
Experimental Cell Models for Investigating Neurodegenerative Diseases


Abstract
1. Introduction
2. Main Text
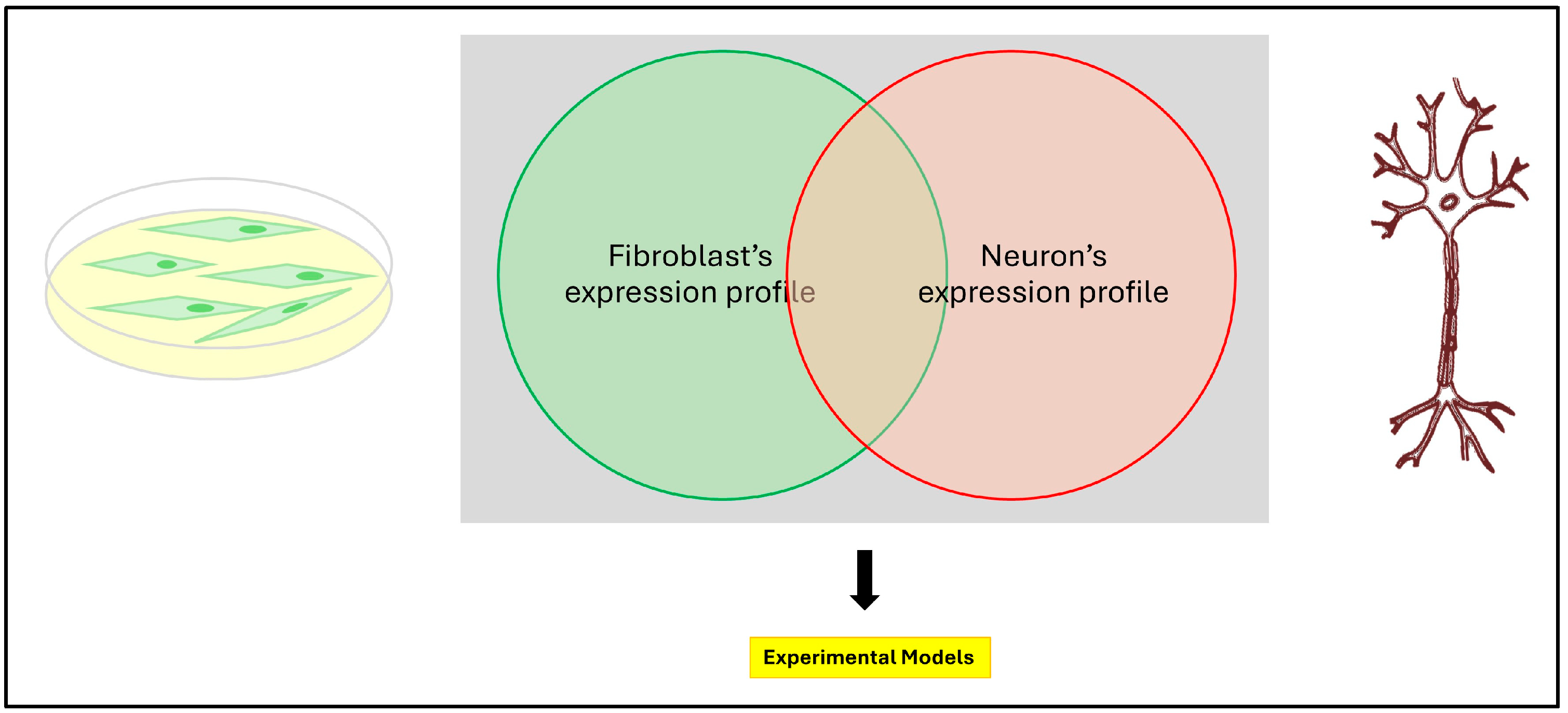
2.1. Primary Patient Cell Lines
2.2. Human Induced Pluripotent Stem Cells (iPSCs)
2.3. Organoids
3. Current Challenges and Future Directions
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Antony, P.M.; Diederich, N.J.; Krüger, R.; Balling, R. The hallmarks of Parkinson’s disease. FEBS J. 2013, 23, 5981–5993. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 2, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 2, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Funayama, M.; Nishioka, K.; Li, Y.; Hattori, N. Molecular genetics of Parkinson’s disease: Contributions and global trends. J. Hum. Genet. 2023, 3, 125–130. [Google Scholar] [CrossRef]
- Williams, E.T.; Chen, X.; Moore, D.J. VPS35, the Retromer Complex and Parkinson’s Disease. J. Park. Dis. 2017, 2, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, J.; Yip, O.; Kim, M.J.; Huang, Y. Cell type-specific roles of APOE4 in Alzheimer disease. Nat. Rev. Neurosci. 2024, 2, 91–110. [Google Scholar] [CrossRef]
- van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-β-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 1, 21–35. [Google Scholar] [CrossRef]
- Talbott, E.O.; Malek, A.M.; Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 138, pp. 225–238. [Google Scholar] [CrossRef]
- Martinello, C.; Panza, E.; Orlacchio, A. Hereditary spastic paraplegias proteome: Common pathways and pathogenetic mechanisms. Expert Rev. Proteom. 2023, 20, 171–188. [Google Scholar] [CrossRef]
- Panza, E.; Meyyazhagan, A.; Orlacchio, A. Hereditary spastic paraplegia: Genetic heterogeneity and common pathways. Exp. Neurol. 2022, 357, 114203. [Google Scholar] [CrossRef]
- Aversano, S.; Caiazza, C.; Caiazzo, M. Induced pluripotent stem cell-derived and directly reprogrammed neurons to study neurodegenerative diseases: The impact of aging signatures. Front. Aging Neurosci. 2022, 14, 1069482. [Google Scholar] [CrossRef]
- Morello, G.; La Cognata, V.; Guarnaccia, M.; La Bella, V.; Conforti, F.L.; Cavallaro, S. A Diagnostic Gene-Expression Signature in Fibroblasts of Amyotrophic Lateral Sclerosis. Cells 2023, 14, 1884. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.C.; Messer, A.; Peterson, A. The motor neuron degeneration (mnd) gene acts intrinsically in motor neurons and peripheral fibroblasts. Mol. Cell. Neurosci. 1997, 3, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Kremerm, L.S.; Baderm, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef]
- Hauser, S.; Schuster, S.; Heuten, E.; Höflinger, P.; Admard, J.; Schelling, Y.; Velic, A.; Macek, B.; Ossowski, S.; Schöls, L. Comparative Transcriptional Profiling of Motor Neuron Disorder-Associated Genes in Various Human Cell Culture Models. Front. Cell Dev. Biol. 2020, 8, 544043. [Google Scholar] [CrossRef]
- Corenblum, M.J.; McRobbie-Johnson, A.; Carruth, E.; Bernard, K.; Luo, M.; Mandarino, L.J.; Peterson, S.; Sans-Fuentes, M.A.; Billheimer, D.; Maley, T.; et al. Parallel neurodegenerative phenotypes in sporadic Parkinson’s disease fibroblasts and midbrain dopamine neurons. Prog. Neurobiol. 2023, 229, 102501. [Google Scholar] [CrossRef] [PubMed]
- Iannello, G.; Patel, A.; Sirabella, D.; Diaz, A.G.; Hoover, B.N.; Sarmah, H.; Corneo, B. Simple, Fast, and Efficient Method for Derivation of Dermal Fibroblasts From Skin Biopsies. Curr. Protoc. 2023, 3, e714. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Yu, X. Fibroblasts: New players in the central nervous system? Fundam. Res. 2023, 4, 262–266. [Google Scholar] [CrossRef]
- Olesen, M.A.; Villavicencio-Tejo, F.; Quintanilla, R.A. The use of fibroblasts as a valuable strategy for studying mitochondrial impairment in neurological disorders. Transl. Neurodegener. 2022, 1, 36. [Google Scholar] [CrossRef]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef]
- Eysert, F.; Kinoshita, P.F.; Lagarde, J.; Lacas-Gervais, S.; Xicota, L.; Dorothée, G.; Bottlaender, M.; Checler, F.; Potier, M.C.; Sarazin, M.; et al. Mitochondrial alterations in fibroblasts from sporadic Alzheimer’s disease (AD) patients correlate with AD-related clinical hallmarks. Acta Neuropathol. Commun. 2024, 1, 90. [Google Scholar] [CrossRef]
- Galvagnion, C.; Marlet, F.R.; Cerri, S.; Schapira, A.H.V.; Blandini, F.; Di Monte, D.A. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. Brain 2022, 3, 1038–1051. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Castanedo-Vázquez, D.; Gil-Bea, F.; Tapia, O.; Arozamena, J.; Durán-Vían, C.; Sedano, M.J.; Berciano, M.T.; Lopez de Munain, A.; Lafarga, M. ALS-derived fibroblasts exhibit reduced proliferation rate, cytoplasmic TDP-43 aggregation and a higher susceptibility to DNA damage. J. Neurol. 2020, 267, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Imberechts, D.; Kinnart, I.; Wauters, F.; Terbeek, J.; Manders, L.; Wierda, K.; Eggermont, K.; Madeiro, R.F.; Sue, C.; Verfaillie, C.; et al. DJ-1 is an essential downstream mediator in PINK1/parkin-dependent mitophagy. Brain 2022, 12, 4368–4384. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Toledo, G.; Silva-Lucero, M.D.; Herrera-Díaz, J.; García, D.E.; Arias-Montaño, J.A.; Cardenas-Aguayo, M.D. Patient-Derived Fibroblasts with Presenilin-1 Mutations, That Model Aspects of Alzheimer’s Disease Pathology, Constitute a Potential Object for Early Diagnosis. Front. Aging Neurosci. 2022, 14, 921573. [Google Scholar] [CrossRef]
- Ibáñez-Salazar, A.; Bañuelos-Hernández, B.; Rodríguez-Leyva, I.; Chi-Ahumada, E.; Monreal-Escalante, E.; Jiménez-Capdeville, M.E.; Rosales-Mendoza, S. Oxidative Stress Modifies the Levels and Phosphorylation State of Tau Protein in Human Fibroblasts. Front. Neurosci. 2017, 11, 495. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 6, 4566–4575. [Google Scholar] [CrossRef]
- Hung, C.L.; Maiuri, T.; Bowie, L.E.; Gotesman, R.; Son, S.; Falcone, M.; Giordano, J.V.; Gillis, T.; Mattis, V.; Lau, T.; et al. A patient-derived cellular model for Huntington’s disease reveals phenotypes at clinically relevant CAG lengths. Mol. Biol. Cell 2018, 23, 2809–2820. [Google Scholar] [CrossRef]
- Månberg, A.; Skene, N.; Sanders, F.; Trusohamn, M.; Remnestål, J.; Szczepińska, A.; Aksoylu, I.S.; Lönnerberg, P.; Ebarasi, L.; Wouters, S.; et al. Altered perivascular fibroblast activity precedes ALS disease onset. Nat. Med. 2021, 4, 640–646. [Google Scholar] [CrossRef]
- Romano, N.; Catalani, A.; Lattante, S.; Belardo, A.; Proietti, S.; Bertini, L.; Silvestri, F.; Catalani, E.; Cervia, D.; Zolla, L.; et al. ALS skin fibroblasts reveal oxidative stress and ERK1/2-mediated cytoplasmic localization of TDP-43. Cell. Signal 2020, 70, 109591. [Google Scholar] [CrossRef]
- Deng, R.; Medico-Salsench, E.; Nikoncuk, A.; Ramakrishnan, R.; Lanko, K.; Kühn, N.A.; van der Linde, H.C.; Lor-Zade, S.; Albuainain, F.; Shi, Y.; et al. AMFR dysfunction causes autosomal recessive spastic paraplegia in human that is amenable to statin treatment in a preclinical model. Acta Neuropathol. 2023, 2, 353–368. [Google Scholar] [CrossRef]
- MIM Number: 272200; Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University: Baltimore, MD, USA, 2020.
- Schlotawa, L.; Tyka, K.; Kettwig, M.; Ahrens-Nicklas, R.C.; Baud, M.; Berulava, T.; Brunetti-Pierri, N.; Gagne, A.; Herbst, Z.M.; Maguire, J.A.; et al. Drug screening identifies tazarotene and bexarotene as therapeutic agents in multiple sulfatase deficiency. EMBO Mol. Med. 2023, 3, e14837. [Google Scholar] [CrossRef]
- Rehman, A.; Fatima, I.; Noor, F.; Qasim, M.; Wang, P.; Jia, J.; Alshabrmim, F.M.; Liao, M. Role of small molecules as drug candidates for reprogramming somatic cells into induced pluripotent stem cells: A comprehensive review. Comput. Biol. Med. 2024, 177, 108661. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 5858, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 5, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Pazzin, D.B.; Previato, T.T.R.; Gonçalves, J.I.B.; Zanirati, G.; Xavier, F.A.C.; da Costa, J.C.; Marinowic, D.R. Induced Pluripotent Stem Cells and Organoids in Advancing Neuropathology Research and Therapies. Cells 2024, 9, 745. [Google Scholar] [CrossRef]
- van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 5, 434–444. [Google Scholar] [CrossRef]
- Ayeni, E.A.; Aldossary, A.M.; Ayejoto, D.A.; Gbadegesin, L.A.; Alshehri, A.A.; Alfassam, H.A.; Afewerky, H.K.; Almughem, F.A.; Bello, S.M.; Tawfik, E.A. Neurodegenerative Diseases: Implications of Environmental and Climatic Influences on Neurotransmitters and Neuronal Hormones Activities. Int. J. Environ. Res. Public Health 2022, 19, 12495. [Google Scholar] [CrossRef]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 2, 278–293.e9. [Google Scholar] [CrossRef]
- Matilla-Dueñas, A.; Corral-Juan, M.; Rodríguez-Palmero Seuma, A.; Vilas, D.; Ispierto, L.; Morais, S.; Sequeiros, J.; Alonso, I.; Volpini, V.; Serrano-Munuera, C.; et al. Rare Neurodegenerative Diseases: Clinical and Genetic Update. Adv. Exp. Med. Biol. 2017, 1031, 443–496. [Google Scholar] [CrossRef]
- Liu, Q.; Waltz, S.; Woodruff, G.; Ouyang, J.; Israel, M.A.; Herrera, C.; Sarsoza, F.; Tanzi, R.E.; Koo, E.H.; Ringman, J.M.; et al. Effect of potent γ-secretase modulator in human neurons derived from multiple presenilin 1-induced pluripotent stem cell mutant carriers. JAMA Neurol. 2014, 12, 1481–1489. [Google Scholar] [CrossRef]
- Martín-Maestro, P.; Gargini, R.A.; Sproul, A.; García, E.; Antón, L.C.; Noggle, S.; Arancio, O.; Avila, J.; García-Escudero, V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer’s Disease-Associated Presenilin 1 Mutation. Front. Mol. Neurosci. 2017, 10, 291. [Google Scholar] [CrossRef] [PubMed]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS ONE 2014, 1, e84547. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, H.; Ma, Y.; Shi, G.; Song, J.; Tang, Y.; Li, S.; Li, T.; Liu, N.; Tang, F.; et al. Early pathogenic event of Alzheimer’s disease documented in iPSCs from patients with PSEN1 mutations. Oncotarget 2017, 5, 7900–7913. [Google Scholar] [CrossRef]
- Budny, V.; Knöpfli, Y.; Meier, D.; Zürcher, K.; Bodenmann, C.; Peter, S.L.; Müller, T.; Tardy, M.; Cortijo, C.; Tackenberg, C. APOE4 Increases Energy Metabolism in APOE-Isogenic iPSC-Derived Neurons. Cells 2024, 14, 1207. [Google Scholar] [CrossRef]
- Wang, C.; Najm, R.; Xu, Q.; Jeong, D.E.; Walker, D.; Balestra, M.E.; Yoon, S.Y.; Yuan, H.; Li, G.; Miller, Z.A.; et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 2018, 5, 647–657. [Google Scholar] [CrossRef]
- Byers, B.; Cord, B.; Nguyen, H.N.; Schüle, B.; Fenno, L.; Lee, P.C.; Deisseroth, K.; Langston, J.W.; Pera, R.R.; Palmer, T.D. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate α-synuclein and are susceptible to oxidative stress. PLoS ONE 2011, 11, e26159. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011, 2, 440. [Google Scholar] [CrossRef] [PubMed]
- MIM Number: 614203; Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University: Baltimore, MD, USA, 2024.
- Bono, K.; Hara-Miyauchi, C.; Sumi, S.; Oka, H.; Iguchi, Y.; Okano, H.J. Endosomal dysfunction in iPSC-derived neural cells from Parkinson’s disease patients with VPS35 D620N. Mol. Brain 2020, 1, 137. [Google Scholar] [CrossRef] [PubMed]
- Yarkova, E.S.; Grigor’eva, E.V.; Medvedev, S.P.; Pavlova, S.V.; Zakian, S.M.; Malakhova, A.A. IPSC-Derived Astrocytes Contribute to In Vitro Modeling of Parkinson’s Disease Caused by the GBA1 N370S Mutation. Int. J. Mol. Sci. 2023, 1, 327. [Google Scholar] [CrossRef]
- Oshkolova, A.A.; Grekhnev, D.A.; Kruchinina, A.A.; Belikova, L.D.; Volovikov, E.A.; Lebedeva, O.S.; Bogomazova, A.N.; Vigont, V.A.; Lagarkova, M.A.; Kaznacheyeva, E.V. Comparison of the calcium signaling alterations in GABA-ergic medium spiny neurons produced from iPSCs of different origins. Biochimie 2024, 222, 63–71. [Google Scholar] [CrossRef]
- Workman, M.J.; Lim, R.G.; Wu, J.; Frank, A.; Ornelas, L.; Panther, L.; Galvez, E.; Perez, D.; Meepe, I.; Lei, S.; et al. Large-scale differentiation of iPSC-derived motor neurons from ALS and control subjects. Neuron 2023, 8, 1191–1204.e5. [Google Scholar] [CrossRef]
- Jothi, D.; Kulka, L.A.M. Strategies for modeling aging and age-related diseases. NPJ Aging 2024, 1, 32. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Xia, C.; Close, J.L.; Zhang, M.; He, J.; Huang, Z.; Halpern, A.R.; Long, B.; Miller, J.A.; Lein, E.S.; et al. Conservation and divergence of cortical cell organization in human and mouse revealed by MERFISH. Science 2022, 6601, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Lyu, Y.; Tan, Y.; Hu, Z. Progress in construction of mouse models to investigate the pathogenesis and immune therapy of human hematological malignancy. Front. Immunol. 2023, 14, 1195194. [Google Scholar] [CrossRef]
- Song, H.W.; Foreman, K.L.; Gastfriend, B.D.; Kuo, J.S.; Palecek, S.P.; Shusta, E.V. Transcriptomic comparison of human and mouse brain microvessels. Sci. Rep. 2020, 1, 12358. [Google Scholar] [CrossRef] [PubMed]
- Bjornson-Hooper, Z.B.; Fragiadakis, G.K.; Spitzer, M.H.; Chen, H.; Madhireddy, D.; Hu, K.; Lundsten, K.; McIlwain, D.R.; Nolan, G.P. A Comprehensive Atlas of Immunological Differences Between Humans, Mice, and Non-Human Primates. Front. Immunol. 2022, 13, 867015. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 1, 267. [Google Scholar] [CrossRef]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef]
- Hulse, J.; Bachstetter, A.; Paul, S.; Bhaskar, K. Editorial: Innate immunity and neurodegenerative diseases—Triggers from self and non-self. Front. Mol. Neurosci. 2023, 16, 1227896. [Google Scholar] [CrossRef]
- Bakken, T.E.; Jorstad, N.L.; Hu, Q.; Lake, B.B.; Tian, W.; Kalmbach, B.E.; Crow, M.; Hodge, R.D.; Krienen, F.M.; Sorensen, S.A.; et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature 2021, 7879, 111–119. [Google Scholar] [CrossRef]
- Pippucci, T.; Panza, E.; Pompilii, E.; Donadio, V.; Borreca, A.; Babalini, C.; Patrono, C.; Zuntini, R.; Kawarai, T.; Bernardi, G.; et al. Autosomal recessive hereditary spastic paraplegia with thin corpus callosum: A novel mutation in the SPG11 gene and further evidence for genetic heterogeneity. Eur. J. Neurol. 2009, 1, 121–126. [Google Scholar] [CrossRef]
- Pérez-Brangulí, F.; Buchsbaum, I.Y.; Pozner, T.; Regensburger, M.; Fan, W.; Schray, A.; Börstler, T.; Mishra, H.; Gräf, D.; Kohl, Z.; et al. Human SPG11 cerebral organoids reveal cortical neurogenesis impairment. Hum. Mol. Genet. 2019, 6, 961–971. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 2, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat. Commun. 2021, 1, 4744. [Google Scholar] [CrossRef]
- Gao, C.; Shi, Q.; Pan, X.; Chen, J.; Zhang, Y.; Lang, J.; Wen, S.; Liu, X.; Cheng, T.L.; Lei, K. Neuromuscular organoids model spinal neuromuscular pathologies in C9orf72 amyotrophic lateral sclerosis. Cell Rep. 2024, 3, 113892. [Google Scholar] [CrossRef] [PubMed]
- Szebényi, K.; Wenger, L.M.D.; Sun, Y.; Dunn, A.W.E.; Limegrover, C.A.; Gibbons, G.M.; Conci, E.; Paulsen, O.; Mierau, S.B.; Balmus, G.; et al. Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat. Neurosci. 2021, 11, 1542–1554. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 7, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Calixto, A.; Mukherjee, A.; Ramirez, S.; Sepulveda, S.; Sinha, T.; Al-Lahham, R.; De Gregorio, N.; Gherardelli, C.; Soto, C. Lewy Body-like Pathology and Loss of Dopaminergic Neurons in Midbrain Organoids Derived from Familial Parkinson’s Disease Patient. Cells 2023, 4, 625. [Google Scholar] [CrossRef] [PubMed]
- Muwanigwa, M.N.; Modamio-Chamarro, J.; Antony, P.M.A.; Gomez-Giro, G.; Krüger, R.; Bolognin, S.; Schwamborn, J.C. Alpha-synuclein pathology is associated with astrocyte senescence in a midbrain organoid model of familial Parkinson’s disease. Mol. Cell. Neurosci. 2024, 128, 103919. [Google Scholar] [CrossRef]
- Su, C.J.; Feng, Y.; Liu, T.T.; Liu, X.; Bao, J.J.; Shi, A.M.; Hu, D.M.; Liu, T.; Yu, Y.L. Thioredoxin-interacting protein induced α-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 9, 717–723. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 3, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.; Bolognin, S.; Poovathingal, S.K.; Magni, S.; Gérard, D.; Antony, P.M.; Nickels, S.L.; Salamanca, L.; Berger, E.; Smits, L.M.; et al. The Parkinson’s-disease-associated mutation LRRK2-G2019S alters dopaminergic differentiation dynamics via NR2F1. Cell Rep. 2021, 3, 109864. [Google Scholar] [CrossRef] [PubMed]
- Morrone Parfitt, G.; Coccia, E.; Goldman, C.; Whitney, K.; Reyes, R.; Sarrafha, L.; Nam, K.H.; Sohail, S.; Jones, D.R.; Crary, J.F.; et al. Disruption of lysosomal proteolysis in astrocytes facilitates midbrain organoid proteostasis failure in an early-onset Parkinson’s disease model. Nat. Commun. 2024, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- MIM Number: 608375; Online Mendelian Inheritance in Man, OMIM®. Johns Hopkins University: Baltimore, MD, USA, 2016.
- Wulansari, N.; Darsono, W.H.W.; Woo, H.J.; Chang, M.Y.; Kim, J.; Bae, E.J.; Sun, W.; Lee, J.H.; Cho, I.J.; Shin, H.; et al. Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci. Adv. 2021, 8, 1540. [Google Scholar] [CrossRef]
- Miura, Y.; Li, M.Y.; Revah, O.; Yoon, S.J.; Narazaki, G.; Pașca, S.P. Engineering brain assembloids to interrogate human neural circuits. Nat. Protoc. 2022, 1, 15–35. [Google Scholar] [CrossRef]
- Reumann, D.; Krauditsch, C.; Novatchkova, M.; Sozzi, E.; Wong, S.N.; Zabolocki, M.; Priouret, M.; Doleschall, B.; Ritzau-Reid, K.I.; Piber, M.; et al. In vitro modeling of the human dopaminergic system using spatially arranged ventral midbrain-striatum-cortex assembloids. Nat. Methods 2023, 12, 2034–2047. [Google Scholar] [CrossRef]
- Ozgun, A.; Lomboni, D.J.; Aylsworth, A.; Macdonald, A.; Staines, W.A.; Martina, M.; Schlossmacher, M.G.; Tauskela, J.S.; Woulfe, J.; Variola, F. Unraveling the assembloid: Real-time monitoring of dopaminergic neurites in an inter-organoid pathway connecting midbrain and striatal regions. Mater. Today Bio 2024, 25, 100992. [Google Scholar] [CrossRef]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 12, 2363–2374. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat. Commun. 2020, 11, 5540. [Google Scholar] [CrossRef]
- Vanova, T.; Sedmik, J.; Raska, J.; Amruz Cerna, K.; Taus, P.; Pospisilova, V.; Nezvedova, M.; Fedorova, V.; Kadakova, S.; Klimova, H.; et al. Cerebral organoids derived from patients with Alzheimer’s disease with PSEN1/2 mutations have defective tissue patterning and altered development. Cell Rep. 2023, 28, 113310. [Google Scholar] [CrossRef]
- Park, J.C.; Jang, S.Y.; Lee, D.; Lee, J.; Kang, U.; Chang, H.; Kim, H.J.; Han, S.H.; Seo, J.; Choi, M.; et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat. Commun. 2021, 12, 280. [Google Scholar] [CrossRef] [PubMed]
- Orlacchio, A.; Bernardi, G.; Orlacchio, A.; Martino, S. Stem cells: An overview of the current status of therapies for central and peripheral nervous system diseases. Curr. Med. Chem. 2010, 17, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.G.; Kriegstein, A.R. Challenges of Organoid Research. Annu. Rev. Neurosci. 2022, 45, 23–39. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Cell Model | Advantage | Disadvantage |
|---|---|---|
| Fibroblasts | Easy to harvest | Lacks dimensional complexity |
| Standardized protocols | Lacks neuron tissue specific features key for deep characterization | |
| Expression profile similar to neurons | ||
| Minimally invasive to collect | ||
| Important for basic characterization | ||
| Useful platform for drug screening | ||
| iPSCs | Derived from somatic cells | Lacks dimensional complexity |
| Can be source of cell therapy and derived directly from the recipient | Poor standardization of protocols | |
| Important for deep characterization thanks to tissue-specific features | Lacks epigenetic age and correlation of phenotypes related to it | |
| Can generate neural cells without invasive surgical approaches | Diseases with complex genetic features are hard to address | |
| Useful platform for drug screening | Expensive maintenance | |
| Useful for early phenotype correlation | ||
| Organoids | Adds dimensional complexity to disease modeling | Core of necrotic cells |
| Can be more reliable than animal models in drug testing and in recapitulating disease features | Poor standardization protocols | |
| Can recapitulate complex interactions and brain physiology | Lacks epigenetic age and correlation between phenotypes related to it | |
| Hard and expensive maintenance |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evangelisti, C.; Ramadan, S.; Orlacchio, A.; Panza, E. Experimental Cell Models for Investigating Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 9747. https://doi.org/10.3390/ijms25179747
Evangelisti C, Ramadan S, Orlacchio A, Panza E. Experimental Cell Models for Investigating Neurodegenerative Diseases. International Journal of Molecular Sciences. 2024; 25(17):9747. https://doi.org/10.3390/ijms25179747
Chicago/Turabian StyleEvangelisti, Cecilia, Sherin Ramadan, Antonio Orlacchio, and Emanuele Panza. 2024. "Experimental Cell Models for Investigating Neurodegenerative Diseases" International Journal of Molecular Sciences 25, no. 17: 9747. https://doi.org/10.3390/ijms25179747
APA StyleEvangelisti, C., Ramadan, S., Orlacchio, A., & Panza, E. (2024). Experimental Cell Models for Investigating Neurodegenerative Diseases. International Journal of Molecular Sciences, 25(17), 9747. https://doi.org/10.3390/ijms25179747