
Synthesis of Ethyl Pyrimidine-Quinolincarboxylates Selected from Virtual Screening as Enhanced Lactate Dehydrogenase (LDH) Inhibitors


Abstract
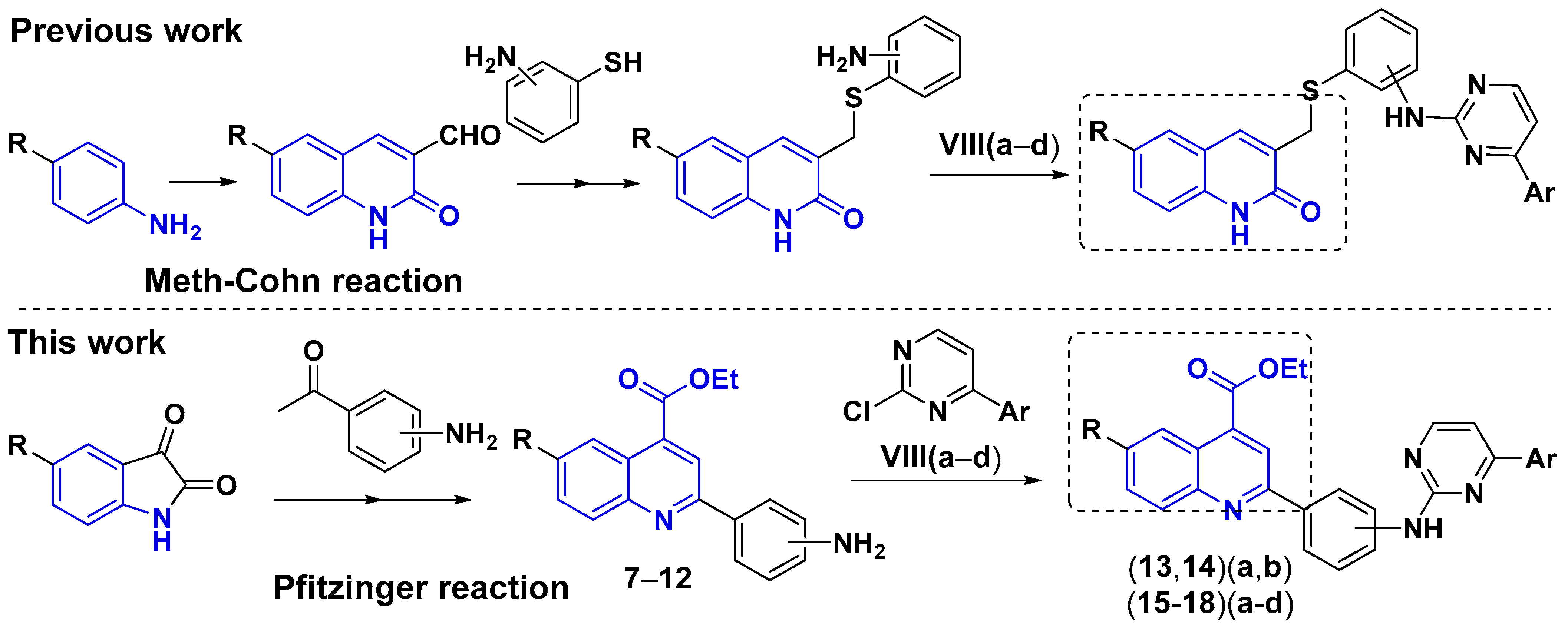
1. Introduction
2. Results and Discussion
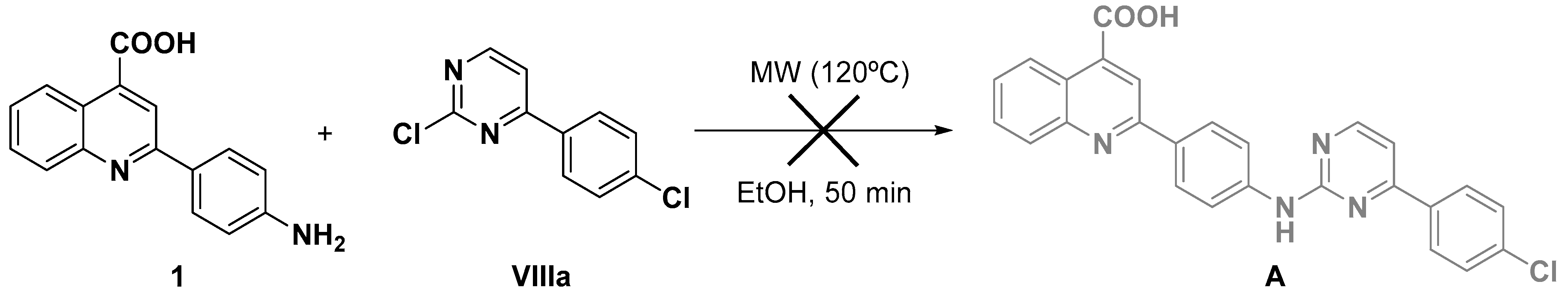
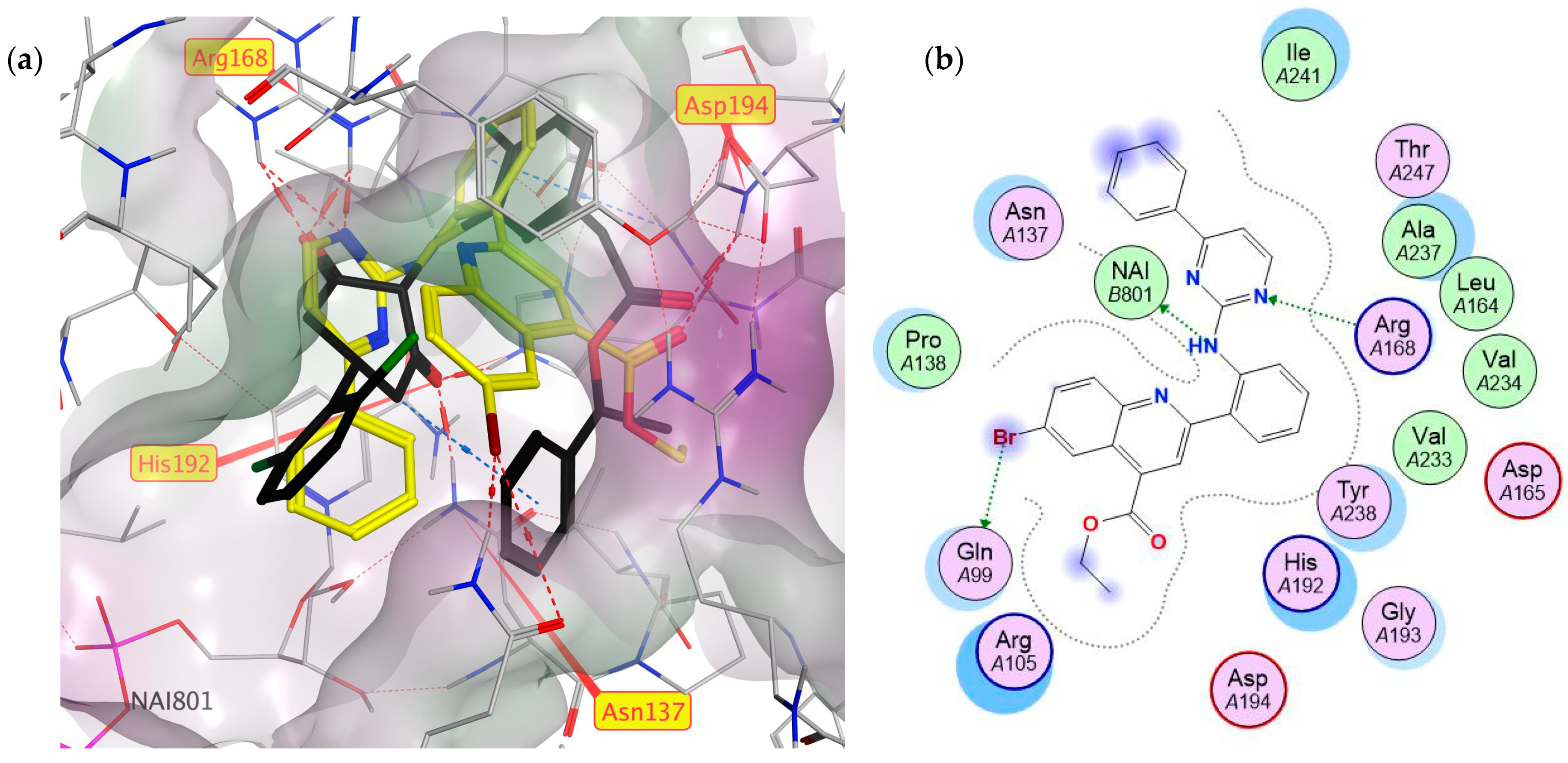
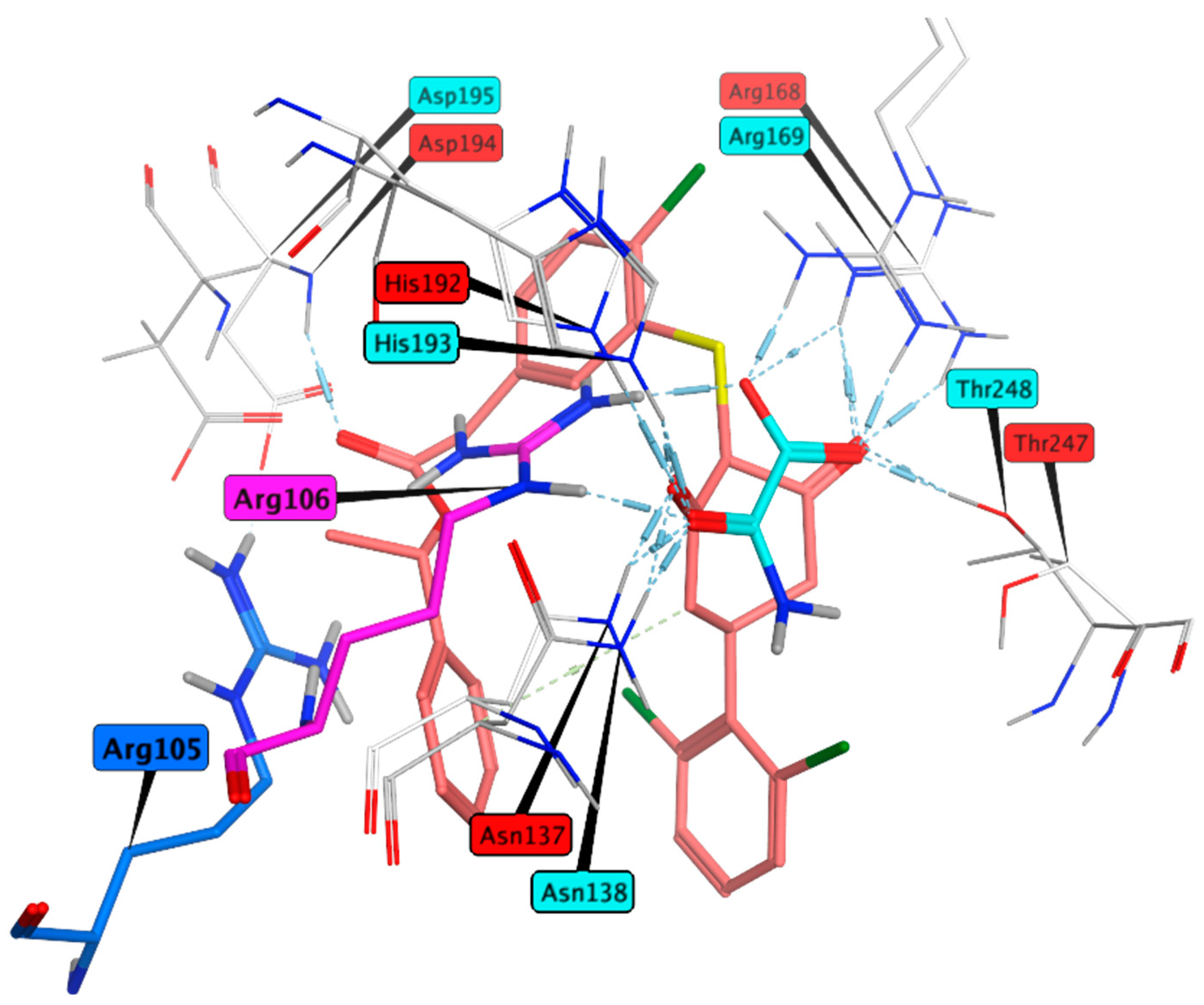
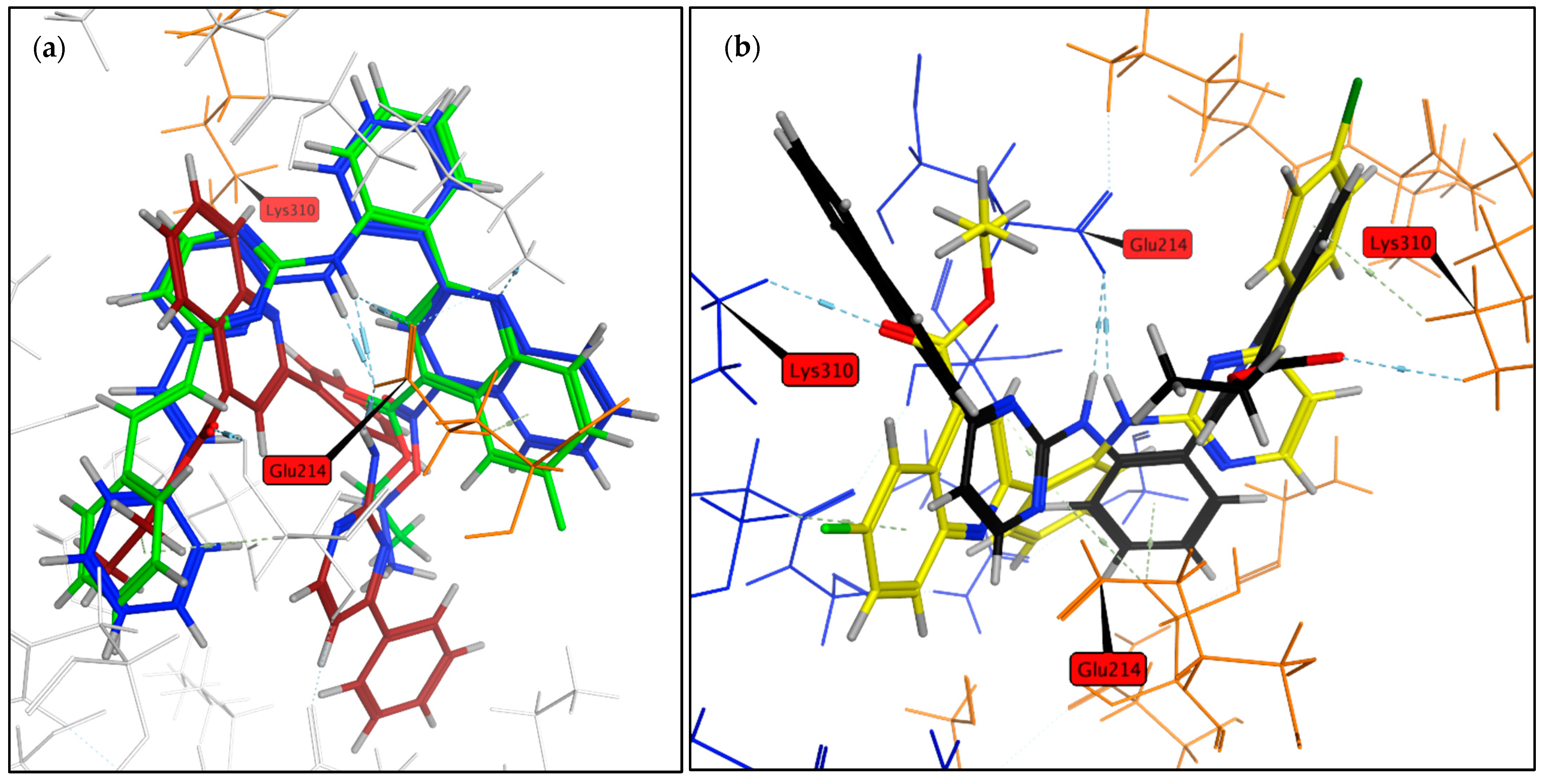
2.1. Virtual Screening
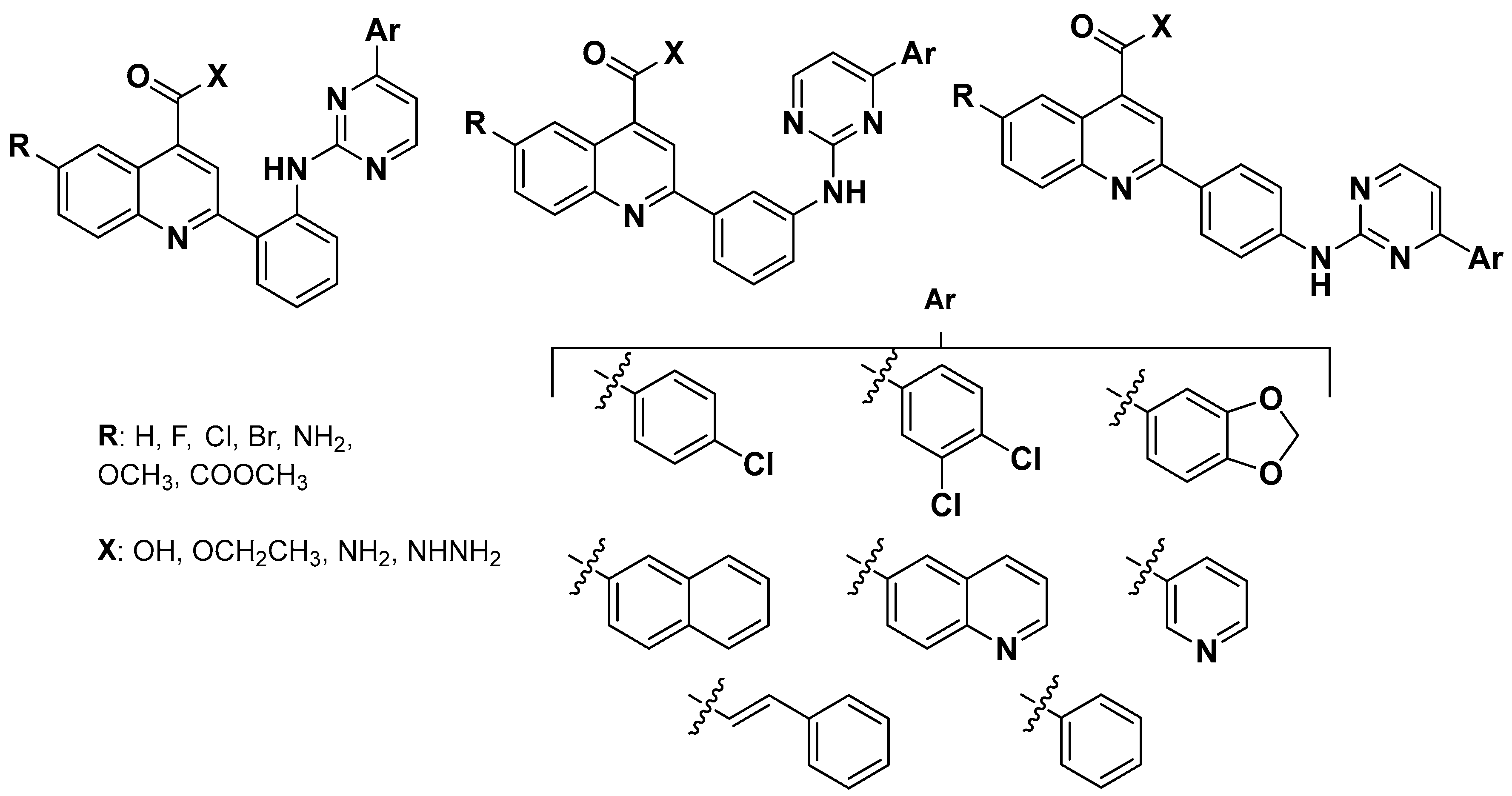
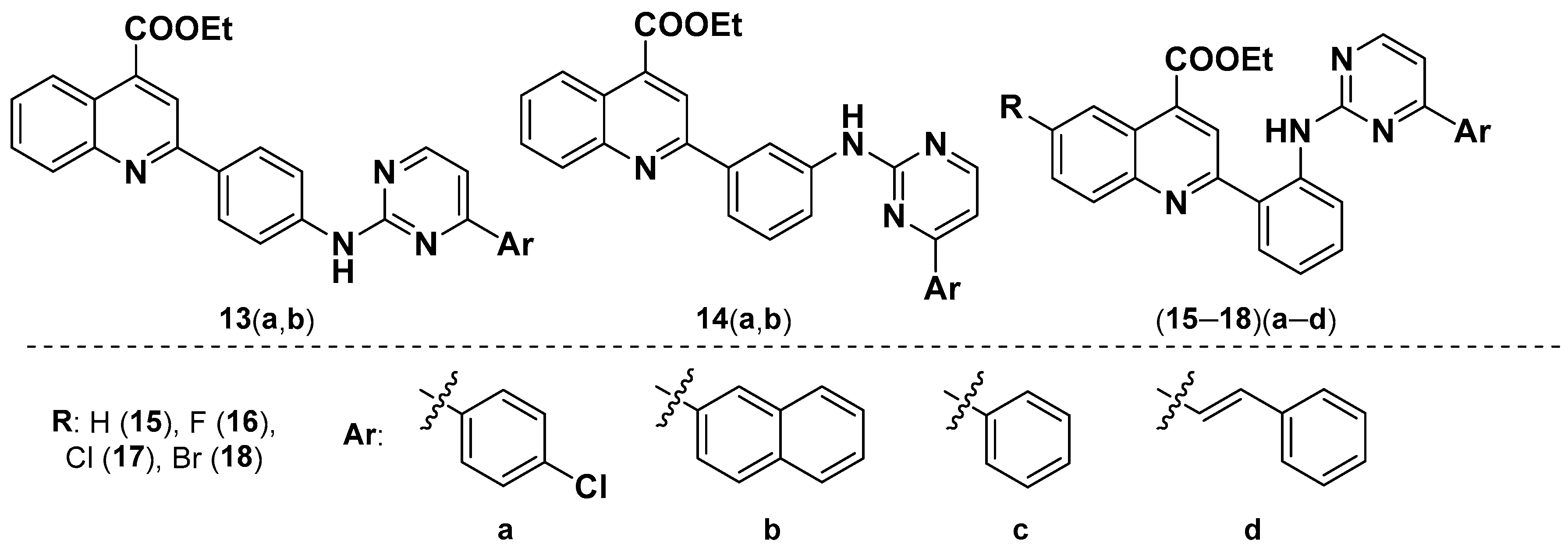
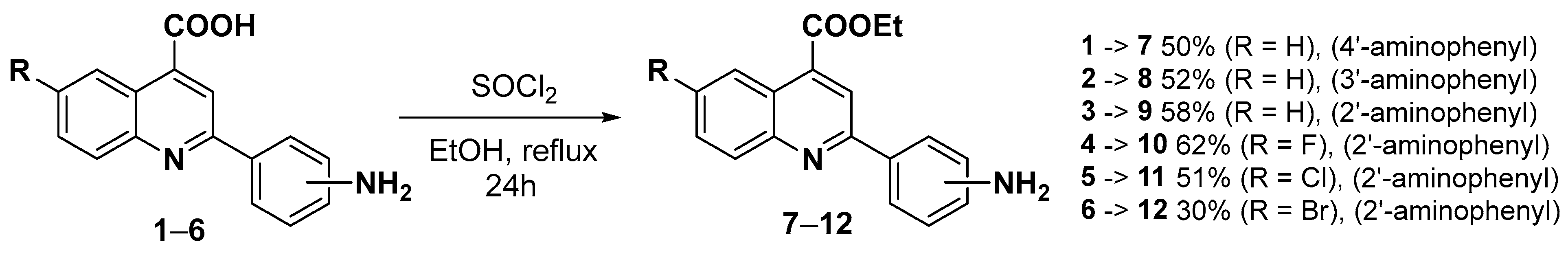
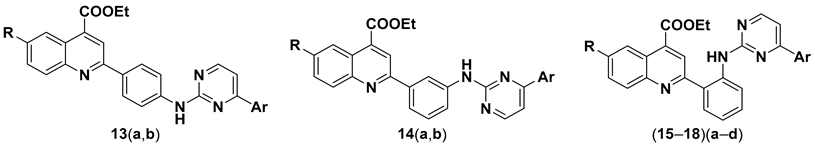
2.2. Chemistry
2.3. hLDH Inhibitory Assays and Preliminary Structure-Activity Relationship
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of 2-Aminophenylquinoline-4-carboxylic Acids (1–6) [97]
- 2-(4-Aminophenyl)quinoline-4-carboxylic acid (1).
- From isatin I and 4-aminoacetophenone V. Red solid (80%) M.p. (193–195) °C. Rf DCM:MeOH (9:1): 0.07. 1H NMR (400 MHz, DMSO-d6) δ 8.57 (d, J = 8.3 Hz, 1H), 8.31 (s, 1H), 8.03 (m, 3H), 7.76 (pt, J = 7.2 Hz, 1H), 7.58 (pt, J = 7.2 Hz, 1H), 6.71 (d, J = 8.6 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.9, 156.2, 151.0, 148.5, 137.0, 129.9, 129.2, 128.4, 126.5, 125.3, 124.9, 122.7, 118.3, 113.8.
- 2-(3-Aminophenyl) quinoline-4-carboxylic acid (2).
- From isatin I and 3-aminoacetophenone VI. White solid (59%) M.p. (179–181) °C. Rf DCM:MeOH (9:1): 0.02. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (d, J = 8.4 Hz, 1H), 8.36 (s, 1H), 8.13 (d, J = 8.3 Hz, 1H), 7.84 (pt, J = 7.5 Hz, 1H), 7.68 (pt, J = 7.5 Hz, 1H), 7.54 (s, 1H), 7.40 (d, J = 7.5 Hz, 1H), 7.21 (t, J = 7.7 Hz, 1H), 6.73 (d, J = 7.5 Hz, 1H). 13C NMR (100 MHz, DMSO d6) δ 167.7, 156.5, 149.2, 148.4, 138.5, 137.1, 130.2, 129.7, 129.6, 127.6, 125.4, 123.5, 119.3, 115.7, 114.9, 112.4.
- Structure appears as patented [84], but no characterization data are reported.
- 2-(2-Aminophenyl)quinoline-4-carboxylic acid (3).
- From isatin I and 2-aminoacetophenone VII. Yellow solid (74%) M.p. (227–229) °C. Rf DCM:MeOH, 9:1: 0.17. 1H NMR (400 MHz, DMSO-d6 at 393 K) δ 8.64 (d, J = 8.3 Hz, 1H), 8.37 (s, 1H), 8.14 (d, J = 8.3 Hz, 1H), 7.89–7.78 (m, 2H), 7.68 (pt, J = 7.3 Hz, 1H), 7.21 (t, J = 7.1 Hz, 1H), 6.92 (d, J = 8.1 Hz, 1H), 6.75 (t, J = 7.2 Hz, 1H). 13C NMR (100 MHz, DMSO) δ 167.6, 158.2, 147.5, 147.1, 137.1, 130.7, 130.2, 129.6, 129.0, 127.4, 125.3, 122.3, 120.8, 119.3, 117.4, 116.7.
- Structure appears as patented [84], but no characterization data are reported.
- 2-(2-Aminophenyl)-6-fluoroquinoline-4-carboxylic acid (4).
- From 5-fluoroisatin II and 2-aminoacetophenone VII. Yellow solid (31%) M.p. (210–212) °C. Rf DCM:MeOH (9:1): 0.11. 1H NMR (400 MHz, DMSO-d6, at 393 K) δ 8.42 (dd, 3JHF = 10.2 Hz, J = 2.9 Hz, 1H), 8.40 (s, 1H), 8.16 (dd, J = 9.2, 4JHF = 5.7 Hz, 1H), 7.72 (dd, J = 8.0, 1.3 Hz, 1H), 7.67 (ddd, J = 9.2, 2.9 Hz, 3JHF = 8.3 Hz, 1H), 7.18 (ddd, J = 8.3, 7.2, 1.3 Hz, 1H), 6.89 (dd, J = 8.3, 1.3 Hz, 1H), 6.72 (ddd, J = 8.0, 7.2, 1.3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.3, 159.8 (d, 1JCF = 245.4 Hz), 157.4 (d, 6JCF = 2.5 Hz), 147.7, 144.1, 136.2 (d, 4JCF = 5.5 Hz), 131.0 (d, 3JCF = 10.9 Hz), 129.7, 128.7, 122.8 (d, 3JCF = 10.9 Hz), 121.2, 119.0 (d, 2JCF = 25.0 Hz), 118.8, 116.4, 115.5, 108.5 (d, 2JCF = 25.0 Hz). IR (ATR, cm−1): (3500–1900) (broad NH2 along with COOH signal), 1712 (C=O), 1549, 1234, 749, 468. EI MS (70 eV): m/z (%): 282 (M+, 78), 281 (62), 237 (100), 236 (64). HRMS (ESI-QTOF) (M+H) calc. for C16H11FN2O2: 283.0877 found: 283.0879. HRMS (ESI-QTOF) (M-H) calc. for C16H11FN2O2: 281.0732 found: 281.0732.
- 2-(2-Aminophenyl)-6-chloroquinoline-4-carboxylic acid (5).
- From 5-chloroisatin III and 2-aminoacetophenone VII. Brown solid (94%) M.p. (215–217) °C. Rf DCM:MeOH (9:1): 0.15. 1H NMR (400 MHz, DMSO-d6, at 393 K) δ 8.78 (d, J = 2.4 Hz, 1H), 8.37 (s, 1H), 8.09 (d, J = 9.0 Hz, 1H), 7.76 (dd, J = 9.0, 2.4 Hz, 1H), 7.73 (dd, J = 8.2, 1.2 Hz, 1H), 7.18 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 6.88 (dd, J = 8.2, 1.2 Hz, 1H), 6.72 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.4, 158.4, 147.9, 145.3, 137.5, 130.9, 130.1, 129.9, 129.4, 128.7, 124.0, 123.0, 120.9, 118.8, 116.5, 115.5. IR (ATR, cm−1): 3460 (NH2), 3330 (NH2), (3200–1900) (broad COOH signal), 1720 (C=O), 1548, 1320, 760, 666. EI MS (70 eV): m/z (%): 298 (M+, 88), 297 (77), 253 (100), 252 (47). HRMS (ESI-QTOF) (M+H) calc. for C16H11ClN2O2: 299.0582 found: 299.0584. HRMS (ESI-QTOF) (M-H) calc. for C16H11ClN2O2: 297.0436 found: 297.0437.
- 2-(2-Aminophenyl)-6-bromoquinoline-4-carboxylic acid (6).
- From 5-bromoisatin IV and 2-aminoacetophenone VII. Brown solid (70%) M.p. (243–245) °C. Rf DCM:MeOH (9:1): 0.15. 1H NMR (400 MHz, DMSO-d6, at 393 K) δ 8.93 (d, J = 2.2 Hz, 1H), 8.37 (s, 1H), 8.02 (d, J = 8.9 Hz, 1H), 7.88 (dd, J = 8.9, 2.2 Hz, 1H), 7.74 (dd, J = 8.0, 1.2 Hz, 1H), 7.18 (ddd, J = 8.3, 7.0, 1.2 Hz, 1H), 6.88 (pd, J = 8.3 Hz, 1H), 6.72 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.3, 158.5, 147.9, 145.5, 136.7, 132.1, 130.2, 129.9, 128.8, 127.2, 123.3, 121.0, 119.5, 118.7, 116.5, 115.5. IR (ATR, cm−1): (3500–1900) (broad, NH2 along with COOH signal), 1712 (C=O), 1544, 1452, 1371, 1319, 758, 661. EI MS (70 eV): m/z (%): 342 (M+, 95), 340 (82), 299 (76), 298 (52), 297 (100), 218 (87), 217 (50). HRMS (ESI-QTOF) (M+H) calc. for C16H11BrN2O2: 343.0077 found: 343.0076. HRMS (ESI-QTOF) (M-H) calc. for C16H11BrN2O2: 340.9931 found: 340.9933.
3.2.2. General Procedure for the Synthesis of Ethyl 2-(2-Aminophenyl)quinoline-4-carboxylates (7–12)
- Ethyl 2-(4-aminophenyl)quinoline-4-carboxylate (7).
- From compound 1. Yellow solid (50%) M.p. (143–145) °C [Lit [97] 418 K]. Rf Hex:AcOEt (7:3): 0.26. 1H NMR (400 MHz, CDCl3) δ 8.70–8.65 (m, 1H), 8.31 (s, 1H), 8.18–8.13 (m, 1H), 8.10–8.04 (m, 2H), 7.72 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.56 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 6.86–6.79 (m, 2H), 4.54 (q, J = 7.1 Hz, 2H), 3.93 (s, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.8, 156.7, 149.4, 148.3, 135.9, 130.1, 129.8, 129.2, 128.9, 127.1, 125.5, 123.6, 119.8, 115.3, 62.0, 14.5.
- Ethyl 2-(3-aminophenyl)quinoline-4-carboxylate (8).
- From compound 2. Yellow oil (52%) Rf Hex:AcOEt (7:3): 0.37. 1H NMR (400 MHz, CDCl3) δ 8.73 (ddd, J = 8.4, 1.4, 0.6 Hz, 1H), 8.35 (s, 1H), 8.21 (ddd, J = 8.5, 1.4, 0.6 Hz, 1H), 7.76 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.62 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.60–7.58 (m, 1H), 7.52 (ddd, J = 7.8, 1.7, 1.0 Hz, 1H), 7.36–7.30 (m, 1H), 6.82 (ddd, J = 7.8, 2.4, 1.0 Hz, 1H), 4.55 (q, J = 7.1 Hz, 2H), 3.85 (s, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.6, 157.0, 149.3, 147.2, 140.1, 136.1, 130.4, 129.98, 129.96, 127.8, 125.5, 124.2, 120.6, 118.0, 116.7, 114.1, 62.0, 14.5.
- Structure appears as patented [98], but no characterization data are reported.
- Ethyl 2-(2-aminophenyl)quinoline-4-carboxylate (9).
- From compound 3. Orange solid (58%) M.p. (71–73) °C Rf Hex:AcOEt (9:1): 0.22. 1H NMR (400 MHz, DMSO-d6) δ 8.51 (d, J = 8.3 Hz, 1H), 8.36 (s, 1H), 8.13 (d, J = 8.3 Hz, 1H), 7.87–7.76 (m, 2H), 7.67 (pt, J = 7.4 Hz, 1H), 7.27–7.03 (m, 3H), 6.87 (pd, J = 8.3 Hz, 1H), 6.69 (pt, J = 7.3 Hz, 1H), 4.48 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 158.3, 148.8, 147.0, 136.1, 130.8, 130.3, 129.5, 129.0, 127.5, 124.9, 121.9, 120.7, 118.3, 116.9, 115.8, 61.9, 14.1. IR (ATR, cm−1): 3429 (NH), 3271 (N-H--NQ, H-bond), 1703 (C=O), 1593, 1544, 1472, 1250, 758, 656. EI MS (70 eV): m/z (%): 292 (M+, 28), 219 (31), 91 (100). HRMS (ESI-QTOF) (M+H) calc. for C18H16N2O2: 293.1285 found: 293.1286.
- Ethyl 2-(2-aminophenyl)-6-fluoroquinoline-4-carboxylate (10).
- From compound 4. Orange solid (62%) M.p. (110–112) °C. Rf Hex:AcOEt (7:3): 0.46. 1H NMR (400 MHz, DMSO-d6) δ 8.41 (s, 1H), 8.29 (dd, 3JHF = 10.8 Hz, J = 2.9 Hz, 1H), 8.21 (dd, J = 9.2 Hz, 4JHF = 5.7 Hz, 1H), 7.78–7.71 (m, 2H), 7.18 (ddd, J = 8.3. 7.0, 1.2 Hz,, 1H), 7.05 (s, 2H), 6.87 (dd, J = 8.3, 1.2 Hz, 1H), 6.69 (ddd, J = 8.0, 7.0, 1.2 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.4, 160.4 (d, 1JCF = 245.4 Hz), 157.8 (d, 6JCF = 3.0 Hz), 148.6, 144.4, 134.9 (d, 4JCF = 5.5 Hz), 131.9 (d, 3JCF = 9.6 Hz), 130.8, 129.4, 122.8 (d, 3JCF = 11.0 Hz), 121.9, 120.1 (d, 2JCF = 25.0 Hz), 118.1, 116.9, 115.8, 108.9 (d, 2JCF = 25.0 Hz), 62.0, 14.0. IR (ATR, cm−1): 3429 (NH), 3267 (N-H--NQ, H-bond), 1702 (C=O), 1595, 1545, 1475, 1227, 740, 645. EI MS (70 eV): m/z (%): 310 (M+, 84), 309 (67), 237 (100), 236 (46). HRMS (ESI-QTOF) (M+H) calc. for C18H15FN2O2: 311.1193 found: 311.1190.
- Ethyl 2-(2-aminophenyl)-6-chloroquinoline-4-carboxylate (11).
- From compound 5. Orange solid (51%) M.p. (145–147) °C. Rf Hex:AcOEt (9:1): 0.20. 1H NMR (400 MHz, DMSO-d6) δ 8.60 (d, J = 2.2 Hz, 1H), 8.41 (s, 1H), 8.15 (d, J = 9.0 Hz, 1H), 7.82 (dd, J = 9.0, 2.2 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.19 (pt, J = 7.8 Hz, 1H), 7.13 (broad s, 2H), 6.87 (d, J = 8.3 Hz, 1H), 6.68 (pt, J = 7.6 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.3, 158.7, 148.8, 145.6, 134.6, 132.0, 131.1, 131.0, 130.6, 129.5, 123.9, 122.7, 121.9, 117.8, 117.0, 115.8, 62.0, 14.0. IR (ATR, cm−1): 3414 (NH), 3231 (N-H--NQ, H-bond), 1699 (C=O), 1587, 1463, 1270, 729, 677. EI MS (70 eV): m/z (%): 326 (M+, 66), 325 (56), 253 (100), 66 (77). HRMS (ESI-QTOF) (M+H) calc. for C18H15ClN2O2: 327.0895 found: 327.0900.
- Ethyl 2-(2-aminophenyl)-6-bromoquinoline-4-carboxylate (12).
- From compound 6. Orange solid (30%) M.p. (148–150) °C. Rf Hex:AcOEt (9:1): 0.17. 1H NMR (400 MHz, DMSO-d6) δ 8.77 (ps, 1H), 8.41 (s, 1H), 8.08 (d, J = 8.8 Hz, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.19 (pt, J = 7.8 Hz, 1H), 7.14 (s, 2H), 6.87 (d, J = 8.3 Hz, 1H), 6.69 (pt, J = 7.6 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 1.41 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.3, 158.8, 148.9, 145.8, 134.5, 133.2, 131.2, 131.0, 129.5, 127.1, 123.1, 121.9, 120.7, 117.8, 117.0, 115.8, 62.0, 14.0. IR (ATR, cm−1): 3417 (NH), 3231 (N-H--NQ, H-bond), 1700 (C=O), 1584, 1462, 729, 668. EI MS (70 eV): m/z (%): 370 (M+, 36), 299 (76), 297 (100), 218 (50), 84 (49), 66 (83). HRMS (ESI-QTOF) (M+H) calc. for C18H15BrN2O2: 371.0390 found: 371.0391.
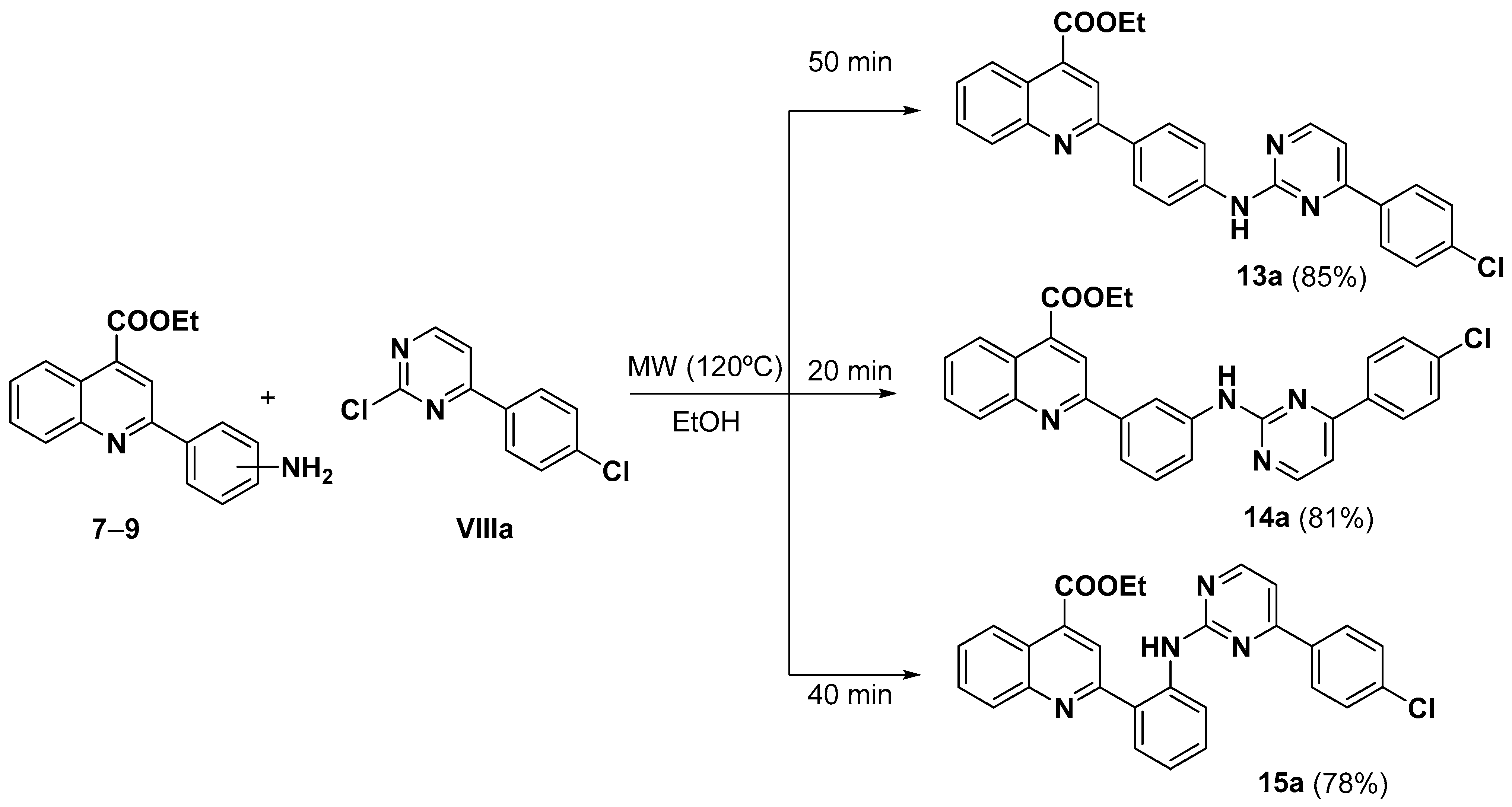
3.2.3. General Procedure for the Synthesis of Ethyl 2-(2-((4-Arylpyrimidin-2-yl)amino) phenyl)quinoline-4-carboxylates (13–18)(a–d)
- Ethyl 2-(4-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (13a).
- From compounds 7 and VIIIa. Reaction time: 50 min. Orange solid (85%) M.p. (192–194) °C. Rf Hex:AcOEt (7:3): 0.39. 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.63 (d, J = 5.2 Hz, 1H), 8.51 (d, J = 8.5 Hz, 1H), 8.45 (s, 1H), 8.30 (d, J = 8.7 Hz, 2H), 8.23 (d, J = 8.5 Hz, 2H), 8.19 (d, J = 8.4 Hz, 1H), 8.06 (d, J = 8.7 Hz, 2H), 7.85 (pt, J = 7.7 Hz, 1H), 7.68 (pt, J = 7.7 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 5.2 Hz, 1H), 4.50 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.9, 162.6, 159.8, 159.2, 155.4, 147.7, 142.8, 137.0, 135.9, 135.3, 130.6, 129.8, 129.1, 129.0, 128.8, 128.0, 127.7, 125.1, 122.8, 119.0, 118.8, 108.5, 62.0, 14.1. IR (ATR, cm−1): 3214 (NH), 1684 (C=O), 1533, 1500, 1387, 1215, 1069, 742. EI MS (70 eV): m/z (%): 480 (M+, 100), 451 (32), 226 (16). HRMS (ESI-QTOF) (M+H) calc. for C28H21ClN4O2: 481.1426 found: 481.1428.
- Ethyl 2-(4-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (13b).
- From compounds 7 and VIIIb. Reaction time: 60 min. Orange solid (86%) M.p. (194–196) °C. Rf Hex:AcOEt (7:3): 0.27. 1H NMR (400 MHz, DMSO-d6) δ 10.13 (s, 1H), 8.80 (s, 1H), 8.66 (d, J = 5.2 Hz, 1H), 8.51 (dd, J = 8.5, 1.0 Hz, 1H), 8.46 (s, 1H), 8.37–8.29 (m, 3H), 8.18–8.08 (m, 5H), 8.02–7.98 (m, 1H), 7.84 (ddd, J = 8.4, 7.0, 1.2 Hz, 1H), 7.67 (ddd, J = 8.5, 7.0, 1.2 Hz, 1H), 7.65–7.59 (m, 3H), 4.50 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.0, 163.8, 159.9, 159.0, 155.5, 147.9, 142.8, 136.8, 134.2, 133.9, 132.8, 130.5, 130.0, 129.2, 129.0, 128.6, 128.0, 127.7, 127.61, 127.56, 127.2, 126.8, 125.1, 124.0, 122.8, 118.9, 118.8, 108.9, 62.0, 14.1. IR (ATR, cm−1): 3219 (NH), 1682 (C=O), 1525, 1387, 1211, 756, 689. EI MS (70 eV): m/z (%): 496 (M+, 100), 467 (31), 234 (22). HRMS (ESI-QTOF) (M+H) calc. for C32H24N4O2: 497.1972 found: 497.1975.
- Ethyl 2-(3-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (14a).
- From compounds 8 and VIIIa. Reaction time: 20 min. Yellow solid (81%) M.p. (214–216) °C. Rf Hex:AcOEt (7:3): 0.40. 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.95 (s, 1H), 8.61 (d, J = 5.3 Hz, 1H), 8.59 (dd, J = 8.5, 1.0 Hz, 1H), 8.43 (s, 1H), 8.25 (d, J = 8.7 Hz, 2H), 8.15 (d, J = 8.4 Hz, 1H), 7.93–7.83 (m, 3H), 7.73 (ddd, J = 8.5, 7.0, 1.0 Hz, 1H), 7.56–7.50 (m, 3H), 7.48 (d, J = 5.3 Hz, 1H), 4.46 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.8, 162.7, 159.8, 159.0, 156.0, 148.2, 141.1, 138.0, 136.6, 135.9, 135.4, 130.5, 129.7, 129.3, 129.0, 128.8, 128.1, 125.2, 123.3, 120.8, 120.7, 119.3, 117.7, 108.1, 62.0, 14.0. IR (ATR, cm−1): 3204 (NH), 1682 (C=O), 1559, 1191, 756. EI MS (70 eV): m/z (%): 480 (M+, 100), 451 (29), 369 (18), 226 (20), 203 (16). HRMS (ESI-QTOF) (M+H) calc. for C28H21ClN4O2: 481.1426 found: 481.1434.
- Ethyl 2-(3-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (14b).
- From compounds 8 and VIIIb. Reaction time: 20 min. Sonication helps the precipitation of 14b. Yellow solid (78%) M.p. (139–141) °C. Rf Hex:AcOEt (7:3): 0.33. 1H NMR (400 MHz, DMSO-d6) δ 10.01 (s, 1H), 9.01 (s, 1H), 8.80 (s, 1H), 8.65 (d, J = 5.2 Hz, 1H), 8.61 (d, J = 8.5 Hz, 1H), 8.47 (s, 1H), 8.37 (d, J = 8.5 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 8.01–7.80 (m, 6H), 7.73 (pt, J = 7.7 Hz, 1H), 7.65–7.53 (m, 3H), 7.49 (pt, J = 7.5 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.8, 163.6, 160.1, 159.1, 156.1, 148.4, 141.3, 138.2, 136.4, 134.2, 134.0, 132.7, 130.4, 129.9, 129.3, 128.9, 128.5, 128.1, 127.6, 127.5, 127.2, 126.7, 125.2, 123.9, 123.3, 120.7, 120.5, 119.3, 117.7, 108.5, 61.8, 13.9. IR (ATR, cm−1): 3210 (NH), 1697 (C=O), 1536, 1513, 1391, 1223, 1173, 756. EI MS (70 eV): m/z (%): 496 (M+, 100), 467 (25), 234 (28), 211 (22), 44 (28). HRMS (ESI-QTOF) (M+H) calc. for C32H24N4O2: 497.1972 found: 497.1967.
- Ethyl 2-(2-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (15a).
- From compounds 9 and VIIIa. Reaction time: 40 min. Purified by FCC using Hex:AcOEt (85:15). Yellow solid (78%) M.p. (118–120) °C. Rf Hex:AcOEt (8:2): 0.22. 1H NMR (400 MHz, CDCl3) δ 12.57 (s, 1H), 8.79 (d, J = 8.4 Hz, 1H), 8.76 (d, J = 8.5 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.44–8.38 (m, 2H), 8.05 (d, J = 8.6 Hz, 2H), 7.91 (dd, J = 8.1, 1.2 Hz, 1H), 7.85 (ddd, J = 8.4, 7.0, 1.2 Hz, 1H), 7.67 (ddd, J = 8.5, 7.0, 1.2 Hz, 1H), 7.53 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 7.47 (d, J = 8.6 Hz, 2H), 7.20 (ddd, J = 8.1, 7.2, 1.2 Hz, 1H), 7.11 (d, J = 5.2 Hz, 1H), 4.55 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.3, 163.6, 160.2, 158.5, 157.7, 147.5, 139.8, 137.1, 136.6, 135.6, 130.50, 130.47, 129.8, 129.7, 129.2, 128.5, 128.2, 125.5, 124.7, 123.4, 122.3, 122.0, 121.2, 108.1, 62.2, 14.5. IR (ATR, cm−1): (3300–2400) (wide NH signal), 1690 (C=O), 1501, 1410, 747. EI MS (70 eV): m/z (%): 480 (M+, 74), 407 (100), 280 (83). HRMS (ESI-QTOF) (M+H) calc. for C28H21ClN4O2: 481.1426 found: 481.1430.
- Ethyl 2-(2-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (15b).
- From compounds 9 and VIIIb. Reaction time: 40 min. Purified by FCC using Hex:AcOEt (85:15). Yellow solid (83%) M.p. (142–144) °C. Rf Hex:AcOEt (85:15): 0.18. 1H NMR (400 MHz, CDCl3) δ 12.66 (s, 1H), 8.94 (pd, J = 8.4 Hz, 1H), 8.78 (pd, J = 8.5 Hz, 1H), 8.64 (s, 1H), 8.55 (d, J = 5.2 Hz, 1H), 8.48–8.43 (m, 2H), 8.24 (dd, J = 8.5, 1.8 Hz, 1H), 8.00–7.85 (m, 5H), 7.69 (ddd, J = 8.5, 7.0, 1.2 Hz, 1H), 7.60–7.53 (m, 3H), 7.30 (d, J = 5.2 Hz, 1H), 7.20 (pt, J = 7.5 Hz, 1H), 4.55 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.3, 164.5, 160.6, 158.7, 157.9, 147.5, 140.3, 136.6, 134.7, 134.6, 133.3, 130.6, 130.4, 129.8, 129.7, 129.1, 128.6, 128.2, 127.9, 127.44, 127.36, 126.6, 125.6, 124.4, 124.2, 123.5, 122.4, 121.6, 121.0, 108.7, 62.2, 14.5. IR (ATR, cm−1): (3200–2400) (wide NH signal), 1683 (C=O), 1508, 1406, 748. EI MS (70 eV): m/z (%): 496 (M+, 70), 423 (83), 296 (100). HRMS (ESI-QTOF) (M+H) calc. for C32H24N4O2: 497.1972 found: 497.1978.
- Ethyl 2-(2-((4-phenylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (15c).
- From compounds 9 and VIIIc. Reaction time: 30 min. Purified by FCC using Hex:AcOEt (9:1). Yellow solid (85%) M.p. (95–97) °C. Rf Hex:AcOEt (8:2): 0.22. 1H NMR (400 MHz, CDCl3) δ 12.53 (s, 1H), 8.89 (dd, J = 8.5, 1.2 Hz, 1H), 8.77 (pd, J = 8.5 Hz, 1H), 8.51 (d, J = 5.2 Hz, 1H), 8.43 (s, 1H), 8.40 (pd, J = 8.4 Hz, 1H), 8.17–8.09 (m, 2H), 7.91 (dd, J = 7.9, 1.5 Hz, 1H), 7.85 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 7.68 (ddd, J = 8.5, 7.0, 1.3 Hz, 1H), 7.59–7.49 (m, 4H), 7.23–7.14 (m, 2H), 4.55 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.3, 164.6, 160.6, 158.7, 157.9, 147.6, 140.2, 137.4, 136.6, 130.8, 130.6, 130.4, 129.8, 129.7, 128.9, 128.2, 127.2, 125.5, 124.5, 123.4, 122.4, 121.6, 121.1, 108.5, 62.2, 14.5. IR (ATR, cm−1): (3200–2600) (wide NH signal), 1722 (C=O), 1529, 1432, 1273, 1192, 761. EI MS (70 eV): m/z (%): 446 (M+, 36), 373 (47), 246 (100), 186 (28). HRMS (ESI-QTOF) (M+H) calc. for C28H22N4O2: 447.1816 found: 447.1815.
- Ethyl (E)-2-(2-((4-styrylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (15d).
- From compounds 9 and VIIId. Reaction temperature: 170 °C. Reaction time: 15 min. Purified by FCC using a gradient of Hex:AcOEt (9:1, 8:2). Yellow solid (65%) M.p. (120–122) °C. Rf Hex:AcOEt (8:2): 0.22. 1H NMR (400 MHz, CDCl3) δ 12.45 (s, 1H), 8.86 (d, J = 8.4 Hz, 1H), 8.77 (d, J = 8.5 Hz, 1H), 8.46–8.38 (m, 3H), 7.92–7.83 (m, 3H), 7.68 (ddd, J = 8.5, 7.0, 1.2 Hz, 1H), 7.63–7.59 (m, 2H), 7.54 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 7.45–7.39 (m, 2H), 7.39–7.33 (m, 1H), 7.18 (ddd, J = 8.1, 7.2, 1.2 Hz, 1H), 7.00 (d, J = 15.9 Hz, 1H), 6.76 (d, J = 5.1 Hz, 1H), 4.55 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 166.4, 163.0, 160.4, 158.6, 157.9, 147.6, 140.2, 136.6, 136.5, 136.1, 130.5, 130.4, 129.8, 129.7, 129.3, 129.0, 128.2, 127.7, 126.6, 125.6, 124.5, 123.4, 122.4, 121.6, 121.0, 110.7, 62.2, 14.5. IR (ATR, cm−1): (3200–2600) (wide NH signal), 2922, 2853, 1717 (C=O), 1529, 1432, 763. EI MS (70 eV): m/z (%): 472 (M+, 36), 399 (45), 272 (100), 128 (33). HRMS (ESI-QTOF) (M+H) calc. for C30H24N4O2: 473.1972 found: 473.1975.
- Ethyl 2-(2-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl)-6-fluoroquinoline-4-carboxylate (16a).
- From compounds 10 and VIIIa. Reaction time: 30 min. Yellow solid (88%) M.p. (139–141) °C. Rf Hex:AcOEt (8:2): 0.16. 1H NMR (400 MHz, CDCl3) δ 12.35 (s, 1H), 8.77 (d, J = 8.4 Hz, 1H), 8.52 (dd, 3JHF = 10.7 Hz, J = 2.8 Hz, 1H), 8.50–8.47 (m, 2H), 8.41 (dd, J = 9.2 Hz, 4JHF = 5.6 Hz, 1H), 8.03 (d, J = 8.6 Hz, 2H), 7.88 (dd, J = 8.1, 1.3 Hz, 1H), 7.61 (ddd, J = 9.2, 2.8 Hz, 3JHF = 8.0 Hz, 1H), 7.53 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.47 (d, J = 8.6 Hz, 2H), 7.21 (pt, J = 7.5 Hz, 1H), 7.12 (d, J = 5.3 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.8, 163.8, 161.8 (d, 1JCF = 249.1 Hz), 160.0, 158.3, 157.1 (d, 6JCF = 2.9 Hz), 144.8, 139.6, 137.2, 135.7 (d, 4JCF = 5.6 Hz), 135.6, 132.1 (d, 3JCF = 9.3 Hz), 130.5, 129.8, 129.2, 128.5, 124.7, 124.4 (d, 3JCF = 11.6 Hz), 123.4, 122.1, 121.3, 120.7 (d, 2JCF = 26.2 Hz), 109.8 (d, 2JCF = 25.2 Hz), 108.1, 62.3, 14.5. IR (ATR, cm−1): (3200–2500) (wide NH signal), 1713 (C=O), 1535, 1440, 1224, 806. EI MS (70 eV): m/z (%): 498 (M+, 35), 425 (30), 280 (100). HRMS (ESI-QTOF) (M+H) calc. for C28H20ClFN4O2: 499.1332 found: 499.1334.
- Ethyl 6-fluoro-2-(2-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (16b).
- From compounds 10 and VIIIb. Reaction time: 20 min. Yellow solid (89%) M.p. (141–143) °C. Rf Hex:AcOEt (8:2): 0.25. 1H NMR (400 MHz, CDCl3) δ 12.44 (s, 1H), 8.92 (dd, J = 8.4, 1.1 Hz, 1H), 8.61 (s, 1H), 8.57–8.49 (m, 3H), 8.47–8.40 (m, 1H), 8.22 (pd, J = 8.5 Hz, 1H), 8.00–7.93 (m, 2H), 7.93–7.87 (m, 2H), 7.68–7.60 (m, 1H), 7.60–7.53 (m, 3H), 7.34–7.27 (m, 1H), 7.21 (pt, J = 7.6 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.9, 164.6, 161.8 (d, 1JCF = 249.0 Hz), 160.5, 158.6, 157.2, 144.8, 140.1, 135.6, 134.7, 134.6, 133.3, 132.0 (d, 3JCF = 9.5 Hz), 130.6, 129.7, 129.1, 128.7, 127.9, 127.5, 127.4, 126.7, 124.4 (d, 3JCF = 10.8 Hz), 124.3, 124.1, 123.4, 121.8, 121.2, 120.6 (d, 2JCF = 25.8 Hz), 109.8 (d, 2JCF = 25.3 Hz), 108.7, 62.3, 14.5. IR (ATR, cm−1): (3300–2600) (wide NH signal), 1718 (C=O), 1535, 1434, 1270, 1222, 808, 747. EI MS (70 eV): m/z (%): 514 (M+, 30), 441 (24), 296 (100). HRMS (ESI-QTOF) (M+H) calc. for C32H23FN4O2: 515.1878 found: 515.1879.
- Ethyl 6-fluoro-2-(2-((4-phenylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (16c).
- From compounds 10 and VIIIc. Reaction time: 20 min. Yellow solid (84%) M.p. (122–124) °C. Rf Hex:AcOEt (8:2): 0.24. 1H NMR (400 MHz, CDCl3) δ 12.35 (s, 1H), 8.87 (pd, J = 8.4 Hz, 1H), 8.53 (dd, 3JHF = 10.7 Hz, J = 2.8 Hz, 1H), 8.51–8.48 (m, 2H), 8.38 (dd, J = 9.2 Hz, 4JHF = 5.6 Hz, 1H), 8.14–8.09 (m, 2H), 7.88 (dd, J = 7.9, 1.4 Hz, 1H), 7.61 (ddd, J = 9.2, 2.8 Hz, 3JHF = 8.0 Hz, 1H), 7.57–7.49 (m, 4H), 7.23–7.13 (m, 2H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.9, 164.7, 161.7 (d, 1JCF = 249.2 Hz), 160.5, 158.7, 157.2 (d, 6JCF = 2.8 Hz), 144.8, 140.1, 137.3, 135.5 (d, 4JCF = 6.1 Hz), 132.0 (d, 3JCF = 9.2 Hz), 130.8, 130.6, 129.7, 128.9, 127.2, 124.4 (d, 3JCF = 11.0 Hz), 124.2, 123.4, 121.7, 121.1, 120.6 (d, 2JCF = 26.1 Hz), 109.8 (d, 2JCF = 25.0 Hz), 108.5, 62.3, 14.4. IR (ATR, cm−1): (3200–2500) (wide NH signal), 1721 (C=O), 1537, 1436, 1271, 1223, 767. EI MS (70 eV): m/z (%): 464 (M+, 33), 391 (28), 246 (100). HRMS (ESI-QTOF) (M+H) calc. for C28H21FN4O2: 465.1721 found: 465.1724.
- Ethyl (E)-6-fluoro-2-(2-((4-styrylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (16d).
- From compounds 10 and VIIId. Reaction temperature: 170 °C. Reaction time: 10 min. Yellow solid (70%) M.p. (147–149) °C. Rf Hex:AcOEt (8:2): 0.24. 1H NMR (400 MHz, CDCl3) δ 12.23 (s, 1H), 8.84 (dd, J = 8.4, 1.1 Hz, 1H), 8.53 (dd, 3JHF = 10.7 Hz, J = 2.8 Hz, 1H), 8.49 (s, 1H), 8.42 (d, J = 5.1 Hz, 1H), 8.39 (dd, J = 9.2 Hz, 4JHF = 5.6 Hz, 1H), 7.91–7.80 (m, 2H), 7.65–7.58 (m, 3H), 7.53 (ddd, J = 8.4, 7.2, 1.5 Hz, 1H), 7.45–7.39 (m, 2H), 7.39–7.33 (m, 1H), 7.18 (ddd, J = 8.1, 7.2, 1.2 Hz, 1H), 6.99 (d, J = 15.9 Hz, 1H), 6.76 (d, J = 5.1 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ 165.9, 163.0, 161.8 (d, 1JCF = 250.4 Hz), 160.3, 158.6, 157.3 (d, 6JCF = 2.8 Hz), 144.8, 140.1, 136.5, 136.1, 135.6 (d, 4JCF = 5.7 Hz), 132.0 (d, 3JCF = 9.7 Hz), 130.6, 129.7, 129.3, 129.0, 127.7, 126.6, 124.4 (d, 3JCF = 11.0 Hz), 124.3, 123.5, 121.7, 121.1, 120.6 (d, 2JCF = 25.8 Hz), 110.8, 109.8 (d, 2JCF = 25,3 Hz), 62.3, 14.5. IR (ATR, cm−1): (3200–2500) (wide NH signal), 2922, 2853, 1713 (C=O), 1520, 1402, 1338, 1278, 1230, 744, 699. EI MS (70 eV): m/z (%): 490 (M+, 35), 417 (18), 272 (100). HRMS (ESI-QTOF) (M+H) calc. for C30H23FN4O2: 491.1878 found: 491.1881.
- Crystals suitable for X-ray single-crystal diffraction were obtained from DMSO solution, and the crystal data for 16d were deposited at CCDC with reference CCDC 2368196: Chemical formula C30H23FN4O2, Mr 490.52; Monoclinic, P21/n; 100 K, Cell dimensions a, b, c (Å) 12.2248(7), 7.8728(5), 24.2984(16) β (°) α, β, γ (°) 90, 99.681(2), 90. V (Å3) 2305.3(2), Z = 4, F (000) = 1024, Dx (Mg m−3) = 1.413, Mo Kα, μ (mm−1) = 0.096, Crystal size (mm) = 0.24 × 0.09 × 0.08. Data collection: Diffractometer Bruker D8 Venture (APEX 3), monochromator multilayer mirror, CCD rotation images, thick slices φ and θ scans, Mo INCOATEC high-brilliance microfocus sealed tube (λ = 0.71073 Å), multiscan absorption correction (SADABS 2016/2), Tmin, Tmax 0.660, 0.746. No. of measured, independent and observed [I > 2σ(I)] reflections 54,633, 5286, 4316, Rint = 0. 087, (sin θ/λ)max (Å−1) 0.411, θ values (°): θmax = 27.5, θmin = 2.0; Range h = −15→15, k = −10→10, l = −31→31, Refinement on F2:R[F2 > 2σ(F2)] = 0. 063, wR(F2) = 0.166, S=1.079. No. of reflections 5286, No. of parameters 339, No. of restraints 0. Weighting scheme: w = 1/σ2(Fo2) + (0.0753P)2 + 2.8379P where P = (Fo2 + 2Fc2)/3. (∆/σ) < 0.012, Δρmax, Δρmin (e Å−3) 0.347, −0.31. Initial R was higher than 0.10, and after running TwinRotMat using PLATON (version 260918), a new hkl file was generated as hklf5 and so refined as two-component nonmerohedral twinning related by (0.689, 0.000, −0.311; 0.000, −1.0000, 0.000; −1.689, 0.000, −0.689) matrix with BASF 0.24.
- Ethyl 6-chloro-2-(2-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (17a).
- From compounds 11 and VIIIa. Reaction time: 20 min. Orange solid (85%) M.p. (140–142) °C. Rf Hex:AcOEt (8:2): 0.28. 1H NMR (400 MHz, CDCl3) δ 12.41 (s, 1H), 8.84 (d, J = 2.3 Hz, 1H), 8.81 (dd, J = 8.4, 1.2 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.45 (s, 1H), 8.28 (d, J = 8.9 Hz, 1H), 8.03 (d, J = 8.7 Hz, 2H), 7.88 (dd, J = 8.1, 1.3 Hz, 1H), 7.76 (dd, J = 8.9, 2.3 Hz, 1H), 7.53 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.47 (d, J = 8.7 Hz, 2H), 7.18 (ddd, J = 8.1, 7.2, 1.2 Hz, 1H), 7.10 (d, J = 5.2 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 163.5, 160.4, 158.8, 158.0, 145.9, 140.0, 137.0, 135.7, 135.3, 134.4, 131.4, 131.0, 130.8, 129.8, 129.2, 128.4, 124.8, 124.1, 124.0, 123.3, 121.8, 121.1, 108.2, 62.4, 14.5. IR (ATR, cm−1): (3200–2700) (wide NH signal), 1715 (C=O), 1535, 1435, 1269, 1178, 803. EI MS (70 eV): m/z (%): 514 (M+, 27), 441 (38), 280 (100). HRMS (ESI-QTOF) (M+H) calc. for C28H20Cl2N4O2: 515.1036 found: 515.1037.
- Ethyl 6-chloro-2-(2-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (17b).
- From compounds 11 and VIIIb. Reaction time: 20 min. Yellow solid (91%) M.p. (165–167) °C. Rf Hex:AcOEt (8:2): 0.25. 1H NMR (400 MHz, CDCl3) δ 12.54 (s, 1H), 8.93 (dd, J = 8.4, 1.3 Hz, 1H), 8.85 (d, J = 2.3 Hz, 1H), 8.60 (s, 1H), 8.53 (d, J = 5.2 Hz, 1H), 8.47 (s, 1H), 8.33 (d, J = 8.9 Hz, 1H), 8.20 (dd, J = 8.6, 1.8 Hz, 1H), 7.98–7.93 (m, 2H), 7.91–7.87 (m, 2H), 7.78 (dd, J = 8.9, 2.3 Hz, 1H), 7.59–7.53 (m, 3H), 7.28 (d, J = 5.2 Hz, 1H), 7.18 (ddd, J = 8.1, 7.2, 1.3 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 164.5, 160.5, 158.7, 158.0, 145.9, 140.4, 135.2, 134.7, 134.5, 134.3, 133.3, 131.3, 131.0, 130.8, 129.7, 129.1, 128.6, 127.9, 127.4, 126.6, 124.8, 124.1, 124.0, 123.8, 123.3, 121.6, 121.0, 108.7, 62.3, 14.5. IR (ATR, cm−1): (3200–2600) (wide NH signal), 2921, 2852, 1721 (C=O), 1535, 1429, 1270, 1183, 808, 744. EI MS (70 eV): m/z (%): 530 (M+, 29), 457 (63), 296 (100). HRMS (ESI-QTOF) (M+H) calc. for C32H23ClN4O2: 531.1582 found: 531.1580.
- Ethyl 6-chloro-2-(2-((4-phenylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (17c).
- From compounds 11 and VIIIc. Reaction time: 20 min. Yellow solid (80%) M.p. (170–172) °C. Rf Hex:AcOEt (8:2): 0.29. 1H NMR (400 MHz, CDCl3) δ 12.40 (s, 1H), 8.90–8.83 (m, 2H), 8.50 (d, J = 5.2 Hz, 1H), 8.47 (s, 1H), 8.30 (d, J = 8.9 Hz, 1H), 8.14–8.08 (m, 2H), 7.88 (dd, J = 8.1, 1.2 Hz, 1H), 7.77 (dd, J = 8.9, 2.3 Hz, 1H), 7.57–7.48 (m, 4H), 7.21–7.14 (m, 2H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 164.7, 160.5, 158.7, 158.1, 146.0, 140.2, 137.3, 135.3, 134.3, 131.4, 131.0, 130.84, 130.80, 129.7, 128.9, 127.2, 124.8, 124.04, 124.02, 123.4, 121.6, 121.1, 108.6, 62.3, 14.5. IR (ATR, cm−1): (3200–2600) (wide NH signal), 1719 (C=O), 1536, 1430, 1334, 1269, 764. EI MS (70 eV): m/z (%): 480 (M+, 28), 407 (60), 246 (100). HRMS (ESI-QTOF) (M+H) calc. for C28H21ClN4O2: 481.1426 found: 481.1428.
- Ethyl (E)-6-chloro-2-(2-((4-styrylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (17d).
- From compounds 11 and VIIId. Reaction temperature: 170 °C. Reaction time: 10 min. Orange solid (72%) M.p. (166–168) °C. Rf Hex:AcOEt (8:2): 0.27. 1H NMR (400 MHz, CDCl3) δ 12.30 (s, 1H), 8.90–8.81 (m, 2H), 8.46 (s, 1H), 8.42 (d, J = 5.1 Hz, 1H), 8.31 (dd, J = 8.9, 0.4 Hz, 1H), 7.89–7.86 (m, 1H), 7.84 (d, J = 16.3 Hz, 2H), 7.77 (dd, J = 8.9, 2.3 Hz, 1H), 7.62–7.58 (m, 2H), 7.53 (ddd, J = 8.4, 7.2, 1.2 Hz, 1H), 7.45–7.39 (m, 2H), 7.38–7.33 (m, 1H), 7.17 (ddd, J = 8.1, 7.2, 1.2 Hz, 1H), 6.98 (d, J = 15.9 Hz, 1H), 6.75 (d, J = 5.1 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 163.0, 160.3, 158.6, 158.1, 145.9, 140.2, 136.5, 136.0, 135.3, 134.3, 131.3, 131.1, 130.7, 129.7, 129.3, 129.0, 127.7, 126.6, 124.8, 124.04, 124.00, 123.3, 121.6, 121.0, 110.8, 62.3, 14.5. IR (ATR, cm−1): (3200–2600) (wide NH signal), 2922, 2852, 1719 (C=O), 1534, 1446, 1432, 1372, 1270, 1180, 746. EI MS (70 eV): m/z (%): 506 (M+, 25), 433 (38), 272 (100), 87 (48). HRMS (ESI-QTOF) (M+H) calc. for C30H23ClN4O2: 507.1582 found: 507.1587.
- Ethyl 6-bromo-2-(2-((4-(4-chlorophenyl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (18a).
- From compounds 12 and VIIIa. Reaction time: 20 min. Purified by FCC using Hex:AcOEt (8:2). Yellow solid (70%) M.p. (161–163) °C. Rf Hex:AcOEt (8:2): 0.22. 1H NMR (400 MHz, CDCl3) δ 12.43 (s, 1H), 9.01 (d, J = 2.1 Hz, 1H), 8.82 (dd, J = 8.4, 1.3 Hz, 1H), 8.49 (d, J = 5.2 Hz, 1H), 8.45 (s, 1H), 8.20 (d, J = 8.9 Hz, 1H), 8.03 (d, J = 8.6 Hz, 2H), 7.92–7.86 (m, 2H), 7.53 (ddd, J = 8.4, 7.2, 1.3 Hz, 1H), 7.47 (d, J = 8.6 Hz, 2H), 7.18 (ddd, J = 8.1, 7.2, 1.3 Hz, 1H, 1H), 7.10 (d, J = 5.2 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 163.4, 160.5, 158.9, 158.1, 146.1, 140.1, 137.0, 135.7, 135.2, 134.0, 131.0, 130.8, 129.8, 129.2, 128.4, 128.1, 124.5, 124.0, 123.3, 122.7, 121.8, 121.0, 108.2, 62.4, 14.5. IR (ATR, cm−1): 3273 (NH), 2922, 2852, 1725 (C=O), 1532, 1415, 1268, 1176, 803, 743. EI MS (70 eV): m/z (%): 558 (M+, 21), 485 (29), 280 (100). HRMS (ESI-QTOF) (M+H) calc. for C28H20BrClN4O2: 559.0531 found: 559.0530.
- Ethyl 6-bromo-2-(2-((4-(naphthalen-2-yl)pyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (18b).
- From compounds 12 and VIIIb. Reaction time: 20 min. Yellow solid (84%) M.p. (170–172) °C. Rf Hex:AcOEt (8:2): 0.22. 1H NMR (400 MHz, CDCl3) δ 12.53 (s, 1H), 9.01 (d, J = 2.1 Hz, 1H), 8.92 (d, J = 8.4 Hz, 1H), 8.59 (s, 1H), 8.52 (d, J = 5.2 Hz, 1H), 8.46 (s, 1H), 8.26 (d, J = 8.9 Hz, 1H), 8.20 (dd, J = 8.6, 1.8 Hz, 1H), 7.99–7.92 (m, 2H), 7.91–7.87 (m, 3H), 7.58–7.53 (m, 3H), 7.29 (d, J = 5.2 Hz, 1H), 7.18 (ddd, J = 8.1, 7.2, 1.3 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 164.5, 160.5, 158.6, 158.2, 146.1, 140.3, 135.1, 134.7, 134.5, 133.9, 133.3, 131.1, 130.9, 129.7, 129.1, 128.6, 128.1, 127.9, 127.4, 126.7, 124.5, 124.1, 123.9, 123.2, 122.7, 121.6, 121.1, 108.7, 62.3, 14.4. IR (ATR, cm−1): (3200–2600) (wide NH signal), 1720 (C=O), 1534, 1427, 1269, 807, 741. EI MS (70 eV): m/z (%): 574 (M+, 20), 501 (19), 296 (100), 152 (29). HRMS (ESI-QTOF) (M+H) calc. for C32H23BrN4O2: 575.1077 found: 575.1075.
- Ethyl 6-bromo-2-(2-((4-phenylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (18c).
- From compounds 12 and VIIIc. Reaction time: 20 min. Yellow solid (76%) M.p. (171–173) °C. Rf Hex:AcOEt (8:2): 0.29. 1H NMR (400 MHz, CDCl3) δ 12.39 (s, 1H), 9.02 (d, J = 2.1 Hz, 1H), 8.88 (dd, J = 8.4, 1.2 Hz, 1H), 8.50 (d, J = 5.2 Hz, 1H), 8.46 (s, 1H), 8.24 (d, J = 8.9 Hz, 1H), 8.14–8.09 (m, 2H), 7.93–7.87 (m, 2H), 7.58–7.48 (m, 4H), 7.23–7.14 (m, 2H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 164.7, 160.5, 158.7, 158.2, 146.2, 140.2, 137.3, 135.2, 134.0, 131.1, 130.86, 130.84, 129.8, 128.9, 128.1, 127.2, 124.5, 124.1, 123.3, 122.7, 121.7, 121.1, 108.6, 62.4, 14.5. IR (ATR, cm−1): (3200–2700) (wide NH signal), 1720 (C=O), 1535, 1429, 1270, 1180, 764. EI MS (70 eV): m/z (%): 524 (M+, 18), 451 (20), 246 (100), 186 (25). HRMS (ESI-QTOF) (M+H) calc. for C28H21BrN4O2: 525.0921 found: 525.0918.
- Ethyl (E)-6-bromo-2-(2-((4-styrylpyrimidin-2-yl)amino)phenyl)quinoline-4-carboxylate (18d).
- From compounds 12 and VIIId. Reaction time: 20 min. Yellow solid (75%) M.p. (198–200) °C. Rf Hex:AcOEt (8:2): 0.28. 1H NMR (400 MHz, CDCl3) δ 12.29 (s, 1H), 9.02 (d, J = 2.1 Hz, 1H), 8.84 (d, J = 8.4 Hz, 1H), 8.45 (s, 1H), 8.42 (d, J = 5.1 Hz, 1H), 8.23 (d, J = 8.9 Hz, 1H), 7.92–7.86 (m, 2H), 7.84 (d, J = 15.9 Hz, 1H), 7.60 (d, J = 7.1 Hz, 2H), 7.54 (pt, J = 7.6 Hz, 1H), 7.45–7.39 (m, 2H), 7.39–7.33 (m, 1H), 7.17 (pt, J = 7.6 Hz, 1H), 6.98 (d, J = 15.9 Hz, 1H), 6.76 (d, J = 5.1 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.7, 163.0, 160.2, 158.6, 158.2, 146.1, 140.2, 136.5, 136.0, 135.2, 133.9, 131.1, 130.8, 129.8, 129.3, 129.0, 128.1, 127.7, 126.5, 124.5, 124.0, 123.3, 122.6, 121.6, 121.0, 110.8, 62.3, 14.5. IR (ATR, cm−1): (3200–2700) (wide NH signal), 1720 (C=O), 1534, 1445, 1270, 745. EI MS (70 eV): m/z (%): 550 (M+, 21), 477 (18), 272 (100), 128 (18), 127 (21). HRMS (ESI-QTOF) (M+H) calc. for C30H23BrN4O2: 551.1077 found: 551.1079.
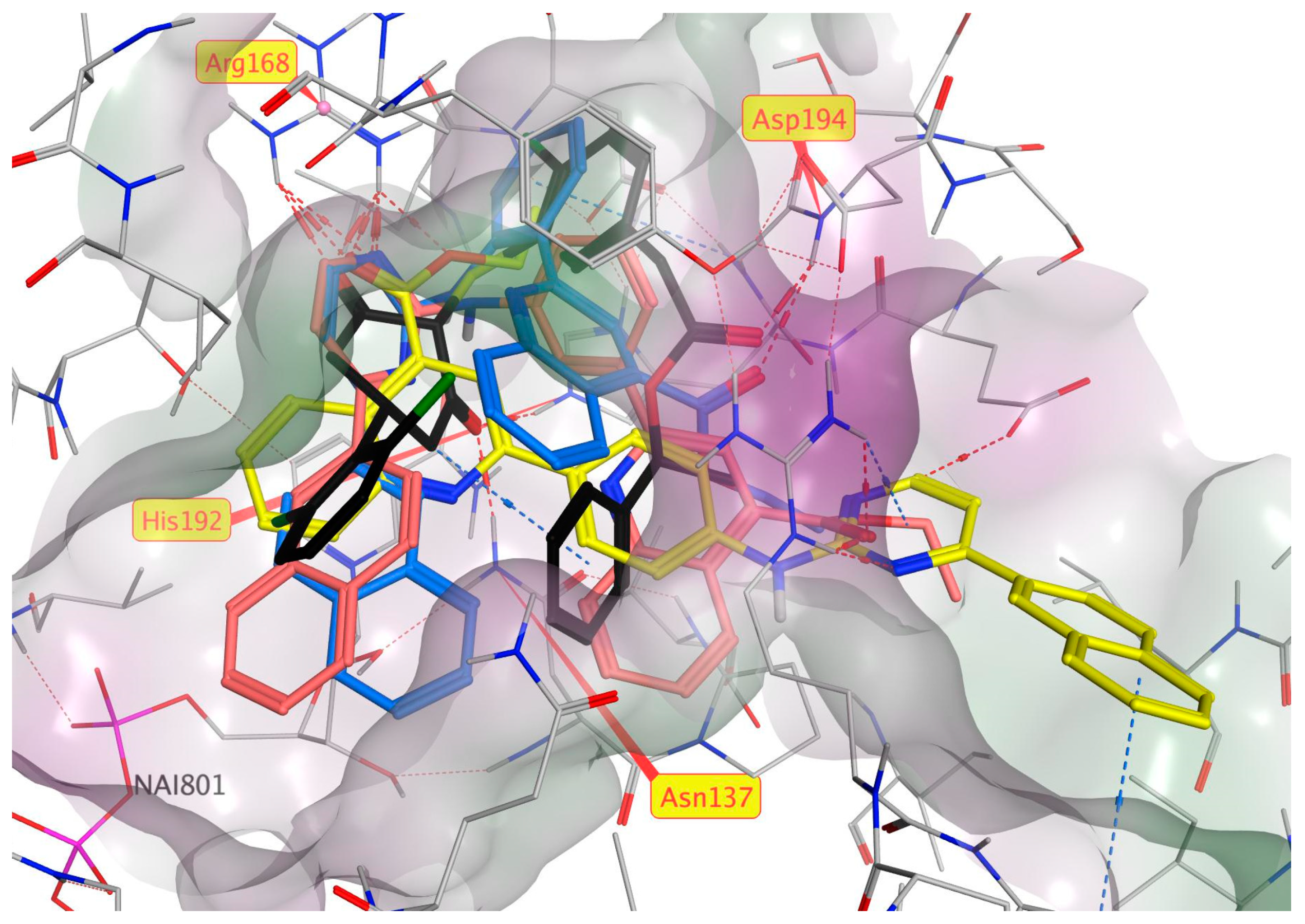
3.3. Molecular Modelling
3.4. Human Lactate Dehydrogenase a Enzymatic Activity Assay
3.5. Human Lactate Dehydrogenase B Enzymatic Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural Basis for Altered Activity of M- and H-Isozyme Forms of Human Lactate Dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Fiume, L.; Manerba, M.; Vettraino, M.; Di Stefano, G. Inhibition of Lactate Dehydrogenase Activity as an Approach to Cancer Therapy. Future Med. Chem. 2014, 6, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Osaka, N.; Sasaki, A.T. Beyond Warburg: LDHA Activates RAC for Tumour Growth. Nat. Metab. 2022, 4, 1623–1625. [Google Scholar] [CrossRef]
- Doherty, J.R.; Cleveland, J.L. Targeting Lactate Metabolism for Cancer Therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2015, 26, 3–17. [Google Scholar] [CrossRef]
- Rani, R.; Kumar, V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (hLDH5) Inhibitors: A Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2016, 59, 487–496. [Google Scholar] [CrossRef]
- Woodford, M.R.; Chen, V.Z.; Backe, S.J.; Bratslavsky, G.; Mollapour, M. Structural and Functional Regulation of Lactate Dehydrogenase-A in Cancer. Future Med. Chem. 2020, 12, 439–455. [Google Scholar] [CrossRef]
- Claps, G.; Faouzi, S.; Quidville, V.; Chehade, F.; Shen, S.; Vagner, S.; Robert, C. The Multiple Roles of LDH in Cancer. Nat. Rev. Clin. Oncol. 2022, 19, 749–762. [Google Scholar] [CrossRef]
- Sharma, D.; Singh, M.; Rani, R. Role of LDH in Tumor Glycolysis: Regulation of LDHA by Small Molecules for Cancer Therapeutics. Semin. Cancer Biol. 2022, 87, 184–195. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate Dehydrogenase A: A Key Player in Carcinogenesis and Potential Target in Cancer Therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Sada, N.; Suto, S.; Suzuki, M.; Usui, S.; Inoue, T. Upregulation of Lactate Dehydrogenase A in a Chronic Model of Temporal Lobe Epilepsy. Epilepsia 2020, 61, e37–e42. [Google Scholar] [CrossRef]
- Yilmaz, M.; Tekten, B.O. Serum Prolactin Level and Lactate Dehydrogenase Activity in Patients with Epileptic and Nonepileptic Seizures: A Cross-Sectional Study. Medicine 2021, 100, e27329. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, A.M.; Smith, C.O.; Shum, L.C.; Awad, H.; Eliseev, R.A. Lactate Dehydrogenase Inhibition with Oxamate Exerts Bone Anabolic Effect. J. Bone Miner. Res. 2020, 35, 2432–2443. [Google Scholar] [CrossRef]
- Kim, J.-H.; Bae, K.-H.; Byun, J.-K.; Lee, S.; Kim, J.-G.; Lee, I.K.; Jung, G.-S.; Lee, Y.M.; Park, K.-G. Lactate Dehydrogenase-A Is Indispensable for Vascular Smooth Muscle Cell Proliferation and Migration. Biochem. Biophys. Res. Commun. 2017, 492, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, H.; Yu, C.; Lu, R.; Song, T.; Wang, X.; Tang, W.; Gao, Y. MiR-638 Repressed Vascular Smooth Muscle Cell Glycolysis by Targeting LDHA. Open Med. 2019, 14, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Bacchetta, J.; Lieske, J.C. Primary Hyperoxaluria Type 1: Novel Therapies at a Glance. Clin. Kidney J. 2022, 15, i17–i22. [Google Scholar] [CrossRef]
- Shee, K.; Stoller, M.L. Perspectives in Primary Hyperoxaluria—Historical, Current and Future Clinical Interventions. Nat. Rev. Urol. 2022, 19, 137–146. [Google Scholar] [CrossRef]
- Hoppe, B.; Martin-Higueras, C. Improving Treatment Options for Primary Hyperoxaluria. Drugs 2022, 82, 1077–1094. [Google Scholar] [CrossRef]
- Li, H.M.; Guo, H.L.; Xu, C.; Liu, L.; Hu, S.Y.; Hu, Z.H.; Jiang, H.H.; He, Y.M.; Li, Y.J.; Ke, J.; et al. Inhibition of Glycolysis by Targeting Lactate Dehydrogenase A Facilitates Hyaluronan Synthase 2 Synthesis in Synovial Fibroblasts of Temporomandibular Joint Osteoarthritis. Bone 2020, 141, 115584–115593. [Google Scholar] [CrossRef] [PubMed]
- Souto-Carneiro, M.M.; Klika, K.D.; Abreu, M.T.; Meyer, A.P.; Saffrich, R.; Sandhoff, R.; Jennemann, R.; Kraus, F.V.; Tykocinski, L.; Eckstein, V.; et al. Effect of Increased Lactate Dehydrogenase A Activity and Aerobic Glycolysis on the Proinflammatory Profile of Autoimmune CD8+ T Cells in Rheumatoid Arthritis. Arthritis Rheum. 2020, 72, 2050–2064. [Google Scholar] [CrossRef]
- Gupta, G.S. The Lactate and the Lactate Dehydrogenase in Inflammatory Diseases and Major Risk Factors in COVID-19 Patients. Inflammation 2022, 45, 2091–2123. [Google Scholar] [CrossRef] [PubMed]
- Vlasiou, M.; Nicolaidou, V.; Papaneophytou, C. Targeting Lactate Dehydrogenase-B as a Strategy to Fight Cancer: Identification of Potential Inhibitors by In Silico Analysis and In Vitro Screening. Pharmaceutics 2023, 15, 2411. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Banerjee, D. Lactate Dehydrogenases as Metabolic Links between Tumor and Stroma in the Tumor Microenvironment. Cancers 2019, 11, 750. [Google Scholar] [CrossRef]
- Shibata, S.; Sogabe, S.; Miwa, M.; Fujimoto, T.; Takakura, N.; Naotsuka, A.; Kitamura, S.; Kawamoto, T.; Soga, T. Identification of the First Highly Selective Inhibitor of Human Lactate Dehydrogenase B. Sci. Rep. 2021, 11, 21353. [Google Scholar] [CrossRef]
- McCleland, M.L.; Adler, A.S.; Shang, Y.; Hunsaker, T.; Truong, T.; Peterson, D.; Torres, E.; Li, L.; Haley, B.; Stephan, J.-P.; et al. An Integrated Genomic Screen Identifies LDHB as an Essential Gene for Triple-Negative Breast Cancer. Cancer Res. 2012, 72, 5812–5823. [Google Scholar] [CrossRef]
- McCleland, M.L.; Adler, A.S.; Deming, L.; Cosino, E.; Lee, L.; Blackwood, E.M.; Solon, M.; Tao, J.; Li, L.; Shames, D.; et al. Lactate Dehydrogenase B Is Required for the Growth of KRAS-Dependent Lung Adenocarcinomas. Clin. Cancer Res. 2013, 19, 773–784. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, H.; Xu, A.; Li, N.; Liu, J.; Liu, C.; Lv, D.; Wu, S.; Huang, L.; Yang, S.; et al. Elevation of Serum L-Lactate Dehydrogenase B Correlated with the Clinical Stage of Lung Cancer. Lung Cancer 2006, 54, 95–102. [Google Scholar] [CrossRef]
- Wang, R.; Li, J.; Zhang, C.; Guan, X.; Qin, B.; Jin, R.; Qin, L.; Xu, S.; Zhang, X.; Liu, R.; et al. Lactate Dehydrogenase B Is Required for Pancreatic Cancer Cell Immortalization Through Activation of Telomerase Activity. Front. Oncol. 2022, 12, 821620. [Google Scholar] [CrossRef]
- Deng, H.; Gao, Y.; Trappetti, V.; Hertig, D.; Karatkevich, D.; Losmanova, T.; Urzi, C.; Ge, H.; Geest, G.A.; Bruggmann, R.; et al. Targeting Lactate Dehydrogenase B-Dependent Mitochondrial Metabolism Affects Tumor Initiating Cells and Inhibits Tumorigenesis of Non-Small Cell Lung Cancer by Inducing mtDNA Damage. Cell. Mol. Life Sci. 2022, 79, 445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wan, Y.; Ma, J.; Gong, J.; Zhong, Z.; Cui, Y.; Zhang, H.; Da, Y.; Ma, J.; Li, C.; et al. Epigenetic Silencing of LDHB Promotes Hepatocellular Carcinoma by Remodeling the Tumor Microenvironment. Cancer Immunol. Immunother. 2024, 73, 127. [Google Scholar] [CrossRef]
- Bockstahler, M.; Salbach, C.; Müller, A.-M.; Kübler, A.; Müller, O.J.; Katus, H.A.; Frey, N.; Kaya, Z. LNA Oligonucleotide Mediates an Anti-Inflammatory Effect in Autoimmune Myocarditis via Targeting Lactate Dehydrogenase B. Immunology 2022, 165, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Saeed, K.; Jo, M.H.; Kim, M.W.; Lee, H.J.; Park, C.-B.; Lee, G.; Kim, M.O. LDHB Deficiency Promotes Mitochondrial Dysfunction Mediated Oxidative Stress and Neurodegeneration in Adult Mouse Brain. Antioxidants 2022, 11, 261. [Google Scholar] [CrossRef]
- Shi, F.; Zhang, G.; Li, J.; Shu, L.; Yu, C.; Ren, D.; Zhang, Y.; Zheng, P. Integrated Analysis of Single Cell-RNA Sequencing and Mendelian Randomization Identifies Lactate Dehydrogenase B as a Target of Melatonin in Ischemic Stroke. CNS Neurosci. Ther. 2024, 30, e14741. [Google Scholar] [CrossRef]
- Hoppe, B.; Koch, A.; Cochat, P.; Garrelfs, S.F.; Baum, M.A.; Groothoff, J.W.; Lipkin, G.; Coenen, M.; Schalk, G.; Amrite, A.; et al. Safety, Pharmacodynamics, and Exposure-Response Modeling Results from a First-in-Human Phase 1 Study of Nedosiran (PHYOX1) in Primary Hyperoxaluria. Kidney Int. 2022, 101, 626–634. [Google Scholar] [CrossRef]
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X. The Growth of siRNA-Based Therapeutics: Updated Clinical Studies. Biocem. Pharmacol. 2021, 189, 114432. [Google Scholar] [CrossRef]
- Lai, C.; Pursell, N.; Gierut, J.; Saxena, U.; Zhou, W.; Dills, M.; Diwanji, R.; Dutta, C.; Koser, M.; Nazef, N.; et al. Specific Inhibition of Hepatic Lactate Dehydrogenase Reduces Oxalate Production in Mouse Models of Primary Hyperoxaluria. Mol. Ther. 2018, 26, 1983–1995. [Google Scholar] [CrossRef] [PubMed]
- Amrite, A.; Fuentes, E.; Marbury, T.C.; Zhang, S. Safety, Pharmacokinetics, and Exposure-Response Modeling of Nedosiran in Participants with Severe Chronic Kidney Disease. Clin. Pharmacol. Drug Dev. 2023, 12, 1164–1177. [Google Scholar] [CrossRef]
- Moya-Garzon, M.D.; Gomez-Vidal, J.A.; Alejo-Armijo, A.; Altarejos, J.; Rodriguez-Madoz, J.R.; Fernandes, M.X.; Salido, E.; Salido, S.; Diaz-Gavilan, M. Small Molecule-Based Enzyme Inhibitors in the Treatment of Primary Hyperoxalurias. J. Pers. Med. 2021, 11, 74. [Google Scholar] [CrossRef]
- Billiard, J.; Dennison, J.B.; Briand, J.; Annan, R.S.; Chai, D.; Colón, M.; Dodson, C.S.; Gilbert, S.A.; Greshock, J.; Jing, J.; et al. Quinoline 3-Sulfonamides Inhibit Lactate Dehydrogenase A and Reverse Aerobic Glycolysis in Cancer Cells. Cancer Metab. 2013, 1, 19. [Google Scholar] [CrossRef]
- Granchi, C.; Paterni, I.; Rani, R.; Minutolo, F. Small-Molecule Inhibitors of Human LDH5. Future Med. Chem. 2013, 5, 1967–1991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.L.; He, Y.; Tam, K.Y. Targeting Cancer Metabolism to Develop Human Lactate Dehydrogenase (hLDH)5 Inhibitors. Drug Discov. Today 2018, 23, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Salido, S.; Alejo-Armijo, A.; Altarejos, J. Synthesis and hLDH Inhibitory Activity of Analogues to Natural Products with 2,8-Dioxabicyclo [3.3.1]Nonane Scaffold. Int. J. Mol. Sci. 2023, 24, 9925. [Google Scholar] [CrossRef] [PubMed]
- Rani, R.; Kumar, V. When Will Small Molecule Lactate Dehydrogenase Inhibitors Realize Their Potential in the Cancer Clinic? Future Med. Chem. 2017, 9, 1113–1115. [Google Scholar] [CrossRef]
- Díaz, I.; Salido, S.; Nogueras, M.; Cobo, J. Design and Synthesis of New Pyrimidine-Quinolone Hybrids as Novel hLDHA Inhibitors. Pharmaceuticals 2022, 15, 792. [Google Scholar] [CrossRef]
- Alejo-Armijo, A.; Cuadrado, C.; Altarejos, J.; Fernandes, M.X.; Salido, E.; Diaz-Gavilan, M.; Salido, S. Lactate Dehydrogenase A Inhibitors with a 2,8-Dioxabicyclo[3.3.1]Nonane Scaffold: A Contribution to Molecular Therapies for Primary Hyperoxalurias. Bioorganic Chem. 2022, 129, 106–127. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Insuasty, D.; Lima, S.; Gutiérrez, L.; Nogueras, M.; Marchal, A.; Abonia, R.; Andújar, S.; Spiegel, S.; Cobo, J.; et al. Design of New Quinolin-2-One-Pyrimidine Hybrids as Sphingosine Kinases Inhibitors. Bioorganic Chem. 2020, 94, 103414. [Google Scholar] [CrossRef]
- Vettorazzi, M.; Díaz, I.; Angelina, E.; Salido, S.; Gutierrez, L.; Alvarez, S.E.; Cobo, J.; Enriz, R.D. Second Generation of Pyrimidin-Quinolone Hybrids Obtained from Virtual Screening Acting as Sphingosine Kinase 1 Inhibitors and Potential Anticancer Agents. Bioorganic Chem. 2024, 144, 107112. [Google Scholar] [CrossRef]
- Wagman, A.S.; Boyce, R.S.; Brown, S.P.; Fang, E.; Goff, D.; Jansen, J.M.; Le, V.P.; Levine, B.H.; Ng, S.C.; Ni, Z.J.; et al. Synthesis, Binding Mode, and Antihyperglycemic Activity of Potent and Selective (5-Imidazol-2-Yl-4-Phenylpyrimidin-2-Yl)[2-(2-Pyridylamino)Ethyl]Amine Inhibitors of Glycogen Synthase Kinase 3. J. Med. Chem. 2017, 60, 8482–8514. [Google Scholar] [CrossRef]
- Taglieri, L.; Saccoliti, F.; Nicolai, A.; Peruzzi, G.; Madia, V.N.; Tudino, V.; Messore, A.; Di Santo, R.; Artico, M.; Taurone, S.; et al. Discovery of a Pyrimidine Compound Endowed with Antitumor Activity. Investig. New Drugs 2020, 38, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Oukoloff, K.; Nzou, G.; Varricchio, C.; Lucero, B.; Alle, T.; Kovalevich, J.; Monti, L.; Cornec, A.S.; Yao, Y.; James, M.J.; et al. Evaluation of the Structure-Activity Relationship of Microtubule-Targeting 1,2,4-Triazolo[1,5- a]Pyrimidines Identifies New Candidates for Neurodegenerative Tauopathies. J. Med. Chem. 2021, 64, 1073–1102. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Moghimi, S.; Toolabi, M.; Foroumadi, A. Pyrimidine-Based EGFR TK Inhibitors in Targeted Cancer Therapy. Eur. J. Med. Chem. 2021, 221, 113523. [Google Scholar] [CrossRef]
- Faraji, A.; Oghabi Bakhshaiesh, T.; Hasanvand, Z.; Motahari, R.; Nazeri, E.; Boshagh, M.A.; Firoozpour, L.; Mehrabi, H.; Khalaj, A.; Esmaeili, R.; et al. Design, Synthesis and Evaluation of Novel Thienopyrimidine-Based Agents Bearing Diaryl Urea Functionality as Potential Inhibitors of Angiogenesis. Eur. J. Med. Chem. 2021, 209, 112942. [Google Scholar] [CrossRef]
- Wang, S.; Yuan, X.H.; Wang, S.Q.; Zhao, W.; Chen, X.B.; Yu, B. FDA-Approved Pyrimidine-Fused Bicyclic Heterocycles for Cancer Therapy: Synthesis and Clinical Application. Eur. J. Med. Chem. 2021, 214, 113218. [Google Scholar] [CrossRef]
- Dinesh, T.V.; Malgija, B.; Ponraj, M.R.; Muralakar, P.; Thathapudi, J.J.; Kandasamy, R.; Alagarmalai, J.; Balakrishnan, A.B.; Ramar, P.S.; James, J.V.; et al. Design of Novel Pyrimidine Based Remdesivir Analogues with Dual Target Specificity for SARS CoV-2: A Computational Approach. Int. J. Biol. Macromol. 2023, 242, 124443. [Google Scholar] [CrossRef]
- Finger, V.; Kufa, M.; Soukup, O.; Castagnolo, D.; Roh, J.; Korabecny, J. Pyrimidine Derivatives with Antitubercular Activity. Eur. J. Med. Chem. 2023, 246, 114946. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Tan, J.; Zhuang, Y.; Zhong, M.; Xiong, Y.; Ma, J.; Yang, Y.; Gao, Z.; Zhao, J.; Ye, Z.; et al. Identification of Crucial Genes of Pyrimidine Metabolism as Biomarkers for Gastric Cancer Prognosis. Cancer Cell Int. 2021, 21, 668. [Google Scholar] [CrossRef]
- Yadav, T.T.; Moin Shaikh, G.; Kumar, M.S.; Chintamaneni, M.; YC, M. A Review on Fused Pyrimidine Systems as EGFR Inhibitors and Their Structure-Activity Relationship. Front. Chem. 2022, 10, 861288. [Google Scholar] [CrossRef]
- Zhou, J.; Fang, S.; Zhang, D.; Qu, Y.; Wang, L.; Pan, S.; Li, L.; Li, J.; Du, W.; Wu, Q. A Novel Pyrimidine-Based Two-Photon Fluorogenic Probe for Rapidly Visualizing Nitroreductase Activity in Hypoxic Cancer Cells and in Vivo. Sens. Actuators B Chem. 2023, 390, 134015. [Google Scholar] [CrossRef]
- Senerovic, L.; Opsenica, D.; Moric, I.; Aleksic, I.; Spasić, M.; Vasiljevic, B. Quinolines and Quinolones as Antibacterial, Antifungal, Anti-Virulence, Antiviral and Anti-Parasitic Agents. Adv. Exp. Med. Biol. 2020, 1282, 37–69. [Google Scholar] [CrossRef] [PubMed]
- Chai, N.; Sun, A.; Zhu, X.; Li, Y.; Wang, R.; Zhang, Y.; Mao, Z. Antifungal Evaluation of Quinoline-Chalcone Derivatives Combined with FLC against Drug-Resistant Candida Albicans. Bioorganic Med. Chem. Lett. 2023, 86, 129242. [Google Scholar] [CrossRef]
- Chen, H.; Mi, J.; Li, S.; Liu, Z.; Yang, J.; Chen, R.; Wang, Y.; Ban, Y.; Zhou, Y.; Dong, W.; et al. Design, Synthesis and Evaluation of Quinoline-O-Carbamate Derivatives as Multifunctional Agents for the Treatment of Alzheimer’s Disease. J. Enzym. Inhib. Med. Chem. 2023, 38, 2169682. [Google Scholar] [CrossRef] [PubMed]
- Elebiju, O.F.; Ajani, O.O.; Oduselu, G.O.; Ogunnupebi, T.A.; Adebiyi, E. Recent Advances in Functionalized Quinoline Scaffolds and Hybrids—Exceptional Pharmacophore in Therapeutic Medicine. Front. Chem. 2023, 10, 1074331. [Google Scholar] [CrossRef] [PubMed]
- Jeleń, M.; Morak-Młodawska, B.; Korlacki, R. Anticancer Activities of Tetra-, Penta-, and Hexacyclic Phenothiazines Modified with Quinoline Moiety. J. Mol. Struct. 2023, 1287, 135700. [Google Scholar] [CrossRef]
- Patel, K.B.; Kumari, P. A Review: Structure-Activity Relationship and Antibacterial Activities of Quinoline Based Hybrids. J. Mol. Struct. 2022, 1268, 133634. [Google Scholar] [CrossRef]
- Lauria, A.; La Monica, G.; Bono, A.; Martorana, A. Quinoline Anticancer Agents Active on DNA and DNA-Interacting Proteins: From Classical to Emerging Therapeutic Targets. Eur. J. Med. Chem. 2021, 220, 113555. [Google Scholar] [CrossRef]
- Uddin, A.; Chawla, M.; Irfan, I.; Mahajan, S.; Singh, S.; Abid, M. Medicinal Chemistry Updates on Quinoline-And Endoperoxide-Based Hybrids with Potent Antimalarial Activity. RSC Med. Chem. 2021, 12, 24–42. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Z.; Chan, D.; Chung, P.Y.; Wang, Y.; Chan, A.S.C.; Law, S.; Lam, K.H.; Tang, J.C.O. The Anticancer Effect of a Novel Quinoline Derivative 91b1 through Downregulation of Lumican. Int. J. Mol. Sci. 2022, 23, 13181. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-Based Drug Discovery—The Importance of High-Quality Molecule Libraries. Mol. Oncol. 2022, 16, 3761–3777. [Google Scholar] [CrossRef]
- Moinul, M.; Khatun, S.; Amin, S.A.; Jha, T.; Gayen, S. Recent Trends in Fragment-Based Anticancer Drug Design Strategies against Different Targets: A Mini-Review. Biochem. Pharmacol. 2022, 206, 115301. [Google Scholar] [CrossRef] [PubMed]
- Villemagne, B.; Faion, L.; Tangara, S.; Willand, N. Recent Advances in Fragment-Based Strategies against Tuberculosis. Eur. J. Med. Chem. 2023, 258, 115569. [Google Scholar] [CrossRef] [PubMed]
- Gaur, A.S.; John, L.; Kumar, N.; Vivek, M.R.; Nagamani, S.; Mahanta, H.J.; Sastry, G.N. Towards Systematic Exploration of Chemical Space: Building the Fragment Library Module in Molecular Property Diagnostic Suite. Mol. Divers. 2023, 27, 1459–1468. [Google Scholar] [CrossRef]
- Davoine, C.; Pochet, L.; Fillet, M. Advances in Analytical Technologies Detecting and Characterizing Noncovalent Interactions for Fragment-Based Drug Discovery. TrAC Trends Anal. Chem. 2023, 166, 117161. [Google Scholar] [CrossRef]
- Fauber, B.P.; Dragovich, P.S.; Chen, J.; Corson, L.B.; Ding, C.Z.; Eigenbrot, C.; Giannetti, A.M.; Hunsaker, T.; Labadie, S.; Liu, Y.; et al. Identification of 2-Amino-5-Aryl-Pyrazines as Inhibitors of Human Lactate Dehydrogenase. Bioorganic Med. Chem. Lett. 2013, 23, 5533–5539. [Google Scholar] [CrossRef]
- Rai, G.; Brimacombe, K.R.; Mott, B.T.; Urban, D.J.; Hu, X.; Yang, S.-M.; Lee, T.D.; Cheff, D.M.; Kouznetsova, J.; Benavides, G.A.; et al. Discovery and Optimization of Potent, Cell-Active Pyrazole-Based Inhibitors of Lactate Dehydrogenase (LDH). J. Med. Chem. 2017, 60, 9184–9204. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Urban, D.J.; Mott, B.T.; Hu, X.; Yang, S.-M.; Benavides, G.A.; Johnson, M.S.; Squadrito, G.L.; Brimacombe, K.R.; Lee, T.D.; et al. Pyrazole-Based Lactate Dehydrogenase Inhibitors with Optimized Cell Activity and Pharmacokinetic Properties. J. Med. Chem. 2020, 63, 10984–11011. [Google Scholar] [CrossRef]
- Hui, Q.; Li, X.; Fan, W.; Gao, C.; Zhang, L.; Qin, H.; Wei, L.; Zhang, L. Discovery of 2-(4-Acrylamidophenyl)-Quinoline-4-Carboxylic Acid Derivatives as Potent SIRT3 Inhibitors. Front. Chem. 2022, 10, 880067. [Google Scholar] [CrossRef]
- Zarghi, A.; Ghodsi, R.; Azizi, E.; Daraie, B.; Hedayati, M.; Dadrass, O.G. Synthesis and Biological Evaluation of New 4-Carboxyl Quinoline Derivatives as Cyclooxygenase-2 Inhibitors. Bioorganic Med. Chem. 2009, 17, 5312–5317. [Google Scholar] [CrossRef]
- Petrović, M.M.; Roschger, C.; Lang, K.; Zierer, A.; Mladenović, M.; Trifunović, S.; Mandić, B.; Joksović, M.D. Synthesis and Biological Evaluation of New Quinoline-4-Carboxylic Acid-Chalcone Hybrids as Dihydroorotate Dehydrogenase Inhibitors. Arch. Pharm. 2023, 356, e2200374. [Google Scholar] [CrossRef]
- Abdelwahid, M.A.S.; Elsaman, T.; Mohamed, M.S.; Latif, S.A.; Mukhtar, M.M.; Mohamed, M.A. Synthesis, Characterization, and Antileishmanial Activity of Certain Quinoline-4-Carboxylic Acids. J. Chem. 2019, 2019, e2859637. [Google Scholar] [CrossRef]
- Dubrovin, A.N.; Mikhalev, A.I.; Ukhov, S.V.; Goldshtein, A.G.; Novikova, V.V.; Odegova, T.F.; Makhmudov, R.R. Synthesis, Properties, and Biological Activities of 2-Methyl- and 2-Styrylquinoline-4-Carboxylic Acids. Pharm. Chem. J. 2015, 49, 309–312. [Google Scholar] [CrossRef]
- Shvekhgeimer, M.G.-A. The Pfitzinger Reaction. (Review). Chem. Heterocycl. Compd. 2004, 40, 257–294. [Google Scholar] [CrossRef]
- Ma, J.-X.; Moran, E.; Deng, G.; Duerfeldt, A.S. Phenylquinoline Compositions for Treatment of Ocular Disorders and Conditions. World Patent WO2017189958, 28 April 2017. [Google Scholar]
- Shvekhgeimer, M.-G.A.; Kondrashova, N.N. Synthesis of Novel Derivatives of 2-Phenylquinoline-4-Carboxylic Acid. Dokl. Chem. 2002, 383, 72–74. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Ramasamy, R.; Shekhtman, A.; Rai, V.; Manigrasso, M.B. Preparation of Quinoline Compounds as Modulators of RAGE Activity. World Patent WO2017184547, 18 April 2017. [Google Scholar]
- Dulla, B.; Wan, B.; Franzblau, S.G.; Kapavarapu, R.; Reiser, O.; Iqbal, J.; Pal, M. Construction and Functionalization of Fused Pyridine Ring Leading to Novel Compounds as Potential Antitubercular Agents. Bioorganic Med. Chem. Lett. 2012, 22, 4629–4635. [Google Scholar] [CrossRef]
- Westermarck, J. Inhibition of Adaptive Therapy Tolerance in Cancer: Is Triplet Mitochondrial Targeting the Key? Mol. Oncol. 2023, 17, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Ždralević, M.; Brand, A.; Di Ianni, L.; Dettmer, K.; Reinders, J.; Singer, K.; Peter, K.; Schnell, A.; Bruss, C.; Decking, S.-M.; et al. Double Genetic Disruption of Lactate Dehydrogenases A and B Is Required to Ablate the “Warburg Effect” Restricting Tumor Growth to Oxidative Metabolism. J. Biol. Chem. 2018, 293, 15947–15961. [Google Scholar] [CrossRef]
- APEX4 v2021.10-0, Bruker AXS Inc.: Madison, WI, USA, 2021.
- Adam, M.; Hovestreydt, E.; Ruf, M.; Kaercher, J. Reaching a new highpoint with crystallography software -APEX3. Acta Crystallogr. A 2015, 71, s194–s195. [Google Scholar] [CrossRef]
- SAINT V8.40B, Bruker AXS Inc.: Madison, WI, USA, 2015.
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. It WinGX and It ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From Visualization to Analysis, Design and Prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- John, H. Chinolinderivate, XLII. Derivate Des 2-Phenyl-4′-Amino-Chinolins. J. Prakt. Chem. 1934, 139, 97–104. [Google Scholar] [CrossRef]
- Woods, D.; Dimitratos, S.; Justice, R. Improved Honeybee Repellents and Uses Thereof. World Patent WO2014179785, 11 June 2014. [Google Scholar]
- Goto, J.; Kataoka, R.; Hirayama, N. Ph4Dock: Pharmacophore-Based Protein—Ligand Docking. J. Med. Chem. 2004, 47, 6804–6811. [Google Scholar] [CrossRef]
- Hajalsiddig, T.T.H.; Saeed, A.E.M. QSAR and Molecular Docking Studies on 4-Quinoline Carboxylic Acid Derivatives as Inhibition of Vesicular Stomatitis Virus Replication. Eur. J. Chem. 2019, 10, 45–51. [Google Scholar] [CrossRef]

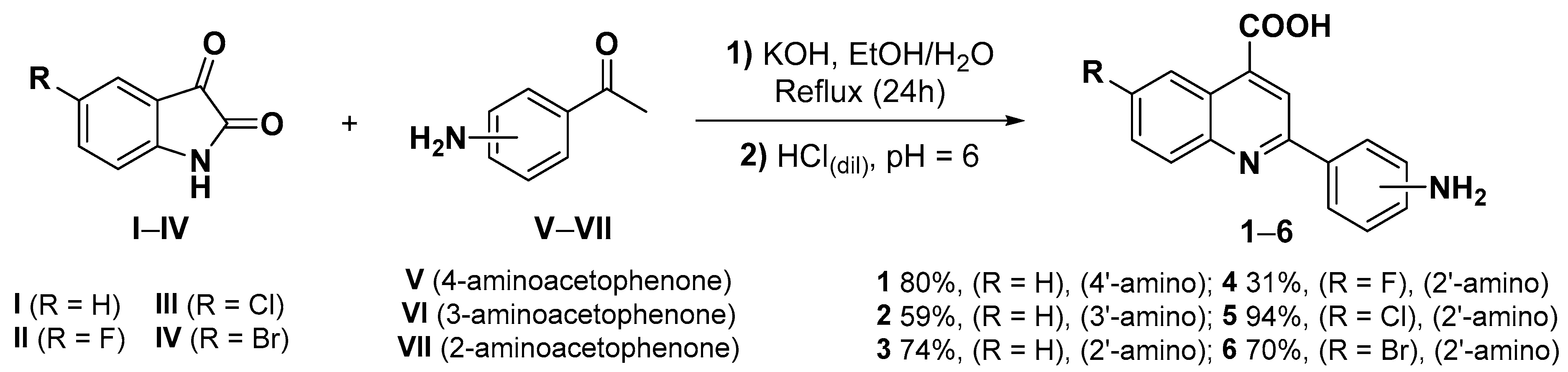




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid | R | Ar | R.T. (min) | Yield | Hybrid | R | Ar | R.T. (min) | Yield |
|---|---|---|---|---|---|---|---|---|---|
| 13a | H | 4-ClC6H4 | 50 | 85% | 16c | F | C6H5 | 20 | 84% |
| 13b | H | Naphth-2-yl | 60 | 86% | 16d | F | Styryl | 10 | 70% |
| 14a | H | 4-ClC6H4 | 20 | 81% | 17a | Cl | 4-ClC6H4 | 20 | 85% |
| 14b | H | Naphth-2-yl | 20 | 78% | 17b | Cl | Naphth-2-yl | 20 | 91% |
| 15a | H | 4-ClC6H4 | 40 | 78% | 17c | Cl | C6H5 | 20 | 80% |
| 15b | H | Naphth-2-yl | 40 | 83% | 17d | Cl | Styryl | 10 | 72% |
| 15c | H | C6H5 | 30 | 85% | 18a | Br | 4-ClC6H4 | 20 | 70% |
| 15d | H | Styryl | 15 | 65% | 18b | Br | Naphth-2-yl | 20 | 84% |
| 16a | F | 4-ClC6H4 | 30 | 88% | 18c | Br | C6H5 | 20 | 76% |
| 16b | F | Naphth-2-yl | 20 | 89% | 18d | Br | Styryl | 20 | 75% |
| Substitution | Ar | Hybrid | IC50 a | R2 b |
|---|---|---|---|---|
| 1,4- | 4-ClC6H4 | 13a | 54.3 ± 2.9 | 0.8430 |
| Naphth-2-yl | 13b | 32.0 ± 4.8 | 0.8485 | |
| 1,3- | 4-ClC6H4 | 14a | 61.5 ± 3.7 | 0.9164 |
| Naphth-2-yl | 14b | 24.1 ± 7.3 | 0.9406 | |
| 1,2- | 4-ClC6H4 | 15a | 22.6 ± 1.1 | 0.9605 |
| Naphth-2-yl | 15b | 2.02 ± 0.73 | 0.9477 |
| hLDHA | hLDHB | |||||
|---|---|---|---|---|---|---|
| R | Ar | Hybrid | IC50 a | R2 b | IC50 a | R2 b |
| H | Naphth-2-yl | 15b | 2.02 ± 0.73 | 0.9477 | 4.41 ± 1.54 | 0.8873 |
| C6H5 | 15c | 9.40 ± 1.28 | 0.9373 | >100 | - | |
| Styryl | 15d | 3.11 ± 0.85 | 0.8914 | >100 | - | |
| F | 4-ClC6H4 | 16a | 4.33 ± 0.47 | 0.9410 | 6.49 ± 1.54 | 0.8117 |
| Naphth-2-yl | 16b | 2.24 ± 0.37 | 0.9561 | 4.19 ± 1.40 | 0.8731 | |
| C6H5 | 16c | 2.94 ± 0.04 | 0.9719 | 6.71 ± 0.72 | 0.8762 | |
| Styryl | 16d | 7.26 ± 1.46 | 0.8416 | >100 | - | |
| Cl | 4-ClC6H4 | 17a | 1.43 ± 0.15 | 0.9474 | 1.67 ± 0.02 | 0.8548 |
| Naphth-2-yl | 17b | 2.36 ± 0.37 | 0.9376 | 3.72 ± 0.57 | 0.9667 | |
| C6H5 | 17c | 4.67 ± 1.72 | 0.9075 | 3.09 ± 0.99 | 0.8586 | |
| Styryl | 17d | 3.50 ± 0.49 | 0.8989 | 8.77 ± 1.06 | 0.8177 | |
| Br | 4-ClC6H4 | 18a | 2.32 ± 0.09 | 0.8976 | 3.47 ± 0.74 | 0.8437 |
| Naphth-2-yl | 18b | 1.71 ± 0.08 | 0.9453 | 1.27 ± 0.35 | 0.8601 | |
| C6H5 | 18c | 1.07 ± 0.41 | 0.9045 | 4.26 ± 0.85 | 0.8538 | |
| Styryl | 18d | 1.57 ± 0.03 | 0.9574 | 1.43 ± 0.02 | 0.8297 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz, I.; Salido, S.; Nogueras, M.; Cobo, J. Synthesis of Ethyl Pyrimidine-Quinolincarboxylates Selected from Virtual Screening as Enhanced Lactate Dehydrogenase (LDH) Inhibitors. Int. J. Mol. Sci. 2024, 25, 9744. https://doi.org/10.3390/ijms25179744
Díaz I, Salido S, Nogueras M, Cobo J. Synthesis of Ethyl Pyrimidine-Quinolincarboxylates Selected from Virtual Screening as Enhanced Lactate Dehydrogenase (LDH) Inhibitors. International Journal of Molecular Sciences. 2024; 25(17):9744. https://doi.org/10.3390/ijms25179744
Chicago/Turabian StyleDíaz, Iván, Sofía Salido, Manuel Nogueras, and Justo Cobo. 2024. "Synthesis of Ethyl Pyrimidine-Quinolincarboxylates Selected from Virtual Screening as Enhanced Lactate Dehydrogenase (LDH) Inhibitors" International Journal of Molecular Sciences 25, no. 17: 9744. https://doi.org/10.3390/ijms25179744
APA StyleDíaz, I., Salido, S., Nogueras, M., & Cobo, J. (2024). Synthesis of Ethyl Pyrimidine-Quinolincarboxylates Selected from Virtual Screening as Enhanced Lactate Dehydrogenase (LDH) Inhibitors. International Journal of Molecular Sciences, 25(17), 9744. https://doi.org/10.3390/ijms25179744