Time-Resolved X-Ray Diffraction Approaches for Protein Dynamics. Annu. Rev. Biophys. 2023, 52, 255–274. [Google Scholar] [CrossRef]
- Wolff, A.M.; Nango, E.; Young, I.D.; Brewster, A.S.; Kubo, M.; Nomura, T.; Sugahara, M.; Owada, S.; Barad, B.A.; Ito, K.; et al. Mapping protein dynamics at high spatial resolution with temperature-jump X-ray crystallography. Nat. Chem. 2023, 15, 1549–1558. [Google Scholar] [CrossRef]
- Palmer, A.G., 3rd. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004, 104, 3623–3640. [Google Scholar] [CrossRef]
- Hellenkamp, B.; Schmid, S.; Doroshenko, O.; Opanasyuk, O.; Kuhnemuth, R.; Rezaei Adariani, S.; Ambrose, B.; Aznauryan, M.; Barth, A.; Birkedal, V.; et al. Precision and accuracy of single-molecule FRET measurements-a multi-laboratory benchmark study. Nat. Methods 2018, 15, 669–676. [Google Scholar] [CrossRef]
- Ando, T. High-speed atomic force microscopy and its future prospects. Biophys. Rev. 2018, 10, 285–292. [Google Scholar] [CrossRef]
- Lerner, E.; Cordes, T.; Ingargiola, A.; Alhadid, Y.; Chung, S.; Michalet, X.; Weiss, S. Toward dynamic structural biology: Two decades of single-molecule Forster resonance energy transfer. Science 2018, 359, eaan1133. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Barredo, P.A.; Balanay, M.P. Recent Advances in Molecular Dynamics Simulations of Tau Fibrils and Oligomers. Membranes 2023, 13, 277. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Grutsch, S.; Bruschweiler, S.; Tollinger, M. NMR Methods to Study Dynamic Allostery. PLoS Comput. Biol. 2016, 12, e1004620. [Google Scholar] [CrossRef]
- Tzeng, S.R.; Kalodimos, C.G. Protein dynamics and allostery: An NMR view. Curr. Opin. Struct. Biol. 2011, 21, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhou, H.X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515. [Google Scholar] [CrossRef]
- Schwartz, S.D. Protein Dynamics and Enzymatic Catalysis. J. Phys. Chem. B 2023, 127, 2649–2660. [Google Scholar] [CrossRef]
- Kohen, A. Role of dynamics in enzyme catalysis: Substantial versus semantic controversies. Acc. Chem. Res. 2015, 48, 466–473. [Google Scholar] [CrossRef]
- McGeagh, J.D.; Ranaghan, K.E.; Mulholland, A.J. Protein dynamics and enzyme catalysis: Insights from simulations. Biochim. Biophys. Acta 2011, 1814, 1077–1092. [Google Scholar] [CrossRef]
- Otten, R.; Liu, L.; Kenner, L.R.; Clarkson, M.W.; Mavor, D.; Tawfik, D.S.; Kern, D.; Fraser, J.S. Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun. 2018, 9, 1314. [Google Scholar] [CrossRef]
- Warshel, A.; Bora, R.P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 2016, 144, 180901. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef]
- Naudi-Fabra, S.; Blackledge, M.; Milles, S. Synergies of Single Molecule Fluorescence and NMR for the Study of Intrinsically Disordered Proteins. Biomolecules 2021, 12, 27. [Google Scholar] [CrossRef]
- van den Bedem, H.; Fraser, J.S. Integrative, dynamic structural biology at atomic resolution--it’s about time. Nat. Methods 2015, 12, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Burnley, B.T.; Afonine, P.V.; Adams, P.D.; Gros, P. Modelling dynamics in protein crystal structures by ensemble refinement. Elife 2012, 1, e00311. [Google Scholar] [CrossRef] [PubMed]
- Rout, M.P.; Sali, A. Principles for Integrative Structural Biology Studies. Cell 2019, 177, 1384–1403. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Wankowicz, S.A.; Ravikumar, A.; Sharma, S.; Riley, B.; Raju, A.; Hogan, D.W.; Flowers, J.; van den Bedem, H.; Keedy, D.A.; Fraser, J.S. Automated multiconformer model building for X-ray crystallography and cryo-EM. Elife 2024, 12, RP90606. [Google Scholar] [CrossRef]
- Verkhivker, G.M.; Agajanian, S.; Hu, G.; Tao, P. Allosteric Regulation at the Crossroads of New Technologies: Multiscale Modeling, Networks, and Machine Learning. Front. Mol. Biosci. 2020, 7, 136. [Google Scholar] [CrossRef]
- Govindaraj, R.G.; Thangapandian, S.; Schauperl, M.; Denny, R.A.; Diller, D.J. Recent applications of computational methods to allosteric drug discovery. Front. Mol. Biosci. 2022, 9, 1070328. [Google Scholar] [CrossRef]
- Hu, G.; Doruker, P.; Li, H.; Demet Akten, E. Editorial: Understanding Protein Dynamics, Binding and Allostery for Drug Design. Front. Mol. Biosci. 2021, 8, 681364. [Google Scholar] [CrossRef]
- Harder, O.F.; Barrass, S.V.; Drabbels, M.; Lorenz, U.J. Fast viral dynamics revealed by microsecond time-resolved cryo-EM. Nat. Commun. 2023, 14, 5649. [Google Scholar] [CrossRef]
- Amann, S.J.; Keihsler, D.; Bodrug, T.; Brown, N.G.; Haselbach, D. Frozen in time: Analyzing molecular dynamics with time-resolved cryo-EM. Structure 2023, 31, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Klebl, D.P.; Aspinall, L.; Muench, S.P. Time resolved applications for Cryo-EM; approaches, challenges and future directions. Curr. Opin. Struct. Biol. 2023, 83, 102696. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, W. Cryo-electron tomography: A long journey to the inner space of cells. Cell 2022, 185, 2649–2652. [Google Scholar] [CrossRef] [PubMed]
- Lucic, V.; Rigort, A.; Baumeister, W. Cryo-electron tomography: The challenge of doing structural biology in situ. J. Cell Biol. 2013, 202, 407–419. [Google Scholar] [CrossRef]
- Golding, C.G.; Lamboo, L.L.; Beniac, D.R.; Booth, T.F. The scanning electron microscope in microbiology and diagnosis of infectious disease. Sci. Rep. 2016, 6, 26516. [Google Scholar] [CrossRef]
- Bauerlein, F.J.B.; Baumeister, W. Towards Visual Proteomics at High Resolution. J. Mol. Biol. 2021, 433, 167187. [Google Scholar] [CrossRef]
- Guaita, M.; Watters, S.C.; Loerch, S. Recent advances and current trends in cryo-electron microscopy. Curr. Opin. Struct. Biol. 2022, 77, 102484. [Google Scholar] [CrossRef]
- Turk, M.; Baumeister, W. The promise and the challenges of cryo-electron tomography. FEBS Lett. 2020, 594, 3243–3261. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Oang, K.Y.; Kim, D.; Ihee, H. A comparative review of time-resolved x-ray and electron scattering to probe structural dynamics. Struct. Dyn. 2024, 11, 031301. [Google Scholar] [CrossRef]
- Schmidt, M. Practical considerations for the analysis of time-resolved x-ray data. Struct. Dyn. 2023, 10, 044303. [Google Scholar] [CrossRef]
- Thorne, R.E. Determining biomolecular structures near room temperature using X-ray crystallography: Concepts, methods and future optimization. Acta Crystallogr. D Struct. Biol. 2023, 79, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Hough, M.A.; Prischi, F.; Worrall, J.A.R. Perspective: Structure determination of protein-ligand complexes at room temperature using X-ray diffraction approaches. Front. Mol. Biosci. 2023, 10, 1113762. [Google Scholar] [CrossRef]
- Shi, D.; Nannenga, B.L.; Iadanza, M.G.; Gonen, T. Three-dimensional electron crystallography of protein microcrystals. Elife 2013, 2, e01345. [Google Scholar] [CrossRef]
- Gallenito, M.J.; Gonen, T. Studying membrane proteins with MicroED. Biochem. Soc. Trans. 2022, 50, 231–239. [Google Scholar] [CrossRef]
- Du, D.X.; Simjanoska, M.; Fitzpatrick, A.W.P. Four-dimensional microED of conformational dynamics in protein microcrystals on the femto-to-microsecond timescales. J. Struct. Biol. 2023, 215, 107941. [Google Scholar] [CrossRef] [PubMed]
- Danelius, E.; Patel, K.; Gonzalez, B.; Gonen, T. MicroED in drug discovery. Curr. Opin. Struct. Biol. 2023, 79, 102549. [Google Scholar] [CrossRef]
- Walinda, E.; Morimoto, D.; Sugase, K. Overview of Relaxation Dispersion NMR Spectroscopy to Study Protein Dynamics and Protein-Ligand Interactions. Curr. Protoc. Protein Sci. 2018, 92, e57. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. NMR spectroscopy, excited states and relevance to problems in cell biology—transient pre-nucleation tetramerization of huntingtin and insights into Huntington’s disease. J. Cell Sci. 2022, 135, jcs258695. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, P.; Lundstrom, P.; Kay, L.E. Relaxation dispersion NMR spectroscopy as a tool for detailed studies of protein folding. Biophys. J. 2009, 96, 2045–2054. [Google Scholar] [CrossRef]
- Dreydoppel, M.; Lichtenecker, R.J.; Akke, M.; Weininger, U. (1)H R(1rho) relaxation dispersion experiments in aromatic side chains. J. Biomol. NMR 2021, 75, 383–392. [Google Scholar] [CrossRef]
- Overbeck, J.H.; Kremer, W.; Sprangers, R. A suite of (19)F based relaxation dispersion experiments to assess biomolecular motions. J. Biomol. NMR 2020, 74, 753–766. [Google Scholar] [CrossRef]
- Xue, Y.; Kellogg, D.; Kimsey, I.J.; Sathyamoorthy, B.; Stein, Z.W.; McBrairty, M.; Al-Hashimi, H.M. Characterizing RNA Excited States Using NMR Relaxation Dispersion. Methods Enzym. 2015, 558, 39–73. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Hansen, D.F.; Kay, L.E. Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2008, 105, 11766–11771. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M.; Iwahara, J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009, 109, 4108–4139. [Google Scholar] [CrossRef] [PubMed]
- Clore, G.M. Practical Aspects of Paramagnetic Relaxation Enhancement in Biological Macromolecules. Methods Enzym. 2015, 564, 485–497. [Google Scholar] [CrossRef]
- Kocman, V.; Di Mauro, G.M.; Veglia, G.; Ramamoorthy, A. Use of paramagnetic systems to speed-up NMR data acquisition and for structural and dynamic studies. Solid. State Nucl. Magn. Reson. 2019, 102, 36–46. [Google Scholar] [CrossRef]
- Lenard, A.J.; Mulder, F.A.A.; Madl, T. Solvent paramagnetic relaxation enhancement as a versatile method for studying structure and dynamics of biomolecular systems. Prog. Nucl. Magn. Reson. Spectrosc. 2022, 132–133, 113–139. [Google Scholar] [CrossRef]
- Schlagnitweit, J.; Tang, M.; Baias, M.; Richardson, S.; Schantz, S.; Emsley, L. Nanostructure of Materials Determined by Relayed Paramagnetic Relaxation Enhancement. J. Am. Chem. Soc. 2015, 137, 12482–12485. [Google Scholar] [CrossRef] [PubMed]
- Bara-Estaun, A.; Harder, M.C.; Lyall, C.L.; Lowe, J.P.; Suturina, E.; Hintermair, U. Paramagnetic Relaxation Agents for Enhancing Temporal Resolution and Sensitivity in Multinuclear FlowNMR Spectroscopy. Chemistry 2023, 29, e202300215. [Google Scholar] [CrossRef]
- Swartjes, A.; White, P.B.; Bruekers, J.P.J.; Elemans, J.; Nolte, R.J.M. Paramagnetic relaxation enhancement NMR as a tool to probe guest binding and exchange in metallohosts. Nat. Commun. 2022, 13, 1846. [Google Scholar] [CrossRef]
- Chen, K.; Tjandra, N. The use of residual dipolar coupling in studying proteins by NMR. Top. Curr. Chem. 2012, 326, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Born, A.; Henen, M.A.; Nichols, P.J.; Vogeli, B. On the use of residual dipolar couplings in multi-state structure calculation of two-domain proteins. Magn. Reson. Lett. 2022, 2, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lemak, A.; Wu, B.; Yee, A.; Houliston, S.; Lee, H.W.; Gutmanas, A.; Fang, X.; Garcia, M.; Semesi, A.; Wang, Y.X.; et al. Structural characterization of a flexible two-domain protein in solution using small angle X-ray scattering and NMR data. Structure 2014, 22, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Traaseth, N.J.; Verardi, R.; Gustavsson, M.; Gao, J.; Veglia, G. Paramagnetic-based NMR restraints lift residual dipolar coupling degeneracy in multidomain detergent-solubilized membrane proteins. J. Am. Chem. Soc. 2011, 133, 2232–2241. [Google Scholar] [CrossRef]
- Poveda, A.; Fittolani, G.; Seeberger, P.H.; Delbianco, M.; Jimenez-Barbero, J. The Flexibility of Oligosaccharides Unveiled Through Residual Dipolar Coupling Analysis. Front. Mol. Biosci. 2021, 8, 784318. [Google Scholar] [CrossRef]
- Sasmal, D.K.; Pulido, L.E.; Kasal, S.; Huang, J. Single-molecule fluorescence resonance energy transfer in molecular biology. Nanoscale 2016, 8, 19928–19944. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Mazal, H.; Haran, G. Single-molecule FRET methods to study the dynamics of proteins at work. Curr. Opin. Biomed. Eng. 2019, 12, 8–17. [Google Scholar] [CrossRef]
- Meszaros, J.; Geggier, P.; Manning, J.J.; Asher, W.B.; Javitch, J.A. Methods for automating the analysis of live-cell single-molecule FRET data. Front. Cell Dev. Biol. 2023, 11, 1184077. [Google Scholar] [CrossRef]
- Agam, G.; Gebhardt, C.; Popara, M.; Machtel, R.; Folz, J.; Ambrose, B.; Chamachi, N.; Chung, S.Y.; Craggs, T.D.; de Boer, M.; et al. Reliability and accuracy of single-molecule FRET studies for characterization of structural dynamics and distances in proteins. Nat. Methods 2023, 20, 523–535. [Google Scholar] [CrossRef]
- Yu, L.; Lei, Y.; Ma, Y.; Liu, M.; Zheng, J.; Dan, D.; Gao, P. A Comprehensive Review of Fluorescence Correlation Spectroscopy. Front. Phys. 2021, 9, 644450. [Google Scholar] [CrossRef]
- Elson, E.L. Fluorescence correlation spectroscopy: Past, present, future. Biophys. J. 2011, 101, 2855–2870. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, H.; Jian, L.; Ding, B.; Huang, K.; Zhang, W.; Xiao, Q.; Huang, S. Principles of fluorescence correlation spectroscopy applied to studies of biomolecular liquid-liquid phase separation. Biophys. Rep. 2022, 8, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Jazani, S.; Sgouralis, I.; Shafraz, O.M.; Levitus, M.; Sivasankar, S.; Presse, S. An alternative framework for fluorescence correlation spectroscopy. Nat. Commun. 2019, 10, 3662. [Google Scholar] [CrossRef]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence lifetime imaging microscopy: Fundamentals and advances in instrumentation, analysis, and applications. J. Biomed. Opt. 2020, 25, 1–43. [Google Scholar] [CrossRef]
- Kaufmann, T.; Herbert, S.; Hackl, B.; Besold, J.M.; Schramek, C.; Gotzmann, J.; Elsayad, K.; Slade, D. Direct measurement of protein-protein interactions by FLIM-FRET at UV laser-induced DNA damage sites in living cells. Nucleic Acids Res. 2020, 48, e122. [Google Scholar] [CrossRef]
- Datta, R.; Gillette, A.; Stefely, M.; Skala, M.C. Recent innovations in fluorescence lifetime imaging microscopy for biology and medicine. J. Biomed. Opt. 2021, 26, 070603. [Google Scholar] [CrossRef]
- Edeling, W.; Vassaux, M.; Yang, Y.; Wan, S.; Guillas, S.; Coveney, P.V. Global ranking of the sensitivity of interaction potential contributions within classical molecular dynamics force fields. npj Comput. Mater. 2024, 10, 87. [Google Scholar] [CrossRef]
- González, M.A. Force fields and molecular dynamics simulations. École Thématique De. La. Société Française De. La. Neutron. 2011, 12, 169–200. [Google Scholar] [CrossRef]
- de Oliveira, C.A.; Hamelberg, D.; McCammon, J.A. Coupling Accelerated Molecular Dynamics Methods with Thermodynamic Integration Simulations. J. Chem. Theory Comput. 2008, 4, 1516–1525. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, S.; Sauceda, H.E.; Muller, K.R.; Tkatchenko, A. Towards exact molecular dynamics simulations with machine-learned force fields. Nat. Commun. 2018, 9, 3887. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Guvench, O.; MacKerell, A.D., Jr. Molecular mechanics. Curr. Pharm. Des. 2014, 20, 3281–3292. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Hospital, A.; Goni, J.R.; Orozco, M.; Gelpi, J.L. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. 2015, 8, 37–47. [Google Scholar] [CrossRef]
- Mouvet, F.; Villard, J.; Bolnykh, V.; Rothlisberger, U. Recent Advances in First-Principles Based Molecular Dynamics. Acc. Chem. Res. 2022, 55, 221–230. [Google Scholar] [CrossRef]
- Bhati, A.P.; Hoti, A.; Potterton, A.; Bieniek, M.K.; Coveney, P.V. Long Time Scale Ensemble Methods in Molecular Dynamics: Ligand-Protein Interactions and Allostery in SARS-CoV-2 Targets. J. Chem. Theory Comput. 2023, 19, 3359–3378. [Google Scholar] [CrossRef] [PubMed]
- Henkelman, G.; Jónsson, H.; Lelièvre, T.; Mousseau, N.; Voter, A.F. Long-Timescale Simulations: Challenges, Pitfalls, Best Practices, for Development and Applications. In Handbook of Materials Modeling; Springer: Berlin/Heidelberg, Germany, 2018; pp. 825–834. [Google Scholar]
- Clayton, J.; Baweja, L.; Wereszczynski, J. Peptide Dynamics and Metadynamics: Leveraging Enhanced Sampling Molecular Dynamics to Robustly Model Long-Timescale Transitions. Methods Mol. Biol. 2022, 2405, 151–167. [Google Scholar] [CrossRef]
- Ray, D.; Parrinello, M. Kinetics from Metadynamics: Principles, Applications, and Outlook. J. Chem. Theory Comput. 2023, 19, 5649–5670. [Google Scholar] [CrossRef]
- Bernardi, R.C.; Melo, M.C.R.; Schulten, K. Enhanced sampling techniques in molecular dynamics simulations of biological systems. Biochim. Biophys. Acta 2015, 1850, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Wei, G.; Ma, B.; Nussinov, R. Replica Exchange Molecular Dynamics: A Practical Application Protocol with Solutions to Common Problems and a Peptide Aggregation and Self-Assembly Example. Methods Mol. Biol. 2018, 1777, 101–119. [Google Scholar] [CrossRef]
- Comer, J.; Gumbart, J.C.; Henin, J.; Lelievre, T.; Pohorille, A.; Chipot, C. The adaptive biasing force method: Everything you always wanted to know but were afraid to ask. J. Phys. Chem. B 2015, 119, 1129–1151. [Google Scholar] [CrossRef] [PubMed]
- Lesage, A.; Lelievre, T.; Stoltz, G.; Henin, J. Smoothed Biasing Forces Yield Unbiased Free Energies with the Extended-System Adaptive Biasing Force Method. J. Phys. Chem. B 2017, 121, 3676–3685. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.E.; Barethiya, S.; Nordquist, E.; Chen, J. Machine Learning Generation of Dynamic Protein Conformational Ensembles. Molecules 2023, 28, 4047. [Google Scholar] [CrossRef]
- Janson, G.; Valdes-Garcia, G.; Heo, L.; Feig, M. Direct generation of protein conformational ensembles via machine learning. Nat. Commun. 2023, 14, 774. [Google Scholar] [CrossRef]
- Mehdi, S.; Smith, Z.; Herron, L.; Zou, Z.; Tiwary, P. Enhanced Sampling with Machine Learning. Annu. Rev. Phys. Chem. 2024, 75, 347–370. [Google Scholar] [CrossRef]
- Majewski, M.; Perez, A.; Tholke, P.; Doerr, S.; Charron, N.E.; Giorgino, T.; Husic, B.E.; Clementi, C.; Noe, F.; De Fabritiis, G. Machine learning coarse-grained potentials of protein thermodynamics. Nat. Commun. 2023, 14, 5739. [Google Scholar] [CrossRef]
- Wang, W.; Gómez-Bombarelli, R. Coarse-graining auto-encoders for molecular dynamics. npj Comput. Mater. 2019, 5, 125. [Google Scholar] [CrossRef]
- Singh, N.; Li, W. Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications. Int. J. Mol. Sci. 2019, 20, 3774. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [PubMed]
- Noid, W.G. Perspective: Coarse-grained models for biomolecular systems. J. Chem. Phys. 2013, 139, 090901. [Google Scholar] [CrossRef] [PubMed]
- Liwo, A.; Czaplewski, C.; Sieradzan, A.K.; Lipska, A.G.; Samsonov, S.A.; Murarka, R.K. Theory and Practice of Coarse-Grained Molecular Dynamics of Biologically Important Systems. Biomolecules 2021, 11, 1347. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M.G.; Voth, G.A. Coarse-graining methods for computational biology. Annu. Rev. Biophys. 2013, 42, 73–93. [Google Scholar] [CrossRef]
- Lee, M. Recent Advances in Deep Learning for Protein-Protein Interaction Analysis: A Comprehensive Review. Molecules 2023, 28, 5169. [Google Scholar] [CrossRef]
- Bhakat, S. Collective variable discovery in the age of machine learning: Reality, hype and everything in between. RSC Adv. 2022, 12, 25010–25024. [Google Scholar] [CrossRef]
- Saharkhiz, S.; Mostafavi, M.; Birashk, A.; Karimian, S.; Khalilollah, S.; Jaferian, S.; Yazdani, Y.; Alipourfard, I.; Huh, Y.S.; Farani, M.R.; et al. The State-of-the-Art Overview to Application of Deep Learning in Accurate Protein Design and Structure Prediction. Top Curr. Chem. 2024, 382, 23. [Google Scholar] [CrossRef] [PubMed]
- Elia Venanzi, N.A.; Basciu, A.; Vargiu, A.V.; Kiparissides, A.; Dalby, P.A.; Dikicioglu, D. Machine Learning Integrating Protein Structure, Sequence, and Dynamics to Predict the Enzyme Activity of Bovine Enterokinase Variants. J. Chem. Inf. Model. 2024, 64, 2681–2694. [Google Scholar] [CrossRef]
- Langmead, C.J. Generative models of conformational dynamics. Adv. Exp. Med. Biol. 2014, 805, 87–105. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Z.; Tong, H.; Lu, Z.; Zhang, N.; Wei, T.; Chen, H.F. Phanto-IDP: Compact model for precise intrinsically disordered protein backbone generation and enhanced sampling. Brief. Bioinform. 2023, 25, bbad429. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Shin, K.; Tran, D.P.; Takemura, K.; Kitao, A.; Terayama, K.; Tsuda, K. Enhancing Biomolecular Sampling with Reinforcement Learning: A Tree Search Molecular Dynamics Simulation Method. ACS Omega 2019, 4, 13853–13862. [Google Scholar] [CrossRef] [PubMed]
- Noe, F.; De Fabritiis, G.; Clementi, C. Machine learning for protein folding and dynamics. Curr. Opin. Struct. Biol. 2020, 60, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, H.; E, W. Reinforced dynamics for enhanced sampling in large atomic and molecular systems. J. Chem. Phys. 2018, 148, 124113. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sidky, H.; Ferguson, A.L. Nonlinear discovery of slow molecular modes using state-free reversible VAMPnets. J. Chem. Phys. 2019, 150, 214114. [Google Scholar] [CrossRef] [PubMed]
- Eaton, W.A. Modern Kinetics and Mechanism of Protein Folding: A Retrospective. J. Phys. Chem. B 2021, 125, 3452–3467. [Google Scholar] [CrossRef]
- Chong, S.H.; Ham, S. Folding Free Energy Landscape of Ordered and Intrinsically Disordered Proteins. Sci. Rep. 2019, 9, 14927. [Google Scholar] [CrossRef]
- Wolynes, P.G. Evolution, energy landscapes and the paradoxes of protein folding. Biochimie 2015, 119, 218–230. [Google Scholar] [CrossRef]
- Nymeyer, H.; Garcia, A.E.; Onuchic, J.N. Folding funnels and frustration in off-lattice minimalist protein landscapes. Proc. Natl. Acad. Sci. USA 1998, 95, 5921–5928. [Google Scholar] [CrossRef]
- Ma, B.; Kumar, S.; Tsai, C.J.; Nussinov, R. Folding funnels and binding mechanisms. Protein Eng. 1999, 12, 713–720. [Google Scholar] [CrossRef]
- Tsai, C.J.; Ma, B.; Nussinov, R. Folding and binding cascades: Shifts in energy landscapes. Proc. Natl. Acad. Sci. USA 1999, 96, 9970–9972. [Google Scholar] [CrossRef]
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins 1995, 21, 167–195. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Nagarajaram, H.A. Intrinsically Disordered Proteins: An Overview. Int. J. Mol. Sci. 2022, 23, 14050. [Google Scholar] [CrossRef]
- DeForte, S.; Uversky, V.N. Not an exception to the rule: The functional significance of intrinsically disordered protein regions in enzymes. Mol. Biosyst. 2017, 13, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Holehouse, A.S.; Kragelund, B.B. The molecular basis for cellular function of intrinsically disordered protein regions. Nat. Rev. Mol. Cell Biol. 2024, 25, 187–211. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef]
- Schuler, B.; Soranno, A.; Hofmann, H.; Nettels, D. Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys. 2016, 45, 207–231. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins and Their “Mysterious” (Meta)Physics. Front. Phys. 2019, 7, 10. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Liberek, K.; Lewandowska, A.; Zietkiewicz, S. Chaperones in control of protein disaggregation. EMBO J. 2008, 27, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Marzano, N.R.; Paudel, B.P.; van Oijen, A.M.; Ecroyd, H. Real-time single-molecule observation of chaperone-assisted protein folding. Sci. Adv. 2022, 8, eadd0922. [Google Scholar] [CrossRef]
- Horowitz, S.; Salmon, L.; Koldewey, P.; Ahlstrom, L.S.; Martin, R.; Quan, S.; Afonine, P.V.; van den Bedem, H.; Wang, L.; Xu, Q.; et al. Visualizing chaperone-assisted protein folding. Nat. Struct. Mol. Biol. 2016, 23, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, Z.; Cheng, K.J.; Shukla, D. Reinforcement Learning Based Adaptive Sampling: REAPing Rewards by Exploring Protein Conformational Landscapes. J. Phys. Chem. B 2018, 122, 8386–8395. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef]
- Tsutsui, Y.; Wintrode, P.L. Hydrogen/deuterium exchange-mass spectrometry: A powerful tool for probing protein structure, dynamics and interactions. Curr. Med. Chem. 2007, 14, 2344–2358. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef]
- Nussinov, R.; Ma, B.; Tsai, C.J. Multiple conformational selection and induced fit events take place in allosteric propagation. Biophys. Chem. 2014, 186, 22–30. [Google Scholar] [CrossRef]
- Paul, F.; Weikl, T.R. How to Distinguish Conformational Selection and Induced Fit Based on Chemical Relaxation Rates. PLoS Comput. Biol. 2016, 12, e1005067. [Google Scholar] [CrossRef]
- Morando, M.A.; Saladino, G.; D’Amelio, N.; Pucheta-Martinez, E.; Lovera, S.; Lelli, M.; Lopez-Mendez, B.; Marenchino, M.; Campos-Olivas, R.; Gervasio, F.L. Conformational Selection and Induced Fit Mechanisms in the Binding of an Anticancer Drug to the c-Src Kinase. Sci. Rep. 2016, 6, 24439. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.; Shao, Y.; Major, D.T.; Wolf-Watz, M. Perspectives on Computational Enzyme Modeling: From Mechanisms to Design and Drug Development. ACS Omega 2024, 9, 7393–7412. [Google Scholar] [CrossRef]
- Wlodarski, T.; Zagrovic, B. Conformational selection and induced fit mechanism underlie specificity in noncovalent interactions with ubiquitin. Proc. Natl. Acad. Sci. USA 2009, 106, 19346–19351. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilser, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef]
- Kern, D.; Zuiderweg, E.R. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 2003, 13, 748–757. [Google Scholar] [CrossRef]
- Tsai, C.J.; Nussinov, R. A unified view of “how allostery works”. PLoS Comput. Biol. 2014, 10, e1003394. [Google Scholar] [CrossRef]
- Klinman, J.P.; Kohen, A. Hydrogen tunneling links protein dynamics to enzyme catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. [Google Scholar] [CrossRef]
- Schramm, V.L.; Schwartz, S.D. Promoting Vibrations and the Function of Enzymes. Emerging Theoretical and Experimental Convergence. Biochemistry 2018, 57, 3299–3308. [Google Scholar] [CrossRef]
- Chalopin, Y.; Piazza, F.; Mayboroda, S.; Weisbuch, C.; Filoche, M. Universality of fold-encoded localized vibrations in enzymes. Sci. Rep. 2019, 9, 12835. [Google Scholar] [CrossRef]
- Sutcliffe, M.J.; Scrutton, N.S. Enzymology takes a quantum leap forward. Philos. Trans. A Math. Phys. Eng. Sci. 2000, 358, 367–386. [Google Scholar] [CrossRef]
- Yang, Z.; Mehmood, R.; Wang, M.; Qi, H.W.; Steeves, A.H.; Kulik, H.J. Revealing quantum mechanical effects in enzyme catalysis with large-scale electronic structure simulation. React. Chem. Eng. 2019, 4, 298–315. [Google Scholar] [CrossRef] [PubMed]
- Corradi, V.; Sejdiu, B.I.; Mesa-Galloso, H.; Abdizadeh, H.; Noskov, S.Y.; Marrink, S.J.; Tieleman, D.P. Emerging Diversity in Lipid-Protein Interactions. Chem. Rev. 2019, 119, 5775–5848. [Google Scholar] [CrossRef] [PubMed]
- Tieleman, D.P.; Sejdiu, B.I.; Cino, E.A.; Smith, P.; Barreto-Ojeda, E.; Khan, H.M.; Corradi, V. Insights into lipid-protein interactions from computer simulations. Biophys. Rev. 2021, 13, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Thienpont, B.; Sapuru, V.; Hite, R.K.; Dittman, J.S.; Sturgis, J.N.; Scheuring, S. Membrane-mediated protein interactions drive membrane protein organization. Nat. Commun. 2022, 13, 7373. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.P.; Jiang, T.; Sun, C.; Lihan, M.; Pant, S.; Mahinthichaichan, P.; Trifan, A.; Tajkhorshid, E. Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation. Chem. Rev. 2019, 119, 6086–6161. [Google Scholar] [CrossRef]
- Gu, R.X.; de Groot, B.L. Lipid-protein interactions modulate the conformational equilibrium of a potassium channel. Nat. Commun. 2020, 11, 2162. [Google Scholar] [CrossRef]
- Marinelli, F.; Faraldo-Gomez, J.D. Conformational free-energy landscapes of a Na(+)/Ca(2+) exchanger explain its alternating-access mechanism and functional specificity. Proc. Natl. Acad. Sci. USA 2024, 121, e2318009121. [Google Scholar] [CrossRef]
- Weyand, S.; Shimamura, T.; Beckstein, O.; Sansom, M.S.; Iwata, S.; Henderson, P.J.; Cameron, A.D. The alternating access mechanism of transport as observed in the sodium-hydantoin transporter Mhp1. J. Synchrotron Radiat. 2011, 18, 20–23. [Google Scholar] [CrossRef]
- Del Alamo, D.; Sala, D.; McHaourab, H.S.; Meiler, J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. Elife 2022, 11, e75751. [Google Scholar] [CrossRef] [PubMed]
- Badiee, S.A.; Isu, U.H.; Khodadadi, E.; Moradi, M. The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs. Membranes 2023, 13, 568. [Google Scholar] [CrossRef]
- Deupi, X.; Kobilka, B.K. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology 2010, 25, 293–303. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 2021, 12, 4721. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. Elife 2019, 8, e50279. [Google Scholar] [CrossRef] [PubMed]
- Alenghat, F.J.; Golan, D.E. Membrane protein dynamics and functional implications in mammalian cells. Curr. Top. Membr. 2013, 72, 89–120. [Google Scholar] [CrossRef]
- Hauser, A.S.; Kooistra, A.J.; Munk, C.; Heydenreich, F.M.; Veprintsev, D.B.; Bouvier, M.; Babu, M.M.; Gloriam, D.E. GPCR activation mechanisms across classes and macro/microscales. Nat. Struct. Mol. Biol. 2021, 28, 879–888. [Google Scholar] [CrossRef]
- Fleetwood, O.; Matricon, P.; Carlsson, J.; Delemotte, L. Energy Landscapes Reveal Agonist Control of G Protein-Coupled Receptor Activation via Microswitches. Biochemistry 2020, 59, 880–891. [Google Scholar] [CrossRef]
- Poudel, H.; Wales, D.J.; Leitner, D.M. Vibrational Energy Landscapes and Energy Flow in GPCRs. J. Phys. Chem. B 2024, 128, 7568–7576. [Google Scholar] [CrossRef]
- Bock, L.V.; Igaev, M.; Grubmuller, H. Single-particle Cryo-EM and molecular dynamics simulations: A perfect match. Curr. Opin. Struct. Biol. 2024, 86, 102825. [Google Scholar] [CrossRef]
- Vant, J.W.; Sarkar, D.; Nguyen, J.; Baker, A.T.; Vermaas, J.V.; Singharoy, A. Exploring cryo-electron microscopy with molecular dynamics. Biochem. Soc. Trans. 2022, 50, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Mockel, C.; Kubiak, J.; Schillinger, O.; Kuhnemuth, R.; Della Corte, D.; Schroder, G.F.; Willbold, D.; Strodel, B.; Seidel, C.A.M.; Neudecker, P. Integrated NMR, Fluorescence, and Molecular Dynamics Benchmark Study of Protein Mechanics and Hydrodynamics. J. Phys. Chem. B 2019, 123, 1453–1480. [Google Scholar] [CrossRef] [PubMed]
- Kovermann, M.; Rogne, P.; Wolf-Watz, M. Protein dynamics and function from solution state NMR spectroscopy. Q. Rev. Biophys. 2016, 49, e6. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Stark, H. Prospects and Limitations of High-Resolution Single-Particle Cryo-Electron Microscopy. Annu Rev Biophys 2023, 52, 391–411. [Google Scholar] [CrossRef]
- Hylton, R.K.; Swulius, M.T. Challenges and triumphs in cryo-electron tomography. iScience 2021, 24, 102959. [Google Scholar] [CrossRef]
- Wilson, M.A. Mapping Enzyme Landscapes by Time-Resolved Crystallography with Synchrotron and X-ray Free Electron Laser Light. Annu. Rev. Biophys. 2022, 51, 79–98. [Google Scholar] [CrossRef]
- Toke, O.; Batta, G. Dynamic Structures of Bioactive Proteins as Determined by Nuclear Magnetic Resonance. Int. J. Mol. Sci. 2023, 25, 295. [Google Scholar] [CrossRef]
- Dos Santos Rodrigues, F.H.; Delgado, G.G.; Santana da Costa, T.; Tasic, L. Applications of fluorescence spectroscopy in protein conformational changes and intermolecular contacts. BBA Adv. 2023, 3, 100091. [Google Scholar] [CrossRef]
- Raghuraman, H.; Chatterjee, S.; Das, A. Site-Directed Fluorescence Approaches for Dynamic Structural Biology of Membrane Peptides and Proteins. Front. Mol. Biosci. 2019, 6, 96. [Google Scholar] [CrossRef]
- Ormeno, F.; General, I.J. Convergence and equilibrium in molecular dynamics simulations. Commun. Chem. 2024, 7, 26. [Google Scholar] [CrossRef]
- Racz, A.; Mihalovits, L.M.; Bajusz, D.; Heberger, K.; Miranda-Quintana, R.A. Molecular Dynamics Simulations and Diversity Selection by Extended Continuous Similarity Indices. J. Chem. Inf. Model. 2022, 62, 3415–3425. [Google Scholar] [CrossRef] [PubMed]
- Janakaloti Narayanareddy, B.R.; Allipeta, N.R.; Allard, J.; Gross, S.P. A new method to experimentally quantify dynamics of initial protein-protein interactions. Commun. Biol. 2024, 7, 311. [Google Scholar] [CrossRef]
- May, A.; Pool, R.; van Dijk, E.; Bijlard, J.; Abeln, S.; Heringa, J.; Feenstra, K.A. Coarse-grained versus atomistic simulations: Realistic interaction free energies for real proteins. Bioinformatics 2014, 30, 326–334. [Google Scholar] [CrossRef] [PubMed]
- van der Kamp, M.W.; Shaw, K.E.; Woods, C.J.; Mulholland, A.J. Biomolecular simulation and modelling: Status, progress and prospects. J. R. Soc. Interface 2008, 5 (Suppl. 3), S173–S190. [Google Scholar] [CrossRef]
- Lapatas, V.; Stefanidakis, M.; Jimenez, R.C.; Via, A.; Schneider, M.V. Data integration in biological research: An overview. J. Biol. Res. 2015, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Walpole, J.; Papin, J.A.; Peirce, S.M. Multiscale computational models of complex biological systems. Annu. Rev. Biomed. Eng. 2013, 15, 137–154. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Snapp, E.; Kenworthy, A. Studying protein dynamics in living cells. Nat. Rev. Mol. Cell Biol. 2001, 2, 444–456. [Google Scholar] [CrossRef]
- Zalejski, J.; Sun, J.; Sharma, A. Unravelling the Mystery inside Cells by Using Single-Molecule Fluorescence Imaging. J. Imaging 2023, 9, 192. [Google Scholar] [CrossRef]
- Feng, R.; Gruebele, M.; Davis, C.M. Quantifying protein dynamics and stability in a living organism. Nat. Commun. 2019, 10, 1179. [Google Scholar] [CrossRef]
- Audagnotto, M.; Czechtizky, W.; De Maria, L.; Kack, H.; Papoian, G.; Tornberg, L.; Tyrchan, C.; Ulander, J. Machine learning/molecular dynamic protein structure prediction approach to investigate the protein conformational ensemble. Sci. Rep. 2022, 12, 10018. [Google Scholar] [CrossRef]
- Ficner, R. Highlight: Integrative structural biology of dynamic macromolecular assemblies. Biol. Chem. 2023, 404, 739. [Google Scholar] [CrossRef] [PubMed]
- Pak, A.J.; Voth, G.A. Advances in coarse-grained modeling of macromolecular complexes. Curr. Opin. Struct. Biol. 2018, 52, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Monachino, E.; Spenkelink, L.M.; van Oijen, A.M. Watching cellular machinery in action, one molecule at a time. J. Cell Biol. 2017, 216, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Prabantu, V.M.; Naveenkumar, N.; Srinivasan, N. Influence of Disease-Causing Mutations on Protein Structural Networks. Front. Mol. Biosci. 2020, 7, 620554. [Google Scholar] [CrossRef]
- Yang, L.Q.; Sang, P.; Tao, Y.; Fu, Y.X.; Zhang, K.Q.; Xie, Y.H.; Liu, S.Q. Protein dynamics and motions in relation to their functions: Several case studies and the underlying mechanisms. J. Biomol. Struct. Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef]
- Pacini, L.; Dorantes-Gilardi, R.; Vuillon, L.; Lesieur, C. Mapping Function from Dynamics: Future Challenges for Network-Based Models of Protein Structures. Front. Mol. Biosci. 2021, 8, 744646. [Google Scholar] [CrossRef]
- Ruiz, C.; Zitnik, M.; Leskovec, J. Identification of disease treatment mechanisms through the multiscale interactome. Nat. Commun. 2021, 12, 1796. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Son, A.; Kim, W.; Park, J.; Lee, W.; Lee, Y.; Choi, S.; Kim, H. Utilizing Molecular Dynamics Simulations, Machine Learning, Cryo-EM, and NMR Spectroscopy to Predict and Validate Protein Dynamics. Int. J. Mol. Sci. 2024, 25, 9725. https://doi.org/10.3390/ijms25179725
Son A, Kim W, Park J, Lee W, Lee Y, Choi S, Kim H. Utilizing Molecular Dynamics Simulations, Machine Learning, Cryo-EM, and NMR Spectroscopy to Predict and Validate Protein Dynamics. International Journal of Molecular Sciences. 2024; 25(17):9725. https://doi.org/10.3390/ijms25179725
Chicago/Turabian StyleSon, Ahrum, Woojin Kim, Jongham Park, Wonseok Lee, Yerim Lee, Seongyun Choi, and Hyunsoo Kim. 2024. "Utilizing Molecular Dynamics Simulations, Machine Learning, Cryo-EM, and NMR Spectroscopy to Predict and Validate Protein Dynamics" International Journal of Molecular Sciences 25, no. 17: 9725. https://doi.org/10.3390/ijms25179725