Abstract
This study aimed to elucidate the genetic causes underlying the juvenile parkinsonism (JP) diagnosed in a girl with several family members diagnosed with spinocerebellar ataxia type 2 (SCA2). To achieve this, whole-exome sequencing, analysis of CAG repeats, RNA sequencing analysis on fibroblasts, and metabolite identification were performed. As a result, a homozygous missense mutation SNP T>C (rs2254562) in synaptojamin 1 (SYNJ1), which has been implicated in the regulation of membrane trafficking in the synaptic vesicles, was identified. Additionally, we observed overexpression of L1 cell adhesion molecule (L1CAM), Cdc37, GPX1, and GPX4 and lower expression of ceruloplasmin in the patient compared to the control. We also found changes in sphingolipid, inositol, and inositol phosphate metabolism. These findings help to clarify the mechanisms of JP and suggest that the etiology of JP in the patient may be multifactorial. This is the first report of the rs2254562 mutation in the SYNJ gene identified in a JP patient with seizures and cognitive impairment.
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder of the central nervous system with multifactorial etiology, and it is estimated to affect approximately 2% of people over 60 years old. The pathogenesis of PD is not fully understood. While 5–10% of patients have a family history of PD, the majority of the cases are idiopathic and are thought to result from a complex interplay of genetic, epigenetic, and environmental factors.
The hallmark clinical features of parkinsonism include movement disorder, such as resting tremor, muscle stiffness, bradykinesia, postural instability, and gait disturbance. In addition, PD patients may also exhibit non-motor symptoms, such as cognitive impairment, olfactory dysfunction, sleeping disturbance, hyposmia, depression, behavior disorder, and autonomic dysfunction [1].
The average age of the onset of PD is 60 years; early-onset PD is defined as the onset of PD symptoms before the age of 40, whereas juvenile parkinsonism (JP) refers to the manifestation of parkinsonian symptoms before the age of 21 years [2,3,4]. Although PD is the second most frequently diagnosed neurological disorder, early-onset parkinsonism is rare, and JP is even rarer.
The clinical features of parkinsonism in infancy, childhood, or adolescence remain controversial due to the development of an immature brain, that is, they are highly heterogeneous both clinically and genetically. Heterogeneous characteristics include resting tremor, bradykinesia, and hypotonia, as well as atypical features, such as epilepsy, early cognitive decline, behavior problems, ataxia, dystonia, and spasticity [3,4]. Diagnosis can therefore be a challenging due to the extensive and complex phenotype, with multiple manifestations and changes over time. A classification system has been proposed based on factors such as age on onset, clinical symptoms, outcome, and etiological background [3].
Neuropathologically, parkinsonism, including PD, is characterized by the loss of dopaminergic neurons in the substantia nigra and the presence of Lewy bodies (LB) in the brain, accompanied by α-synuclein (α-syn) aggregation. Neurodegeneration is also associated with iron and calcium deposition in the brain as well as metabolic and mitochondrial disorders [5]. However, this pathology is not always present in all cases of parkinsonism, and it may also be present in several other neurological diseases.
In the last 20 years, advances in next-generation sequencing (NGS) have led to the identification of an increasing number of genes linked to JP. The most prevalent gene associated with autosomal dominant JP is SNCA, whereas PARK2, PINK1, and DJ1 are related to autosomal recessive cases. Recently, other atypical mutations in genes, such as ATP13A2, DCTN1, VPS13C, PTRHD1, PLA2G6, FBXO7, DNAJC6, and SYNJ1 have been associated with JP, causing more severe disease [3,6,7,8]. However, currently known mutations do not account for all JP cases. Hence, the search for additional genes contributing to the disorder is not complete.
Synaptojanin 1, encoded by SYNJ1, is a lipid phosphatase that regulates phosphoinositide phosphatase enzymes and is expressed in several tissues, including the brain, where it has a role in the endocytosis and recycling of synapsis vesicles, which is essential for preventing neurodegeneration [9,10]. Recently, accumulating evidence indicates that mutations and variants of the SYNJ1 gene are linked to many neurological disorders, including early-onset PD and atypical JP [8,11,12,13,14,15,16], together with ataxia and seizures [11,13,15,16]. In 2013, the same homozygous missense mutation (c.773G>A, p.Arg258Gln) in the SYNJ1 gene was described in two independent families from Italy and Iran with atypical autosomal recessive JP [11,15]. One year later, the same mutation was described in two brothers affected with JP in another Italian family [12]. In 2016, a second novel homozygous missense mutation (c.1377C>A, p.Arg459Pro) in the SYNJ1 gene was reported in a consanguineous Indian family with autosomal recessive JP [14]. A third novel mutation (c.2515C>T) in the SYNJ1 gene was identified in a patient from Iran who presented asymmetric parkinsonism and seizures at 24 years of age [16]. Another novel heterozygous mutation (p.Leu1406Phefs*42 and p.Lys1321Glu) in the SYNJ1 gene was identified in two siblings with mild JP and seizures in a Tunisian family [13]. Recently, in 2021, four mutations were detected in Algeria, Senegal, and France, expanding the mutational variety of SYNJ1-associated JP [8]. The different mutations in the SYNJ1 gene suggest that alterations in synapsis trafficking may play a role in the pathogenesis of the spectrum of synaptopathies. Although patients carrying these mutations display different phenotypes, mutations in SYNJ1 might identify a risk factor for the development of neurological diseases.
In this study, we present the case of a girl who first sought medical attention at the age of 18 months due to global developmental delay and fluctuating distal tremor. She was diagnosed with JP and seizures. Family history revealed the presence of movement disorders on both maternal and paternal sides. For instance, cases of hand tremors, epilepsy, and spinocerebellar ataxia (SCA) type 2 (SCA2) were identified among family members. Given the family history and the phenotype observed in the girl, a diagnosis of SCA2 was initially considered. The aim of this study was to identify the genetic cause underlying the diagnosis of JP and seizures. Therefore, this case required a multiomics approach involving the patient and several family members to confirm which of the different variants carried could be responsible for the patient’s phenotype. Whole-exome sequencing (WES), transcriptome analysis, and metabolite identification were performed in the patient and other available family members.
2. Results
2.1. Case Description
The patient described in this study is a girl from a non-consanguineous family. She was born from a triplet pregnancy with a birth weight of 1.535 Kg. The patient first sought medical attention at the age of 18 months because of global developmental delay and fluctuating distal tremor. She started having multifocal seizures at the age of 2.5, with up to 120 seizures per day, with an electrical status epilepticus during sleep (ESES) pattern. The seizures were eventually controlled with antiepileptic treatment. However, she developed a fluctuating movement disorder characterized by dystonia, tremor, and rigid-akinetic symptoms at the age of 7. She also experienced cognitive difficulties and behavioral alterations.
Biochemical analysis revealed normal levels of amino acids in both blood and urine samples. In addition, the levels of lactic acid, ammonia, lactate, pyruvate, and beta-hydroxybutyrate were all within the expected range in blood samples. Organic acid in urine and sialotransferrin levels were also found to be normal. Hematological analysis showed a normal complete blood count and a normal peripheral blood smear with non-vacuolated lymphocytes. The patient’s immunoglobulin levels showed a slight decrease in IgA and normal levels of IgG and IgM. Apolipoproteins A and B were within the normal ranges. Serological tests for cytomegalovirus, toxoplasma, Epstein-Barr virus, syphilis, borrelia, and viral infections were all negative.
Levodopa, acetazolamide, flunarizine, and immunotherapy were administered in an attempt to manage the symptoms without success. Multiple electroencephalograms (EEGs) were conducted and revealed notable peak wave activity in the posterior temporal and right occipital regions, forming an ESES pattern that resolved.
The analysis of the cerebrospinal fluid showed normal levels of amino acids, organic acids, lactate, protein profile, cultures, biochemistry, and neurotransmitters. Furthermore, abdominal ultrasound and complete radiological examination yielded normal findings. Complete ophthalmological examinations were also normal. Posterior tibial nerve somatosensory evoked potentials were normal, indicating no involvement of the posterior spinal cord. Electromyography with voluntary contraction indicated the presence of axonal motor neuropathy without signs of myopathy. Repeated stimulation showed normal results.
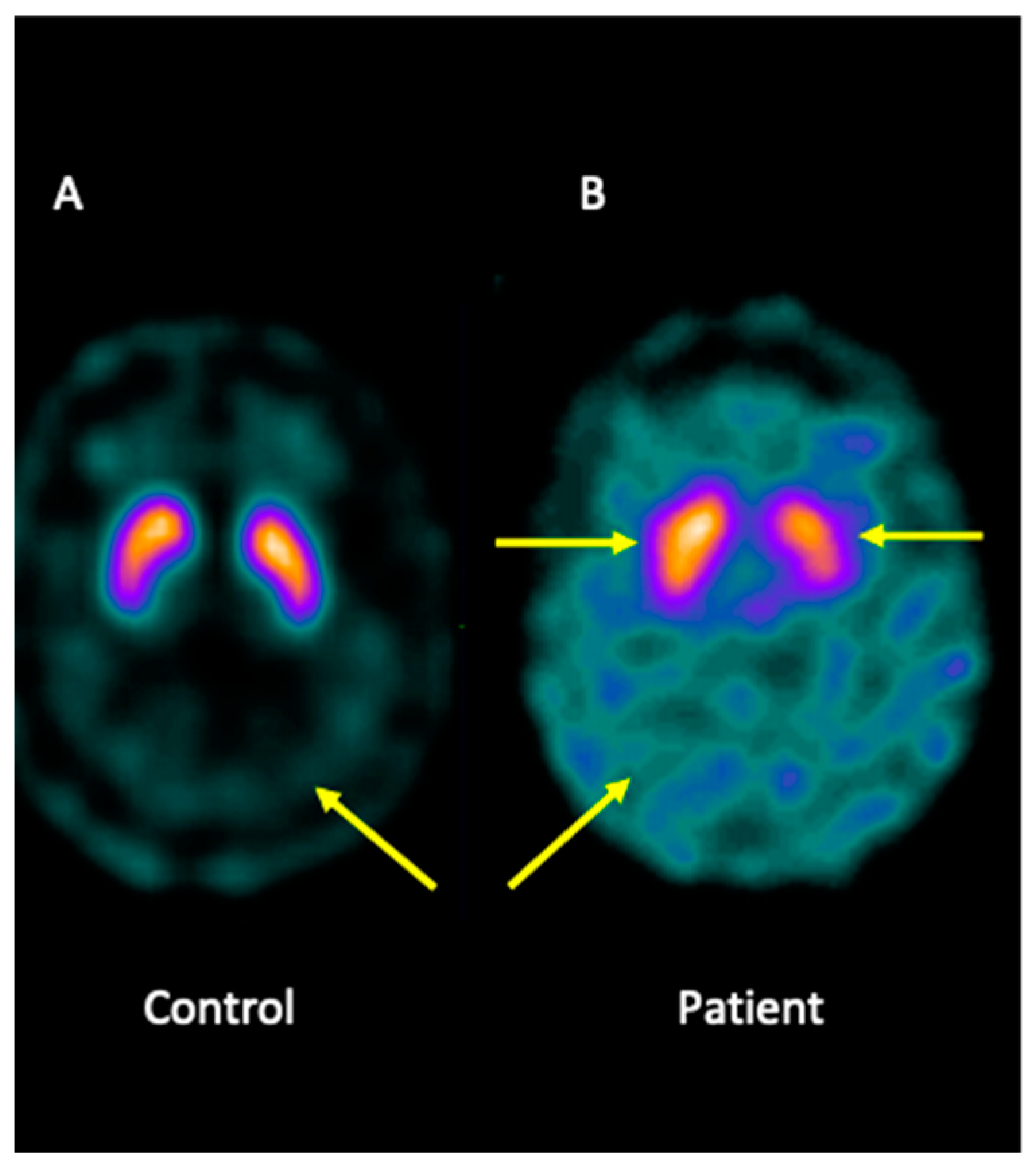
Analysis of brain imaging with SPECT using I-123-Ioflupane showed that the patient displayed asymmetric striata, with the left striata being smaller in size and oval in appearance, together with increased background activity. Furthermore, the analysis was compatible with a loss of dopaminergic activity in both striata. Overall, the pattern may be consistent with JP (Figure 1).
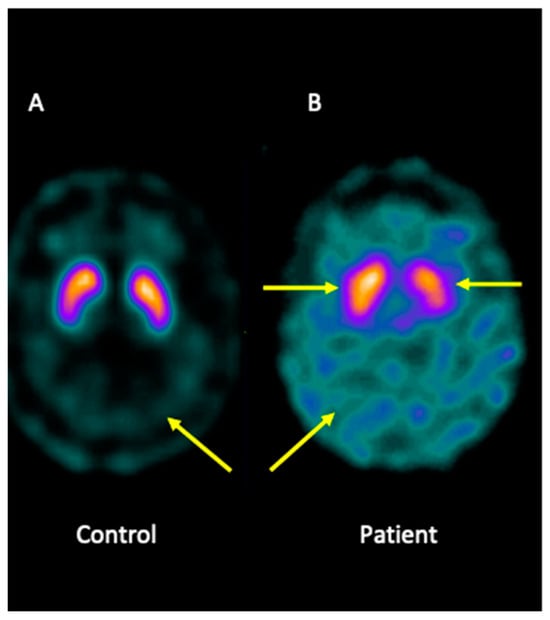
Figure 1.
I-123-Ioflupane SPECT image of the brain patient. (A) Healthy control showing both striata with a normal comma shape and sharp borders, presenting low background activity. (B) The patient presented asymmetric striata, with a lower size in the left striata, an oval appearance, and increased background activity.
2.2. Analysis of SCA2
The father of the case was diagnosed with SCA2, an hereditary neurodegenerative disease characterized by progressive cerebellar ataxia caused by abnormal expansion of the cytosine-adenine-guanine (CAG) repeat in the ATXN2 gene. The father presented a mild phenotype, while other relatives on the paternal side, including the father’s sister and two additional female and male relatives, had more severe symptoms of SCA2 (Supplementary Figure S2). At the age of 7, the patient presented fluctuating movement disorder characterized by dystonia, tremor, and rigid-akinetic. Given the family history and the phenotype observed in the girl, an initial diagnosis of SCA2 was considered. To determine whether these signs were related to the ataxia genotype, we analyzed CAG repeat expansion not only in the patient but also in the mother and both brothers. However, no pathological genotypes were found, and the CAG repeat expansions were within the normal range (Figure S1).
2.3. Whole-Exome Analysis
To determine the cause of the patient’s disease, WES was performed on the patient and available family members, including both parents and both siblings. Variant filtering, such as frequency < 2% and by type, synonymous and UTR variants, and non-coding intronic variants, were used to find the genes responsible for the phenotype. Missense and loss of function variants, nonsense and frameshift mutations, splice site alterations, stop codon losses, codon insertions and deletions, and non-synonymous substitutions were selected. Several family members showed symptoms related to SCA2, including seizures, mental retardation, and early onset of cerebellar ataxia. Initial analysis of genes associated with these diseases, including SCA2, GLUT1, and ATP1A3, revealed no mutations. Regarding the gene associated with JP, the patient did not carry the main mutations known to cause JP in the PARKIN, PINK, and DJ1 genes. In addition, the patient did not present mutations in the GLUT1 and ATP1A3 genes. However, sequence analysis revealed genetic variants in three genes in the patient: SYNJ1, ACOT4, and ACAD9.
We identified a homozygous missense mutation in the SYNJ1 gene in our proband. This mutation was a SNP T>C (rs2254562) located in exon 8 of SYNJ1, resulting in a missense substitution (G = Arg, A = Lys, Lys295Arg). The SYNJ1 gene is located on the long arm of chromosome 21, 21q22.11, position 32,687,042. Analysis of the patient’s family members revealed that both parents and one of the patient’s brothers carried a heterozygous missense mutation (TC) in SYNJ1. Notably, the other sibling was also found to have the same homozygous mutation, but he did not currently exhibit any neurological symptoms. The patient did not have any of the previously reported mutations in the SYNJ1 gene related to JP [8,11,12,13,14,15,16].
WES analysis identified three mutations in the patient’s ACOT4 (acyl-CoA thioesterase) gene. This gene encodes a peroxisomal enzyme involved in the hydrolysis of acyl-coenzyme A to the free fatty acid and coenzyme A [17,18,19,20]. Analysis of the patient’s ACOT4 gene showed a homozygous TCCA insertion (rs373880503). Both parents, aunt, and grandfather carried the same TCCA insertion in a heterozygous state. Additionally, a second mutation, a CTTA deletion (rs375801976), was found in this gene. Moreover, a missense variant, rs35724886 (C>A), resulting in an alanine to aspartic acid change, was also detected, although to date there is no information in the literature relating these mutations to PD [20].
Our analyses also revealed a mutation in the ACAD9 (Acyl-CoA-dehydrogenase 9) gene, which is a flavoprotein implicated in the process of beta-oxidation of unsaturated long-chain acyl-CoAs. A mutation in the ACAD9 gene, homozygous deletion GA, which has not been previously described, was found in the patient’s exome analysis. The study of this mutation in the patient’s family showed that both parents, the patient’s grandfather, and three sisters of the father carried heterozygous deletion GA in the ACAD9 gene. Mutations in ACAD9 have been associated with conditions such as cardiomyopathy, muscular weakness, and exercise intolerance, and in rare cases, patients may also present with other symptoms, such as epilepsy, intellectual deficits, and severe developmental delays [21].
2.4. Transcriptome Analysis
To assess the impact of the mutations identified in the patient’s genotype, we conducted next-generation transcriptome sequencing (RNA-seq) on fibroblast cell culture derived from the patient’s skin. A human foreskin fibroblast (HFF) cell line was used as a control. To identify differentially expressed genes between the patient and the control, we compared gene expression levels. We then focused on analyzing the role of the ten most overexpressed genes and the ten genes with the lowest expression in the patient compared to the control. The ten genes with the highest expression in the patient’s fibroblast cells were RPS4Y1, L1CAM, GPX1, Cdc37, GPX4, EMP3, S100A16, RPS21, PSMD8, and EDF1. The ten genes with the lowest expression in the patient compared to the control were SP9, CD160, EYS, AC09985.2, EPHA3, C4orf47, ceruloplasmin (CP), SFRP2, EMCN, and LHX8 (Table 1). Of the above 20 genes differentially expressed in the patient, 5 genes, L1CAM, Cdc37, GPX1, GPX4, and CP, warrant further investigation as they may be linked to the patient’s phenotype.

Table 1.
Transcriptome analysis. The expression differences between the top ten overexpressed genes and the ten genes with lowest expression in the patient compared with the control.
2.5. Metabolome Analysis
Untargeted metabolomics approaches were used to analyze changes in metabolites in plasma and urine samples. A total of 25 metabolites were identified in plasma, and 16 metabolites were identified in urine. Table 2 and Table 3 show the main percentage changes in 41 metabolites in plasma and urine, respectively. Compared with the control, levels of 11 plasma metabolites and 16 urine metabolites were increased in the patient, and levels of 14 plasma metabolites were decreased in the patient.
Table 2.
Significant metabolites in samples of plasma from the patient.
Table 3.
Significant metabolites in samples of urine from the patient.
Plasma metabolite analysis showed that levels of several metabolites involved in sphingolipid metabolism, including O-phosohoethanolamine, sphingosine, sphingosine 1-phosphate (S1P), sphinganine 1-phosphate, and N-methylalanine were decreased in the patient. In addition, levels of myo-inositol and phosphoric acid were found to be increased in the patient, suggesting a disturbance in inositol phosphate and inositol metabolism. Furthermore, a significant increase was observed in the level of 3-carboxy-4-methyl-5-propyl-2-furanpropionic acid, a metabolite related to lipid and fatty acid metabolites, along with a slight increase in levels of phosphatidylcholines PC(18:3) and PC (20:3), and a decrease in levels of PC(18:0), PC(22:4), and arachidonic acid in the patient. With regard to phospholipid metabolism, the levels of PE (16:0), PE(18:0), and PE(20:3) were lower in the patient than in the control. Levels of uric acid, which is involved in purine metabolism, was found to be elevated in our case, while guanosine levels were decreased. Regarding amino acid metabolism, the patient had elevated levels of pipecolic acid or N-methyl-L-proline. Moreover, levels of 3’-AMP and N-Acetylneuraminic acid were slightly decreased, and those of bilirubin, 1-methylnicotinamide, and hydroxymethylbilane were increased (Table 2).
In urine analysis, we observed a high increase in the level of glucose 6-phosphate, which is related to inositol and inositol phosphate metabolism. Glucose 6-phosphate also plays a role in nucleotide sugar metabolism and is a participant in glycolysis. In addition, levels of dimethylallylpyrophosphate and isopentenyl pyrophosphate, which are involved in steroid biosynthesis, were increased in the patient. With regard to amino acid metabolism, we observed increased levels of Ne,Ne dimethyllysine, some dipeptides, and γ-glutamyl-β-aminopropiononitrile in the patient. We also found an increase in levels of metabolites related to lipid and fatty acid metabolism in the patient, including 2-hydroxymyristoylcarnitine, phosphatidylinositol PI(36:1), 3-methyladipic acid or pimelic acid, droxy-4-(methylthio) butanoic acid, dimethylallyl pyrophosphate or isopentenyl pyrophosphate, nonanoylcarnitine, and 6-keto-decanoylcarnitine. Pipecolic acid was also detected at elevated levels in the patient (Table 3).
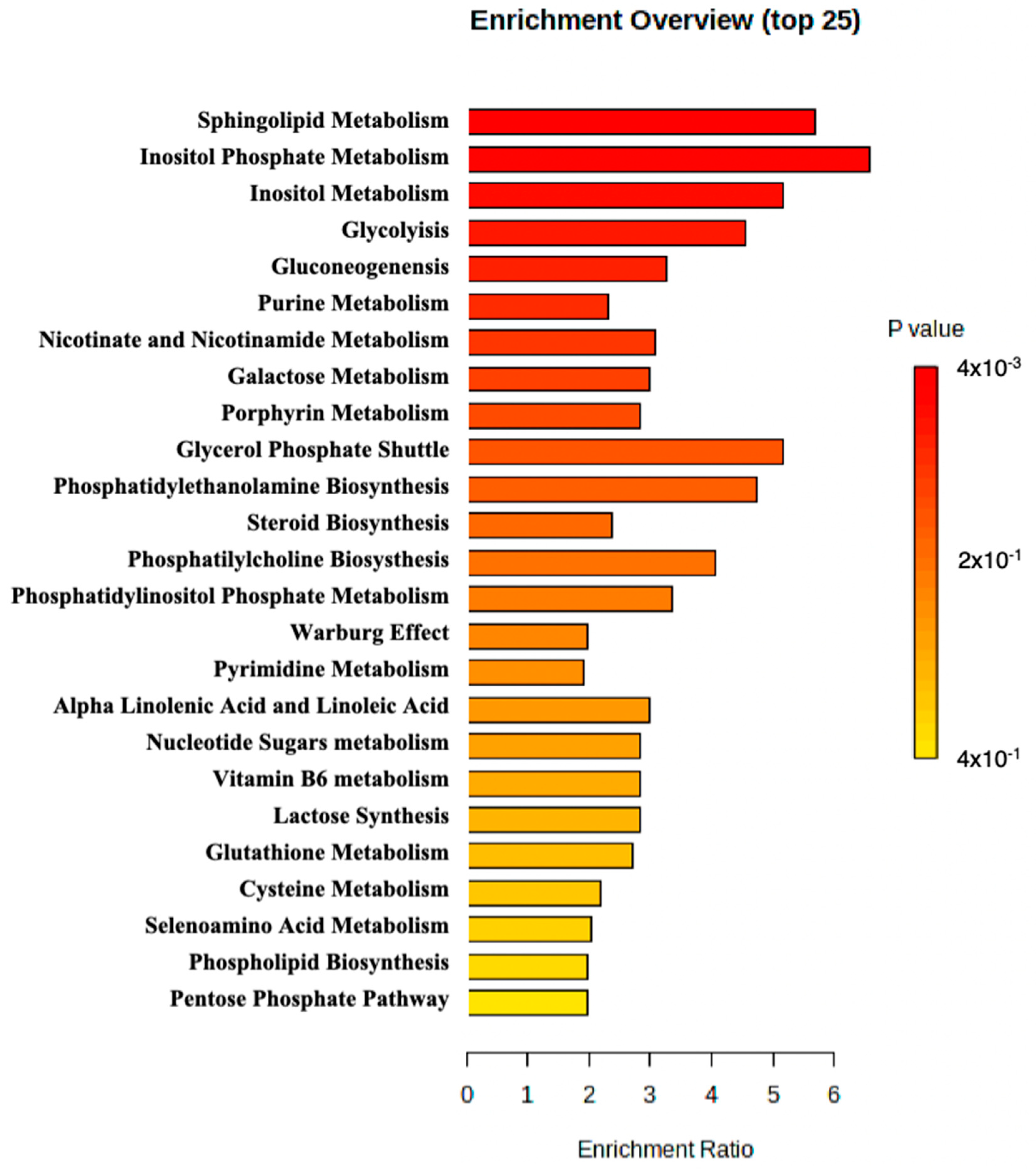
MetaboAnalyst 5.0 was used to perform enrichment analysis of the available metabolite. An overview of the top 25 enriched metabolite sets is shown in Figure 2. The results suggested that sphingolipid metabolism, inositol phosphate metabolism, and inositol metabolism may be altered in the patient and potentially contribute to the pathology observed in the patient.
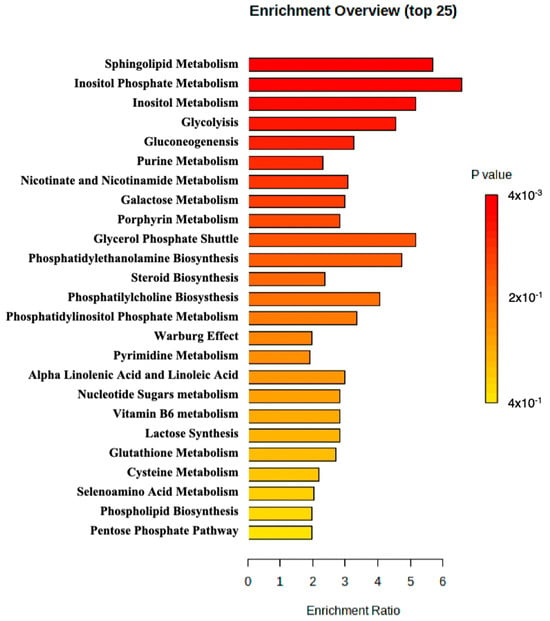
Figure 2.
Metabolite set enrichment analysis perform by MetaboAnalyst.
3. Discussion
In this report, we conducted a genetic study in a patient with seizures and a suspected diagnosis of JP. Our main focus was the elucidation of the genetic etiology of JP. Several family members displayed symptoms related to SCA2, including seizures, mental retardation, and early onset of cerebellar ataxia. Initial analysis of the genes associated with these diseases, including SCA2, GLUT1, and ATP1A3, did not reveal any mutations in the patient. Hence, whole-exome sequencing was performed. We identified a homozygous missense mutation (rs2254562) in the SYNJ1 gene, three different mutations in the ACOT4 gene, and a homozygous deletion in the ACAD9 gene in the diagnosed case. We also observed overexpression of L1CAM, Cdc37, GPX1, and GPX4 in the patient compared to the control, as well as lower expression of CP. In addition, we detected alterations in sphingolipid, inositol, and inositol phosphate metabolism, suggesting their potential pathogenetic role.
To date, several different mutations in the SYNJ1 gene have been described in several families from different countries, including Iran, Italy, India, Tunisia, Algeria, Senegal, Germany, China, and France, presenting early-onset PD, atypical parkinsonism, and JP [8,11,12,13,14,15,16], suggesting a mutational hotspot. In our case, we identified a novel homozygous missense mutation SNP T>C (rs2254562) (G = Arg, A = Lys, Lys295Arg) in the SYNJ1 gene. This mutation was previously reported before by Yuam L et al. [22]. In this study, 35 variants in 22 genes related to PD, including SYNJ1 variant rs2254562, were analyzed in 512 PD patients. However, no statistically significant differences in rs2254562 were observed in that cohort between PD patients and controls in that cohort. The same research team also studied the rs2254562 variant in 200 patients with essential tremor. Again, no statistically significant differences were found in the rs2254562 variant between essential tremor patients and the controls [23]. Therefore, our report provides the first description of the rs2254562 mutation in the SYNJ1 gene in a patient with JP.
SYNJ1 is a phosphoinositide phosphatase that regulates the levels of various phosphatidylinositols (PIs), including phosphatidylinositol 3 phosphate (PI(3)P), PI(4)P, PI(3,5)P2, and PI(3,4,5)P3, in the cellular and intracellular membranes, such as the autophagosome, endoplasmic reticulum, and Golgi apparatus [24]. Robust evidence suggests that SYNJ1 plays a role in regulating membrane trafficking in synaptic vesicles, which affects the release of neurotransmitters [9,10]. Furthermore, emerging evidence suggests its involvement in endosomal trafficking and autophagy [25,26,27]. Our analysis revealed a disruption in inositol phosphate metabolism. Altered metabolism of PIs could impact synaptic transmission and potentially affect autophagic function, thereby potentially contributing to the development of JP mediated by SYNJ1. Further research is needed to elucidate the precise mechanism of SYNJ1.
SYNJ1 is composed of three domains, two of which contain enzymatic domains crucial for lipid homeostasis and membrane trafficking: the suppressor of actin 1 (SAC1) and 5′-phosphatase, which hydrolyzes inositol species such as PI(3)P, PI(4)P, PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3. The third domain is a proline-rich domain (PRD). All three domains are important in the regulation synaptic activity [26]. Mutations in both phosphatase domains of SYNJ1, R258Q, R459P, and R839C have been associated with JP or early-onset of PD, suggesting the involvement of lipid dysregulation in the pathogenesis of PD [11,12,14,28,29]. We hypothesize that the rs2254562 mutation located in exon 8 may affect the stability of SYNJ1 and its function, determining alterations in inositol metabolism, inositol phosphate metabolism, and phosphatidylinositol phosphate metabolism.
For instance, other studies have identified additional variants in SYNJ1, including R136*, Y888C, W843*, Q647R, and S1112T, which can lead to truncation of the protein or loss of protein expression [26,30,31].
Moreover, sphingolipids play an important role in neuron function by regulating neural growth and differentiation, mitochondrial function, autophagy, and endosomal trafficking, and these processes have been showed to be affected in PD pathogenesis [32]. For instance, S1P, a bioactive sphingolipid, acts as a neuroprotective factor by regulating neuronal cell proliferation and differentiation, stress tolerance, and protects cells from apoptosis [33,34]. Decreased plasma levels of S1P have been found in idiopathic PD, dementia with LB, multiple system atrophy, and progressive supranuclear palsy [35]. Consistent with these finding, our study showed a decrease in S1P levels in the patient compared to the control. Furthermore, in an animal model of PD, administration of fingolimod, a S1P agonist, was found to ameliorate neurodegeneration [36]. These findings suggest that abnormal sphingolipid metabolism may contribute to neurodegeneration.
Although the exact mechanisms by which SYNJ1 is involved in the pathogenesis of the disease are not fully understood, it is believed that SYNJ1 does not act alone in this process [26], and other associated factors, such as epigenetic modifications like DNA methylation or microRNAs or other molecules, may also contribute to the pathogenesis of the disease. More research is needed to elucidate the exact mechanism of SYNJ1 and the associated factors involved in the development and the progression of the disease.
Although this novel mutation in SYNJ1 may partly be responsible for the phenotype observed in this case, it is worth noting that the same mutation was found in the patient’s brother, who is currently asymptomatic. Therefore, we cannot rule out the possibility that the brother may develop the disease later in life. In fact, patients with the homozygous SYNJ1 c.773G>A mutation have developed parkinsonism symptoms in their third decade of life [8,11,12,15]. Furthermore, there is phenotypic variability and a difference in the age of onset between patients with the same mutations [11,12,13,14,15]. Another factor that may contribute to the development of the disease in the patient is the altered expression of L1CAM, Cdc37, GPX1, GPX4, and CP proteins, as well as the altered sphingolipid, inositol, and inositol phosphate metabolism observed in the patient. Based on these findings, we propose that JP observed in the patient could have multiple origins, and that the combination of features observed in this case could have led to the development of the disease.
It is well established that the accumulation and aggregation of α-syn is a major cause of the development of LB and has been implicated in the pathogenesis of PD [37,38]. Recently, it has been suggested that lipid dysregulation may play a crucial role in the abnormal aggregation of α-syn in patients with PD [39,40]. Along this line, a study conducted on postmortem PD brain samples, a cell line, and a Caenorhabditis elegans model provided evidence that the accumulation of PI(3,4,5)P3 leads to the aggregation of α-syn and contributes to neurodegeneration, a pathological process that has been attributed to the loss of function of a mutated SYNJ1 [41]. SYNJ1 is also associated with changes in the aggregation and phosphorylation of α-syn in mice [42].
Several studies have suggested that the spread of α-syn in extracellular vesicles in the brain may contribute to the pathogenesis of PD by affecting synaptic vesicle endocytosis [43,44,45]. Hence, high levels of α-syn have been found in extracellular L1CAM exosomes in patients with PD and atypical parkinsonism [46,47]. The L1CAM gene belongs to the immunoglobulin supergene family and encodes a protein that plays an essential role in the development of the nervous system. Neurological diseases such as hydrocephalus have been linked to mutations in L1CAM [48,49]. In addition, L1CAM exosomes containing α-syn may be involved in the pathogenesis of PD [50,51]. Our findings showed increased expression of L1CAM in our patient, indicating that it could contribute to the parkinsonism pathology due to the propagation of altered α-syn [52]. We also found overexpression of Cdc37, a co-chaperone of the cell division cycle 37, which may contribute to the phosphorylation of α-syn, affecting to its aggregation and stability [52]. It has also been reported that overexpression of Cdc37 stabilizes tau and reduces its clearance, and pathological aggregates of tau have been found in many neurological diseases, including sporadic PD [53,54]. Therefore, Cdc37 has been identified as a potential target in neurological disorders such as Alzheimer’s disease and PD [52].
Transcriptome analysis revealed overexpression of GPX1 and GPX4 in the patient compared to the control. GPX1 and GPX4 are selenoenzymes that require selenium for their antioxidant function and play an essential role in alleviating oxidative stress caused by ROS [55]. Reduced GPX activity has been reported in the serum of PD patients [56]. Overexpression of GPX enzymes such as GPX1 and GPX4 is thought to protect against neurodegenerative processes, including convulsive disorders, Alzheimer’s disease, and PD [57]. We hypothesize that increased activity of GPX1 and GPX4 was induced in response to oxidative damage, which contributed to the development of the disease in the patient. GPX1 and GPX4 are therefore attempting to compensate for an oxidative environment. This is supported by the finding that overexpression of GPX protects against neurons loss under neurotoxic conditions [58]. An experimental study using GPX knockout mice showed no neuropathological lesions. However, when the mice were injected with MPTP, a toxin that induces oxidative stress, lesions increased in homozygous GPX knockout mice, indicating that GPX is involved in antioxidant function [59]. In line with these data, GPX4 prevents neuronal death and ferroptosis by inhibiting lipid peroxidation, which has been implicated in the cell death observed in PD, ataxia, and other degenerative diseases [60,61]. Furthermore, GPX1 activity may be used as a biomarker for Friedreich’s ataxia, a fatal neurodegenerative disease [62].
Iron accumulation in the brain has been described as potential contributor to the pathogenesis of PD [63,64], and impaired iron metabolism may increase ROS levels and cause brain damage [65]. Our results showed low expression of CP, a ferroxidase enzyme that regulates brain iron homeostasis, transport, and accumulation in the brain, in the patient [66,67]. This finding suggests that CP impairment may play a role in neurodegenerative diseases characterized by oxidative damage [68,69]. Indeed, reduced serum CP levels have been identified as a risk factor for the development of PD [67]. However, no iron deposition was observed in the patient’s brain.
4. Materials and Methods
4.1. Consent and Approval
Informed consent was obtained from the adult participants and informed consent for children was obtained from the parents of all three child participants. The project received approval from the Biomedical Research Ethics Committee of the Andalusian Public Health System in Granada, Spain (1147-N-15). Sample collection and in vitro experiments were performed in accordance with ethical guidelines following the Nuremberg Code, Belmont Report, and the Declaration of Helsinki.
4.2. Analysis of the Brain Imaging with Single-Photon Emission Computed Tomography
Brain imaging analysis using single-photon emission computed tomography (SPECT) with I-123-Ioflupane was performed on the patient and a control. SPECT images were acquired using a General Electric gamma camera (Siemens Intevo-T6) and processed with Syngo MI-VA60B VA60B software to assess the image quality. The processing of the images was carried out with Workstation Syngovia VB40B-VS 06.04.0000.0040. The images were visually analyzed by a specialist.
4.3. Analysis of CAG Repeats in Spinocerebellar Ataxia
Analysis of CAG repeat expansion to determine SCA2 was performed in the patient, the father, the mother, both brothers, and all of the available members of the family. DNA extraction of samples was performed using the commercial QIAamp DNA Blood Mini Kit (Qiagen). Quantification of the obtained DNA was carried out using the Qubit 2.0 fluorometer (Invitrogen, Waltham, MA, USA). Amplification of the region of interest in the ATXN2 gene was performed. Detection of the amplified fragment was performed by capillary electrophoresis under denaturing conditions in an AB PRISM® 3130 Genetic Analyzer using fluorescent labeling. The results were visualized using GeneMapper 3.2.1 software.
4.4. WES for Candidate Gene Identification
WES analysis was conducted for the patient and all family members who were available. Genomic DNA was isolated and extracted from peripheral blood leucocytes utilizing the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and the QIAcube instrument (Qiagen, Hilden, Germany), following the manufacturer’s protocol. DNA was then fragmented to 180–220 base pair lengths using the Covaris E220 Focused-ultrasonicator (Covaris, Woburn, MA, USA). To capture exomes and prepare the library for target enrichment, 500 ng of sheared original DNA and in-solution hybridization technology were used (Roche NimbleGen SeqCap EZ MedExome; Roche Sequencing Solution, Pleasanton, CA, USA), and processing was performed using the HyperCap workflow (Roche Sequencing Solution, Pleasanton, CA, USA). MedExome design was used to obtain whole exome information, including complex regions, thereby facilitating the identification of a greater number of disease-associated variants. Then, libraries were sequences on the NextSeq 500 system (Illumina, CA, USA) using the NextSeq 500/550 High Output Kit v2.5 (300 cycles) to generate 150 × 2 paired sequences.
Reads were aligned to the human reference genome, and variant identification and and filtering were performed using the Genome Analysis Toolkit (GATK) and SAMtools mpileup pipeline. AMNOVAR and Ensembl were used for annotation, and PollyPhen-2 and SIFT were used as tools to evaluate the pathogenicity of the variants.
DNA was extracted using the DNA Blood Mini Kit (Qiagen, Hilden, Germany) by automated extraction on a Qiacube robot (Qiagen, Hilden, Germany). A total of 0.5 µg of DNA was taken from all cases and their relatives after quantification using Qubit v4 (Thermo Fisher Scientific, Waltham, MA, USA) to start the library. DNA from all samples was fragmented on the Covaris E200 (Covaris, Woburn, MA, USA) according to the Nimblegen-Roche protocol recommendations. Libraries were generated following the ‘Hybridisation-Based Target Enrichment’ protocol for exome capture using the HyperCap Workflow v2.0 and SeqCap® EZ MedExome (Nimblegen Roche, Basil, Switzerland) reagents for exome library generation.
4.5. RNA-Seq for Gene Expression Analysis
RNA sequencing analysis was performed on fibroblasts from both patient and control samples. The patient’s fibroblast cells were obtained via skin biopsy while fibroblasts from a human cell culture line of foreskin fibroblasts (HFF cell line) were used as a control. Cells were cultured in DMEM high glucose medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum and glutamine. RNA was isolated from primary fibroblasts using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The concentration and quality of the RNA were determined using the Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and the 2100 Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA, USA). To prepare libraries, 1 μg of RNA was used with the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. Then, the NexSeq 500 system (Illumina, San Diego, CA, USA) was used to sequence the mRNA libraries utilizing the highest output mode and paired-end 75 bp read lengths. The RNA-seq data were aligned to the human reference genome GRCh38 by RSEM v1 (https://deweylab.github.io/RSEM/, accessed on 15 March 2021) [70]. RSEM was used to quantify the sequence alignment at the transcript level, and DESeq was used to measure differential gene expression (https://www.bioconductor.org/packages//2.10/bioc/html/DESeq.html, accessed on 15 June 2021) [71].
A functional analysis was performed using IPA (Ingenuity Pathway Analysis) [72] and DAVID Bioinformatics Resources V6.8 (https://david.ncifcrf.gov/, accessed on 1 February 2022) to obtain the role of the gene pool, clinical implication, ontology, and involved metabolic pathways. Moreover, the STRING search tool was used to represent a protein–protein interaction (PPI) network including our target genes (https://string-db.org/, accessed on 16 June 2023) [73].
4.6. Validation by Sanger Sequencing
Selected single nucleotide polymorphism (SNP) variants (SYNJ1, ACOT4, and ACAD9) were validated using Sanger sequencing.
To validate some of the identified variants detected in the exome and identified in the transcriptome, DNA and RNA (cDNA) were amplified for subsequent Sanger sequencing.
4.7. Metabolite Identification
Metabolite extraction was performed on plasma and urine samples from the patient, family members, and healthy controls, with the same age, sex, and weight, according to standard protocols [74]. Liquid chromatography-mass spectrometry (LC-MS) analyses were performed using an UHPLC system (Agilent 1290 Infinity LC System, Waldbronn, Germany) coupled to an LC-QTOF-MS analyzer (6520) (Agilent Technologies, Santa Clara, CA, USA). Gas chromatography-mass spectrometry (GC-MS) analyses were performed using an Agilent Technologies 7890A instrument (Waldbronn, Germany) coupled to a 5975C mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). Capillary electrophoresis-mass spectrometry (CE-MS) analyses were performed on a 7100 CE system coupled to a TOF MS 6224 (Agilent Technologies, Santa Clara, CA, USA). Details on the analytical aspects of this study can be found in [74]. For data analysis, Agilent MassHunter Profinder version B.08.00 was used for the data obtained from the LC-MS and CE-MS analyses, while Agilent MassHunter Unknowns and Agilent MassHunter Qualitative Analysis were used for the data obtained from the GC analyses [74]. The obtained features were filtered by coefficient of variation of the signal (CV) in quality controls. The features were compared by their absolute differences in abundance, expressed as the logarithm of the fold change (log2FC). Those characteristics or metabolites that had a log2FC < 1.5 were selected and their percentage of change was indicated [74]. Databases such as METLIN, HMDB, LipidMaps, and CEU Mass Mediator were used for the identification of the features found to be significant in the LC-MS and CE-MS analyses, using their MS/MS spectra to support the identification. An in-house built library was used to identify the features obtained in the GC-MS analyses.
5. Conclusions
This study helps to elucidate the mechanisms underlying JP, although the cause of JP is multifactorial and not fully understood. For the first time, a novel mutation in the SYNJ1 gene is reported as a potential risk factor for the disease. This feature, together with overexpression of L1CAM, Cdc37, GPX1, and GPX4, reduced CP expression, and alterations in sphingolipid, inositol, and inositol phosphate metabolism, may also play a role in neurodegeneration.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms25179754/s1.
Author Contributions
Sample and clinical data collection: S.R., I.M., P.G.-R. and L.J.M.-G.; conceptualization, methodology: P.G.-R., L.J.M.-G. and L.A.; sample and data analysis: C.A.-D., V.G.-C., J.S. and C.B.; analysis of results, interpretation, and statistical analysis: E.L.-D., L.A., S.R., I.M., C.A.-D., V.G.-C., C.E.-B., F.F.-R., M.J.S., J.A.L., L.J.M.-G. and P.G.-R.; creation of figures: E.L.-D., L.A., P.G.-R. and L.J.M.-G.; writing—original draft of the manuscript, review, and editing: E.L.-D., L.A., L.J.M.-G. and P.G.-R.; coordination: L.J.M.-G., P.G.-R., M.J.S., L.A. and E.L.-D. All of the authors discussed the results, the interpretation, and implications, and revised the manuscript at all stages. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundación Mutua Madrileña, Spain (XIII Call on Research Grants 2016), reference number AP163052016.
Institutional Review Board Statement
The project was approved by the Biomedical Research Ethics Committee of the Andalusian Public Health System in Granada, Spain, on October 29, 2015 (project identification code AP163052016). Data recording, sample collection, and all in vitro experiments were conducted in accordance with ethical guidelines following the Nuremberg Code, Belmont Report, and the Declaration of Helsinki.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Sequence data have been deposited at the European Genome-phenome Archive (EGA), which is hosted by the EBI and CRG, under accession number EGAS00001007686.
Acknowledgments
We are grateful for the support and cooperation of the subject, as well as her family. We would also like to acknowledge the support of Lorgen.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Halliday, G.; Lees, A.; Stern, M. Milestones in Parkinson’s disease-Clinical and pathologic features. Mov. Disord. 2011, 26, 1015–1021. [Google Scholar] [CrossRef]
- Guadagnolo, D.; Piane, M.; Torrisi, M.R.; Pizzuti, A.; Petrucci, S. Genotype-Phenotype Correlations in Monogenic Parkinson Disease: A Review on Clinical and Molecular Findings. Front. Neurol. 2021, 12, 648588. [Google Scholar] [CrossRef] [PubMed]
- Niemann, N.; Jankovic, J. Juvenile parkinsonism: Differential diagnosis, genetics, and treatment. Park. Relat. Disord. 2019, 67, 74–89. [Google Scholar] [CrossRef]
- Leuzzi, V.; Nardecchia, F.; Pons, R.; Galosi, S. Parkinsonism in children: Clinical classification and etiological spectrum. Park. Relat. Disord. 2021, 82, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Franco, G.; Lazzeri, G.; di Fonzo, A. Parkinsonism and ataxia. J. Neurol. Sci. 2022, 433, 120020. [Google Scholar] [CrossRef] [PubMed]
- Morales-Briceño, H.; Mohammad, S.S.; Post, B.; Fois, A.F.; Dale, R.C.; Tchan, M.; Fung, V.S.C. Clinical and neuroimaging phenotypes of genetic parkinsonism from infancy to adolescence. Brain 2020, 143, 751–770. [Google Scholar] [CrossRef]
- Jia, F.; Fellner, A.; Kumar, K.R. Monogenic Parkinson’s Disease: Genotype, Phenotype, Pathophysiology, and Genetic Testing. Genes 2022, 13, 471. [Google Scholar] [CrossRef]
- Lesage, S.; Mangone, G.; Tesson, C.; Bertrand, H.; Benmahdjoub, M.; Kesraoui, S.; Arezki, M.; Singleton, A.; Corvol, J.-C.; Brice, A. Clinical Variability of SYNJ1-Associated Early-Onset Parkinsonism. Front. Neurol. 2021, 12, 648457. [Google Scholar] [CrossRef]
- Verstreken, P.; Koh, T.-W.; Schulze, K.L.; Zhai, R.; Hiesinger, P.; Zhou, Y.; Mehta, S.Q.; Cao, Y.; Roos, J.; Bellen, H.J.; et al. Synaptojanin Is Recruited by Endophilin to Promote Synaptic Vesicle Uncoating. Neuron 2003, 40, 733–748. [Google Scholar] [CrossRef]
- Harris, T.W.; Hartwieg, E.; Horvitz, H.R.; Jorgensen, E.M. Mutations in Synaptojanin Disrupt Synaptic Vesicle Recycling. J. Cell Biol. 2000, 150, 589–600. [Google Scholar] [CrossRef]
- Krebs, C.E.; Karkheiran, S.; Powell, J.C.; Cao, M.; Makarov, V.; Darvish, H.; Di Paolo, G.; Walker, R.H.; Shahidi, G.A.; Buxbaum, J.D.; et al. The sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive parkinsonism with generalized seizures. Hum. Mutat. 2013, 34, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; De Rosa, A.; Quadri, M.; Criscuolo, C.; Breedveld, G.J.; Picillo, M.; Pappatà, S.; Quarantelli, M.; Barone, P.; De Michele, G.; et al. PARK20 caused by SYNJ1 homozygous Arg258Gln mutation in a new Italian family. Neurogenetics 2014, 15, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Ben Romdhan, S.; Sakka, S.; Farhat, N.; Triki, S.; Dammak, M.; Mhiri, C. A Novel SYNJ1 Mutation in a Tunisian Family with Juvenile Parkinson’s Disease Associated with Epilepsy. J. Mol. Neurosci. 2018, 66, 273–278. [Google Scholar] [CrossRef]
- Kirola, L.; Behari, M.; Shishir, C.; Thelma, B.K. Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile Parkinsonism. Park. Relat. Disord. 2016, 31, 124–128. [Google Scholar] [CrossRef]
- Quadri, M.; Fang, M.; Picillo, M.; Olgiati, S.; Breedveld, G.J.; Graafland, J.; Wu, B.; Xu, F.; Erro, R.; Amboni, M.; et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset parkinsonism. Hum. Mutat. 2013, 34, 1208–1215. [Google Scholar] [CrossRef]
- Taghavi, S.; Chaouni, R.; Tafakhori, A.; Azcona, L.J.; Firouzabadi, S.G.; Omrani, M.D.; Jamshidi, J.; Emamalizadeh, B.; Shahidi, G.A.; Ahmadi, M.; et al. A Clinical and Molecular Genetic Study of 50 Families with Autosomal Recessive Parkinsonism Revealed Known and Novel Gene Mutations. Mol. Neurobiol. 2018, 55, 3477–3489. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, Y.; Jiang, X.; Jin, Y.; Wu, J.; Qin, Y.; Qi, X.; Cheng, Y.; Mao, Y.; Hua, D. The combined expressions of B7H4 and ACOT4 in cancer-associated fibroblasts are related to poor prognosis in patients with gastric carcinoma. Int. J. Clin. Exp. Pathol. 2019, 12, 2672–2681. [Google Scholar] [PubMed]
- Oliver, K.L.; Lukic, V.; Freytag, S.; Scheffer, I.E.; Berkovic, S.F.; Bahlo, M. In silico prioritization based on coexpression can aid epileptic encephalopathy gene discovery. Neurol. Genet. 2016, 2, e51. [Google Scholar] [CrossRef]
- Ni, C.; Zheng, K.; Gao, Y.; Chen, Y.; Shi, K.; Ni, C.; Jin, G.; Yu, G. ACOT4 accumulation via AKT-mediated phosphorylation promotes pancreatic tumourigenesis. Cancer Lett. 2021, 498, 19–30. [Google Scholar] [CrossRef]
- Auer, P.L.; Nalls, M.; Meschia, J.F.; Worrall, B.B.; Longstreth, W.T.; Seshadri, S.; Kooperberg, C.; Burger, K.M.; Carlson, C.S.; Carty, C.L.; et al. Rare and Coding Region Genetic Variants Associated With Risk of Ischemic Stroke. JAMA Neurol. 2015, 72, 781. [Google Scholar] [CrossRef]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet. J. Rare Dis. 2018, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Song, Z.; Deng, X.; Zheng, W.; Guo, Y.; Yang, Z.; Deng, H. Systematic analysis of genetic variants in Han Chinese patients with sporadic Parkinson’s disease. Sci. Rep. 2016, 6, 33850. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Deng, X.; Song, Z.; Deng, S.; Zheng, W.; Mao, P.; Deng, H. Systematic analysis of genetic variants in patients with essential tremor. Brain Behav. 2018, 8, e01100. [Google Scholar] [CrossRef] [PubMed]
- Balla, T. Phosphoinositides: Tiny Lipids With Giant Impact on Cell Regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef]
- Fasano, D.; Parisi, S.; Pierantoni, G.M.; De Rosa, A.; Picillo, M.; Amodio, G.; Pellecchia, M.T.; Barone, P.; Moltedo, O.; Bonifati, V.; et al. Alteration of endosomal trafficking is associated with early-onset parkinsonism caused by SYNJ1 mutations. Cell Death Dis. 2018, 9, 385. [Google Scholar] [CrossRef]
- Choudhry, H.; Aggarwal, M.; Pan, P.Y. Mini-review: Synaptojanin 1 and its implications in membrane trafficking. Neurosci. Lett. 2021, 765, 136288. [Google Scholar] [CrossRef]
- Vanhauwaert, R.; Kuenen, S.; Masius, R.; Bademosi, A.; Manetsberger, J.; Schoovaerts, N.; Bounti, L.; Gontcharenko, S.; Swerts, J.; Vilain, S.; et al. The SAC1 domain in synaptojanin is required for autophagosome maturation at presynaptic terminals. EMBO J. 2017, 36, 1392–1411. [Google Scholar] [CrossRef]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in cell regulation and membrane dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]
- Mani, M.; Lee, S.Y.; Lucast, L.; Cremona, O.; Di Paolo, G.; De Camilli, P.; Ryan, T.A. The Dual Phosphatase Activity of Synaptojanin1 Is Required for Both Efficient Synaptic Vesicle Endocytosis and Reavailability at Nerve Terminals. Neuron 2007, 56, 1004–1018. [Google Scholar] [CrossRef]
- Hardies, K.; Cai, Y.; Jardel, C.; Jansen, A.C.; Cao, M.; May, P.; Djémié, T.; Le Camus, C.H.; Keymolen, K.; Deconinck, T.; et al. Loss of SYNJ1 dual phosphatase activity leads to early onset refractory seizures and progressive neurological decline. Brain 2016, 139, 2420–2430. [Google Scholar] [CrossRef]
- Dyment, D.A.; Smith, A.C.; Humphreys, P.; Schwartzentruber, J.; Beaulieu, C.L.; Bulman, D.E.; Majewski, J.; Woulfe, J.; Michaud, J.; Boycott, K.M. Homozygous nonsense mutation in SYNJ1 associated with intractable epilepsy and tau pathology. Neurobiol. Aging 2015, 36, 1222.e1–1222.e5. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Klein, C.; Hicks, A.A. Role of Ceramides and Sphingolipids in Parkinson’s Disease. J. Mol. Biol. 2023, 435, 168000. [Google Scholar] [CrossRef]
- Jęśko, H.; Stępień, A.; Lukiw, W.J.; Strosznajder, R.P. The Cross-Talk Between Sphingolipids and Insulin-Like Growth Factor Signaling: Significance for Aging and Neurodegeneration. Mol. Neurobiol. 2019, 56, 3501–3521. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Oizumi, H.; Sugimura, Y.; Totsune, T.; Kawasaki, I.; Ohshiro, S.; Baba, T.; Kimpara, T.; Sakuma, H.; Hasegawa, T.; Kawahata, I.; et al. Plasma sphingolipid abnormalities in neurodegenerative diseases. PLoS ONE 2022, 17, e0279315. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, X.; Yang, L.; Li, M.; Wood, K.; Liu, Q.; Zhu, X. Neuroprotective effects of fingolimod in mouse models of Parkinson’s disease. FASEB J. 2017, 31, 172–179. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Spillantini, M.G. Physiological and pathological properties of α-synuclein. Cell. Mol. Life Sci. 2007, 64, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Suzuki, M.; Sango, K.; Wada, K.; Nagai, Y. Pathological role of lipid interaction with α-synuclein in Parkinson’s disease. Neurochem. Int. 2018, 119, 97–106. [Google Scholar] [CrossRef]
- Kiechle, M.; Grozdanov, V.; Danzer, K.M. The Role of Lipids in the Initiation of α-Synuclein Misfolding. Front. Cell Dev. Biol. 2020, 8, 562241. [Google Scholar] [CrossRef]
- Choong, C.-J.; Aguirre, C.; Kakuda, K.; Beck, G.; Nakanishi, H.; Kimura, Y.; Shimma, S.; Nabekura, K.; Hideshima, M.; Doi, J.; et al. Phosphatidylinositol-3,4,5-trisphosphate interacts with alpha-synuclein and initiates its aggregation and formation of Parkinson’s disease-related fibril polymorphism. Acta Neuropathol. 2023, 145, 573–595. [Google Scholar] [CrossRef] [PubMed]
- Pan, P.-Y.; Sheehan, P.; Wang, Q.; Zhu, X.; Zhang, Y.; Choi, I.; Li, X.; Saenz, J.; Zhu, J.; Wang, J.; et al. Synj1 haploinsufficiency causes dopamine neuron vulnerability and alpha-synuclein accumulation in mice. Hum. Mol. Genet. 2020, 29, 2300–2312. [Google Scholar] [CrossRef]
- Lööv, C.; Scherzer, C.R.; Hyman, B.T.; Breakefield, X.O.; Ingelsson, M. α-Synuclein in Extracellular Vesicles: Functional Implications and Diagnostic Opportunities. Cell Mol. Neurobiol. 2016, 36, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, C.; Cook, T.J.; Bullock, K.M.; Zhao, Y.; Ginghina, C.; Li, Y.; Aro, P.; Dator, R.; He, C.; et al. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson’s disease. Acta Neuropathol. 2014, 128, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Tian, Y.; Zhang, Z. Dysfunction of Synaptic Vesicle Endocytosis in Parkinson’s Disease. Front. Integr. Neurosci. 2021, 15, 619160. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef]
- Si, X.; Tian, J.; Chen, Y.; Yan, Y.; Pu, J.; Zhang, B. Central Nervous System-Derived Exosomal Alpha-Synuclein in Serum May Be a Biomarker in Parkinson’s Disease. Neuroscience 2019, 413, 308–316. [Google Scholar] [CrossRef]
- Shaheen, R.; Sebai, M.A.; Patel, N.; Ewida, N.; Kurdi, W.; Altweijri, I.; Sogaty, S.; Almardawi, E.; Seidahmed, M.Z.; Alnemri, A.; et al. The genetic landscape of familial congenital hydrocephalus. Ann. Neurol. 2017, 81, 890–897. [Google Scholar] [CrossRef]
- Finckh, U.; Schröder, J.; Ressler, B.; Veske, A.; Gal, A. Spectrum and detection rate of L1CAM mutations in isolated and familial cases with clinically suspected L1-disease. Am. J. Med. Genet. 2000, 92, 40–46. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Berg, D.; Hu, M.T.; Pilotto, A.; Borroni, B.; Davis, J.J.; Tofaris, G.K. Validation of α-Synuclein in L1CAM-Immunocaptured Exosomes as a Biomarker for the Stratification of Parkinsonian Syndromes. Mov. Disord. 2021, 36, 2663–2669. [Google Scholar] [CrossRef]
- Zou, J.; Guo, Y.; Wei, L.; Yu, F.; Yu, B.; Xu, A. Long Noncoding RNA POU3F3 and α-Synuclein in Plasma L1CAM Exosomes Combined with β-Glucocerebrosidase Activity: Potential Predictors of Parkinson’s Disease. Neurotherapeutics 2020, 17, 1104–1119. [Google Scholar] [CrossRef]
- Gracia, L.; Lora, G.; Blair, L.J.; Jinwal, U.K. Therapeutic Potential of the Hsp90/Cdc37 Interaction in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1263. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Trotter, J.H.; Abisambra, J.F.; Koren, J.; Lawson, L.Y.; Vestal, G.D.; O’Leary, J.C.; Johnson, A.G.; Jin, Y.; Jones, J.R.; et al. The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J. Biol. Chem. 2011, 286, 16976–16983. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Tinggi, U. Selenium: Its role as antioxidant in human health. Environ. Health Prev. Med. 2008, 13, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Çokal, B.G.; Yurtdaş, M.; Güler, S.K.; Güneş, H.N.; Uçar, C.A.; Aytaç, B.; Durak, Z.E.; Yoldaş, T.K.; Durak, I.; Çubukçu, H.C. Serum glutathione peroxidase, xanthine oxidase, and superoxide dismutase activities and malondialdehyde levels in patients with Parkinson’s disease. Neurol. Sci. 2017, 38, 425–431. [Google Scholar] [CrossRef]
- Sharma, G.; Shin, E.-J.; Sharma, N.; Nah, S.-Y.; Mai, H.N.; Nguyen, B.T.; Jeong, J.H.; Lei, X.G.; Kim, H.-C. Glutathione peroxidase-1 and neuromodulation: Novel potentials of an old enzyme. Food Chem. Toxicol. 2021, 148, 111945. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, E.; Brooke, S.; Chang, P.; Sapolsky, R. Over-expression of antioxidant enzymes protects cultured hippocampal and cortical neurons from necrotic insults. J. Neurochem. 2003, 87, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Mueller, G.; Lancelot, E.; Bogdanov, M.; Andersen, J.K.; Jiang, D.; Beal, M.F. Mice Deficient in Cellular Glutathione Peroxidase Show Increased Vulnerability to Malonate, 3-Nitropropionic Acid, and 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyridine. J. Neurosci. 2000, 20, 1–7. [Google Scholar] [CrossRef]
- SStockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Wu, J.R.; Tuo, Q.Z.; Lei, P. Ferroptosis, a Recent Defined Form of Critical Cell Death in Neurological Disorders. J. Mol. Neurosci. 2018, 66, 197–206. [Google Scholar] [CrossRef] [PubMed]
- McMackin, M.Z.; Durbin-Johnson, B.; Napierala, M.; Napierala, J.S.; Ruiz, L.; Napoli, E.; Perlman, S.; Giulivi, C.; Cortopassi, G.A. Potential biomarker identification for Friedreich’s ataxia using overlapping gene expression patterns in patient cells and mouse dorsal root ganglion. PLoS ONE 2019, 14, e0223209. [Google Scholar] [CrossRef] [PubMed]
- Sivagurunathan, N.; Gnanasekaran, P.; Calivarathan, L. Mitochondrial Toxicant-Induced Neuronal Apoptosis in Parkinson’s Disease: What We Know so Far. Degener. Neurol. Neuromuscul. Dis. 2023, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Mills, E.; Dong, X.-P.; Wang, F.; Xu, H. Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Med. Chem. 2010, 2, 51–64. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef]
- Guan, X.; Bai, X.; Zhou, C.; Guo, T.; Wu, J.; Gu, L.; Gao, T.; Wang, X.; Wei, H.; Zhang, Y.; et al. Serum Ceruloplasmin Depletion is Associated With Magnetic Resonance Evidence of Widespread Accumulation of Brain Iron in Parkinson’s Disease. J. Magn. Reson. Imaging 2021, 54, 1098–1106. [Google Scholar] [CrossRef]
- Olivieri, S.; Conti, A.; Iannaccone, S.; Cannistraci, C.V.; Campanella, A.; Barbariga, M.; Codazzi, F.; Pelizzoni, I.; Magnani, G.; Pesca, M.; et al. Ceruloplasmin oxidation, a feature of Parkinson’s disease CSF, inhibits ferroxidase activity and promotes cellular iron retention. J. Neurosci. 2011, 31, 18568–18577. [Google Scholar] [CrossRef]
- Barbariga, M.; Curnis, F.; Andolfo, A.; Zanardi, A.; Lazzaro, M.; Conti, A.; Magnani, G.; Volontè, M.A.; Ferrari, L.; Comi, G.; et al. Ceruloplasmin functional changes in Parkinson’s disease-cerebrospinal fluid. Mol. Neurodegener. 2015, 10, 59. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Ching-López, A.; Martinez-Gonzalez, L.J.; Arrabal, L.; Sáiz, J.; Gavilán, Á.; Barbas, C.; Lorente, J.A.; Roldán, S.; Sánchez, M.J.; Gutierrez-Ríos, P. Combined Genome, Transcriptome and Metabolome Analysis in the Diagnosis of Childhood Cerebellar Ataxia. Int. J. Mol. Sci. 2021, 22, 2990. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).